

Abstract Abstract
Pulmonary hypertension is recognized as a leading cause of morbidity and mortality in patients with sickle cell disease (SCD). We now report benchtop phenotyping from the explanted lungs of the first successful lung transplant in SCD. Pulmonary artery smooth muscle cells (PASMCs) cultured from the explanted lungs were analyzed for proliferate capacity, superoxide (O2•−) production, and changes in key pulmonary arterial hypertension (PAH)–associated molecules and compared with non-PAH PASMCs. Upregulation of several pathologic processes persisted in culture in SCD lung PASMCs in spite of cell passage. SCD lung PASMCs showed growth factor– and serum-independent proliferation, upregulation of matrix genes, and increased O2•− production compared with control cells. Histologic analysis of SCD-associated PAH arteries demonstrated increased and ectopically located extracellular matrix deposition and degradation of elastin fibers. Biomechanical analysis of these vessels confirmed increased arterial stiffening and loss of elasticity. Functional analysis of distal fifth-order pulmonary arteries from these lungs demonstrated increased vasoconstriction to an α1-adrenergic receptor agonist and concurrent loss of both endothelial-dependent and endothelial-independent vasodilation compared with normal pulmonary arteries. This is the first study to evaluate the molecular, cellular, functional, and mechanical changes in end-stage SCD-associated PAH.
Keywords: sickle cell disease, pulmonary arterial hypertension, lung transplant, CD47, thrombospondin 1, endothelin 1, superoxide, matrix
Introduction
Sickle cell disease (SCD) is an autosomal recessive hemoglobinopathy that is characterized by chronic hemolytic anemia and cycles of microvascular vaso-occlusion and that results in a diffuse and progressive vasculopathy.1 The complications of SCD are wide ranging and include acute chest syndrome,2 vaso-occlusive crisis, organ failure,3 bone infarction,4 and stroke.5 Pulmonary arterial hypertension (PAH) is a common chronic pulmonary complication of SCD in autopsy studies6,7 and has a prevalence ranging from 6% to 11% when measured by the gold standard, right heart catheterization.8-11 Elevated tricuspid regurgitant jet velocity (TRV), an echocardiographic surrogate of pulmonary artery systolic pressure elevation, is associated with increased mortality in SCD,12,13 and in a recent series where patients with elevated TRV had confirmatory right heart catheterization, pulmonary hypertension resulted in a 37% 6-year mortality rate.10 Treatment of SCD-associated PAH is limited, and attempts at expanding accepted PAH treatments to this subset of patients has led to unexpected complications.14
PAH is epidemiologically linked to a high baseline rate of intravascular hemolysis in SCD.15 Preclinical and human vascular studies have confirmed that hemolytic anemia leads to vasculopathy through decreased NO bioavailability due to NO scavenging by cell-free plasma hemoglobin, increased arginase 1 levels in plasma, and increased xanthine oxidase–mediated superoxide production.16-18 Similar to patients with SCD, sickle cell mice with high baseline hemolysis have diminished responses to NO and NO donors and develop PAH as they age.19 Preclinical studies also suggest that an increased inflammatory response, cell adhesion,20,21 and pathologic reactive oxygen species (ROS) production22 contribute to SCD-associated vasculopathy. Interestingly, the proadhesive protein thrombospondin 1 (TSP1) was recently reported to inhibit NO signaling23,24 while promoting PAH in preclinical models25 and to be upregulated in SCD patient plasma.26
Although studies have documented the hemodynamics and outcomes of SCD-associated PAH and interrogated the systemic vascular function of these patients, there have been no human studies directly evaluating the pulmonary vasculature. We herein describe ex vivo benchtop profiling that includes molecular, cellular, mechanical, genetic, and functional signatures of the explanted SCD-associated PAH lungs from a patient who underwent successful lung transplantation. Although personalized, this multifaceted analysis provides insights into the pathophysiology of this disease.
Material and methods
Reagents
TSP1 was purchased from Athens Research (Athens, GA). CD47 antibody (clone B6H12) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). TSP1 (clone 6.1), endothelin 1 (ET-1) (polyclonal, Ab117757), 3-nitrotyrosine (3NT, clone 39B6), and endothelin receptor A (ETA; polyclonal ab117521) antibodies were purchased from Abcam (Cambridge, MA).
Subject characteristics
The analysis conducted herein was based on human tissue samples, including lung parenchymal biopsies, pulmonary arteries, and pulmonary artery smooth muscle cells (PASMCs) from a group of control patients (n = 3) without PAH or overt lung disease, patients with PAH (n = 4), and 1 patient with SCD-associated PAH. The clinical characteristics of these subjects were as follows: controls, 52-year-old female and 38-year-old and 49-year-old males; SCD-associated PAH, 19-year-old male; and non-SCD-associated PAH, 66-year-old female and 69-year-old, 64-year-old, and 25-year-old males. All PAH patients (SCD associated and not SCD associated) had mean pulmonary arterial pressures >25 mmHg. In cell experiments, PASMCs were harvested from pulmonary arterial biopsy samples from SCD-associated lungs and from a control lung.
Primary PASMC cultures
The freshly harvested proximal (main) pulmonary artery from an explanted control lung and a SCD-associated PAH lung (1 control and 1 SCD) were rinsed with growth medium, and under sterile conditions the endothelial lining was mechanically removed. Arteries were then placed luminal side down in 6-well culture plates (Nalgene Nunc; Sigma-Aldrich, St. Louis, MO). Medium 231 with smooth muscle growth supplements (Invitrogen, Grand Island, NY) and penicillin/streptomycin were added to wells. Cultures were maintained through addition of medium daily until cells were observed growing from the tissue segments. Tissue segments were removed and cells were allowed to reach surface saturation, at which point the cells were trypsinized and plated in a T25 flask (Nalgene Nunc) and maintained with Medium 231 with smooth muscle cell growth supplements (SMGS; Invitrogen) and penicillin/streptomycin. Cell culture experiments were performed at 80% surface saturation and weaned over 48 hours from serum and growth factors. Cells were treated in basal medium lacking serum and growth factors and containing 0.1% bovine serum albumin (BSA). In some experiments, cells were challenged with hypoxia (1% FiO2) or normoxia (21% FiO2) for 3 or 24 hours with or without a CD47 antagonist antibody or were cocultured with exogenous TSP1 (2.2 nM).
Human tissue
Freshly explanted control non-PAH and end-stage PAH lungs were obtained under an ongoing University of Pittsburgh Institutional Review Board protocol (970946). Under sterile conditions and employing magnification, the proximal pulmonary artery and distal fifth-order pulmonary arteries were dissected from the lung parenchyma using a minimal “touch” technique to prevent injury to the endothelial and smooth muscle cell layers.
Cell proliferation
PASMCs were obtained from the pulmonary artery from a SCD-associated PAH (n = 1) or a control non-PAH (n = 1) lung, cryopreserved at passage 4, and thawed and cultured in Medium 231 with SMGS. Additionally, control cells were purchased from Gibco and handled similarly. Cells were expanded, trypsinized, and plated onto a 96-well plate in triplicate, with 10,000 cells in 200 μL of medium per well. Cells were incubated at 37°C in 5% CO2 for 48 h under serum-replete or serum-starved conditions. At 48 hours, 90 μL of medium was removed from the top of each well, and 20 μL of CellTiter Blue reagent (Promega, Madison, WI) was added per well. Plates were read on an enzyme-linked immunosorbent assay plate reader at 560 nm/590 nm to quantify the degree of cell proliferation, as per the CellTiter Blue protocol. Experiments were performed twice in triplicate.
Histology and 2-photon second-harmonic-generation microscopy
Hematoxylin-eosin and Verhoeff’s staining were performed following the protocols of the Department of Pathology, University of Pittsburgh Medical Center. Immunofluorescent staining and imaging was performed at the Center for Biologic Imaging of the University of Pittsburgh. Tissue sections were snap-frozen and fixed in 2% paraformaldehyde; cryostat sections (5 μm) were cut and washed 3 times with phosphate-buffered saline (PBS), followed by 3 washes with a solution of 0.5% BSA in PBS. Sections were blocked with 2% normal goat serum in BSA solution for 30 minutes. The slides were incubated for 1 hour at room temperature with primary antibodies for TSP1 (1∶200; PA1-29196, ThermoFisher, Waltham, MA) combined with ET-1 (1∶250; MA3-005, Pierce, ThermoFisher) or CD47 (1∶100; SC-12730, Santa Cruz Biotechnology) combined with ETA (1∶100; ab117521, Abcam) in 0.5% BSA solution. Slides were washed 3 times with BSA solution and incubated for 1 hour at room temperature with CY3 goat anti-rabbit secondary antibody (Jackson Immuno Reasearch, West Grove, PA) diluted 1∶500 in BSA solution, in combination with a 1∶1,000 dilution of goat anti-mouse Alexa 488 (Life Technologies, Invitrogen). Nuclei were stained with Hoescht’s dye (1 mg of bisbenzamide/100 mL of water) for 30 seconds. After 3 rinses with PBS, sections were coverslipped with Gelvatol mounting medium. Fluorescent images were captured with an Olympus Fluoview 1000 confocal microscope (software ver. 1.7a). For 2-photon second-harmonic-generation microscopy, specimens were fixed in 10% buffered formalin and embedded into paraffin. Sections (5 μm) were then processed for modified Movat’s pentachrome histologic 5-color stain. Stained sections were scanned using the Aperio digital imaging system (Aperio ScanScope XT). To characterize collagen and elastin structure throughout the thickness of the pulmonary artery, we used 2-photon second-harmonic-generation microscopy. Images were obtained using Zen 2011 software (Carl Zeiss Microimaging) on a Zeiss LSM 510 META Axiovert 200M instrument with the following: Axiovert 200M, 800-nm laser, HQ575/250m-2p and HQ400/20m-2p filters, and C-Apochromat 40×/1.2 objective. Digital image processing was done using custom-written code in MatLab (MathWorks, Natick, MA).
Western immunoblot
Cells were rinsed with ice-cold PBS and then lysed at 4°C in buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mM ethylene glycol tetraacetic acid (EGTA) plus 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1× protease inhibitor cocktail (Sigma-Aldrich), and 1× phosphatase inhibitor cocktail (Roche Applied Science, Hercules, CA). Cell lysates were centrifuged at 17,000 g for 20 minutes. A DC assay (BioRad, Hercules, CA) was used to quantify total protein. Cell lysates mixed with sodium dodecyl sulfate sample buffer were boiled at 95°C for 5 minutes, electrophoretically separated on 7.5% polyacrylamide gel electrophoresis gels for ∼1 hour at 150 V, and transferred to nitrocellulose membrane (BioRad) for 2 h at 400 mA. Membranes were blocked in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) and incubated overnight at 4°C with primary antibodies. Appropriate secondary antibodies were added for 1 hour at room temperature, and membranes were visualized using an Odyssey CLx (LI-COR). The intensity of the bands was quantified using Odyssey software or ImageJ (http://rsbweb.nih.gov/ij/). All immunoblots were performed on 3 sequential cell passages.
RNA extraction and quantification by real-time polymerase chain reaction (PCR)
Total RNA was extracted from cells using Qiagen RNeasy Mini Kits (Qiagen, Hilden, Germany), as per the manufacturer’s instructions, with on-column DNaseI treatment. RNA was quantified using the Take3 Gen5 spectrophotometer (BioTek, Winooski, VT). One microgram of RNA was then reverse transcribed using Superscript III First Strand Synthesis SuperMix (Invitrogen). Complementary DNA was amplified using Platinum Quantitative PCR SuperMix-UDG (Invitrogen) in 10-μL volumes in triplicate with gene-specific primers using an ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). Thermal cycling conditions were 50°C for 2 minutes and 95°C for 2 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Data were analyzed using the 2−ΔΔCt method with expression normalized to the housekeeping gene. The following primer sequences for human TSP1 (Hs00962908_m1), CD47 (Hs00179953_m1), ET-1 (Hs00174961_m1), EDNRA (03988672_m1), collagen Iα1 (Hs00943809_m1), collagen IIIα1 (Hs00164004_m1), collagen IVα1 (Hs00266237_m1), and hypoxanthine phosphoribosyltransferase 1 (HPRT1; HS99999909_m1) were employed (Taqman, Applied Biosystems). All reverse-transcription PCR (RT-PCR) was performed on 3 independent samples from sequential cell passages, and results are expressed as mean 2−ΔΔCt ± standard deviation.
Biaxial stress analysis of proximal pulmonary arteries
Proximal pulmonary arterial segments (one from the SCD vessel and one from a control vessel) measuring 1.25 cm × 1.25 cm were labeled on the endothelial side with 4 black dots and mounted trampoline fashion on the biaxial testing system using thin threads, as described elsewhere.27 This allows for the tissue to move freely in both directions, circumferential and longitudinal. The strain was measured optically by software that keeps track of the relative distance between the black dots on the sample (optical markers). Two load cells record the force required to stretch the tissue in both directions. The biaxial testing system employs software to control the 4 motors in charge of pulling the tissue (stretching and stretching rate) by closely monitoring the signals coming from the 2 load cells. For each sample, the protocol involved 1 preload and 9 load testing sessions. The 9 load sessions were 0% load in the longitudinal direction–100% load in the circumferential direction (uniaxial testing), 25%-100%, 50%-100%, 75%-100%, 100%-100%, 100%-75%, 100%-50%, 100%-25%, and 100%-0% (uniaxial testing). The preload session was performed using a load slightly lower than the maximum calculated testing load (same load in both directions). Testing was performed at 25°C with the sample in PBS without calcium and magnesium. Data analysis was performed using proprietary software written in MatLab (MathWorks). Analysis details have been reported elsewhere.27 The analysis software accesses testing data from a proprietary database.
Cytochrome c analysis of superoxide (O2•−) production
PASMCs isolated from control (n = 1) or SCD-associated PAH (n = 1) lungs were grown to 80% confluence, serum starved overnight, and challenged with hypoxia (1% FiO2) or normoxia (21% FiO2) for 3 hours. Cells were then washed with ice-cold phosphate buffer (PBS), scraped in lysis buffer (8 mM potassium, sodium phosphate buffer [pH 7.0], 131 mM NaCl, 340 mM sucrose, 2 mM NaN3, 5 mM MgCl2, 1 mM EGTA, 1 mM ethylenediaminetetraacetic acid, and protease inhibitors), and lysed by 5 freeze/thaw cycles and 5 passes through a 30-gauge needle. The cell lysate was centrifuged at 1,000 g for 5 minutes at 4°C to remove any remaining cellular material. To obtain the membrane fraction, the lysates were spun at 28,000 g for 15 minutes at 4°C. The supernatants were removed, and the membrane fractions were suspended in lysis buffer. The protein concentration was measured by the Bradford method. Superoxide production in the membrane fractions (20 μg/mL) was measured in 100 μL of oxidase assay buffer (65 mM sodium phosphate buffer [pH 7.0], 1 mM EGTA, 10 μM flavin adenine dinucleotide [FAD], 1 mM MgCl2, 2 mM NaN3, and 0.2 mM cytochrome c). Superoxide production from 3 successive cell passages was initiated by the addition of 180 μM nicotinamide adenine dinucleotide phosphate (NADPH) and was calculated from the initial linear rate of superoxide dismutase (SOD)–inhibitable (150 U/mL) cytochrome c reduction quantified at 550 nm using an extinction coefficient of 21.1 mM−1 cm−1 (Biotek Synergy 4 Hybrid Multi-Mode Microplate Reader).
Genomewide RNA expression profiling and validation
RNA was isolated from previously frozen lung parenchyma tissue specimens from the SCD subject (n = 1) and normal control subjects (n = 10) using Qiazol reagent (Qiagen) and was purified using the miRNeasy Kit (Qiagen). Microarray experiments were performed as described elsewhere.28 In brief, RNA quality was assessed on an Agilent 2100 bioanalyzer, and the quantity was measured on an Agilent Nanodrop. Fifty nanograms of total RNA was Cy3 labeled using the Agilent One Color Low Input Quick Amp Labeling Kit, hybridized to an Agilent Whole Genome 8 × 60K microarray, and scanned using an Agilent DNA microarray scanner. Expression data were extracted and analyzed using Agilent Feature Extraction software. RT-PCR was performed as described elsewhere28 using an ABI Prism 7000 Sequence Detection System with an Express SYBR GreenER Kit with ROX (Invitrogen). Threshold cycles were then normalized to glyceraldehyde-3-phosphate dehydrogenase. Microarray data were analyzed using the methods described elsewhere.28 In brief, data were log2 transformed and normalized using cyclic loess and Agilent Feature Extraction, and they were expressed as the difference between the log of gProcessed signal and the log of the geometric mean of controls. Fold changes of individual genes in the SCD subject were calculated by dividing the processed normalized expression ratio of the probes by the mean of the respective probe across all control subjects. Quantitative PCR data were analyzed by analysis of variance (ANOVA) with post hoc Bonferroni correction, with P < 0.05 considered significant.
Arterial myography
Distal fifth-order pulmonary arteries were dissected from 3 control and 1 SCD-associated PAH lungs, mounted on a dual-pin myography apparatus (MultiMyograph Model 610M) in standard incubation buffer and maintained at 37°C and pH 7.4, gassed with 95% O2 and 5% CO2, and brought to an optimal resting tension by increasing force by 500 mg every 10–15 minutes to an end point of 2 g. Rings were allowed to stabilize for 1 hour, with the incubation buffer replaced every 20 minutes. Viability of vessels was confirmed by a contractile response to potassium chloride (100 mM KCl) until contractions reached plateau. Rings were then washed 3 times with buffer and allowed to stabilize to baseline. Phenylephrine (PE; Sigma-Aldrich) concentration-response curves (10−9 to 10−5 M) were generated by measuring contraction plateaus at each concentration. After vessel segments reached a stable plateau phase induced by a PE dose producing 80% maximum contraction (EC80), log dose concentrations of acetylcholine (Ach; 10−8 to 10−5 M; Sigma-Aldrich) or sodium nitroprusside (SNP; 10−9 to 10−6 M; Sigma-Aldrich) were then tested to assess endothelial-dependent and endothelial-independent vasodilation, respectively.
Statistics
Statistics were performed using GraphPad Prism software (GraphPad Software, La Jolla, CA). Data were analyzed by one-way ANOVA followed by the Tukey test for multiple comparisons. For grouped analysis, data were analyzed by two-way ANOVA followed by the Bonferroni post hoc test. A P value of <0.05 was assumed to be significant.
Results
SCD lungs have increased expression of PAH-promoting signal transduction pathways
Loss of vasodilator signaling, increased vasoconstrictor activity, and arterial smooth muscle cell hypertrophy/proliferation are appreciated in preclinical and human PAH.29 Dysregulation of signal transduction pathways is associated with this process, including ET-1.30,31 We assessed this pathway in lung parenchyma from control, SCD-associated, and non-SCD PAH lungs and found upregulation of ET-1 (Fig. 1A). PASMCs express ETA,30,32 and ETA was upregulated in PAH lungs compared with controls and further overexpressed in SCD (Fig. 1A). The secreted protein TSP1 and its receptor, CD47, have been reported to promote preclinical PAH,25 and both TSP1 and CD47 protein was significantly increased in PAH and SCD-associated PAH lungs (Fig. 1A).
Figure 1.

Sickle cell disease (SCD) lungs overexpress markers of pulmonary arterial hypertension (PAH). A, Western immunoblot of lysates from lung tissue were performed. Representative blots from 3 control (CTRL), 1 SCD-associated PAH, and 4 non-SCD PAH lungs are presented. Densitometry is presented as the mean ratio of total protein compared with β-actin ± standard deviation (SD). Single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL, and double asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL and SCD PAH. B, Pulmonary arterial smooth muscle cells from healthy CTRL and SCD pulmonary arteries were serum starved and treated in growth factor– and serum-free medium containing 0.1% bovine serum albumin with thrombospondin 1 (TSP1; 2.2 nmol/L) or hypoxia (1% O2) with or without a CD47 blocking antibody (Ab; clone B6H12, 1 μg/mL). Cell lysates were prepared and Western immunoblots preformed. Representative data from 3 separate experiments are shown. Densitometry is presented as the mean ratio of total protein to β-actin ± SD. For TSP1, single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL, and the asterisk with a pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL and SCD-normoxia (Nx); for CD47, single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL; for endothelin receptor A (ETA), single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, the pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-hypoxia (Hx), and the pound sign with a percentage sign indicates a statistically significant difference (P < 0.05) compared with SCD-Nx, SCD-TSP1, and SCD-Hx; and for endothelin 1 (ET-1), single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, the pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-Hx, and double asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL. C, Total RNA was extracted and reverse transcribed to complementary DNA, and reverse-transcription polymerase chain reaction was performed. Results were normalized to the housekeeping gene hypoxanthine phosphoribosyltransferase 1 and are presented as mean  ± SD (from 3 experiments). For TSP1 messenger RNA (mRNA), single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, double asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL, the single pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-TSP1 and CTRL-Hx, and the double pound sign indicates a statistically significant difference (P < 0.05) compared with SCD-Nx, SCD-TSP1, and SCD-Hx; for CD47 mRNA, asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx; for ET-1 mRNA, single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, and double asterisks indicate a statistically significant difference (P < 0.05) compared with SCD-Nx; and for EDNRA mRNA, the single asterisk indicates a statistically significant difference (P < 0.05) compared with CTRL-Nx, the pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-TSP1, and double asterisks indicate a statistically significant difference (P < 0.05) compared with SCD-Nx and SCD-TSP1.
± SD (from 3 experiments). For TSP1 messenger RNA (mRNA), single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, double asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL, the single pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-TSP1 and CTRL-Hx, and the double pound sign indicates a statistically significant difference (P < 0.05) compared with SCD-Nx, SCD-TSP1, and SCD-Hx; for CD47 mRNA, asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx; for ET-1 mRNA, single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, and double asterisks indicate a statistically significant difference (P < 0.05) compared with SCD-Nx; and for EDNRA mRNA, the single asterisk indicates a statistically significant difference (P < 0.05) compared with CTRL-Nx, the pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-TSP1, and double asterisks indicate a statistically significant difference (P < 0.05) compared with SCD-Nx and SCD-TSP1.
PAH-associated signal transduction mechanisms are induced in cultured PASMCs from SCD lungs
As PASMCs are target cells for PAH,33 we assessed signal transduction activity in cultures of PASMCs from control pulmonary arterial and SCD-associated PAH pulmonary arterial vessels under normoxic and hypoxic conditions. PASMCs from SCD lung vessels displayed induction of the ET-1–ETA and the TSP1-CD47 signaling axis under normoxia compared with control PASMCs, and this was further increased by hypoxia (1% O2, 24 hours; Fig. 1B). Expression levels of PAH markers demonstrated persistent elevation even at high passage (≥7). Treating PASMCs with exogenous TSP1 (2.2 nmol/L, 24 hours) increased ET-1, ETA, and CD47 protein expression. Conversely, a CD47 antibody (clone B6H12, 1 μg/mL) that blocks TSP1 binding34 inhibited expression of TSP1, ET-1, and ETA in SCD-associated PASMCs (Fig. 1B). Paralleling upregulation of PAH-associated signaling markers, SCD PASMCs displayed increased messenger RNA (mRNA) levels of TSP1 but decreased CD47, ET-1, and EDNRA (ETA) transcript compared with control cells (Fig. 1C). Likewise, treatment of PASMCs with the CD47 antibody inhibited the hypoxia-mediated increase in TSP1 and EDNRA.
SCD-mediated PAH promotes vasculopathy in pulmonary arteries
As a result of PAH, the pulmonary vasculature undergoes progressive alterations in structure.35,36 Standard hematoxylin-eosin (Fig. 2A, right) and Verhoeff’s (Fig. 2A, left) staining of lung tissue sections prepared from explanted SCD-associated PAH lungs displayed marked thickening of the pulmonary arteries and excessive collagen.
Figure 2.
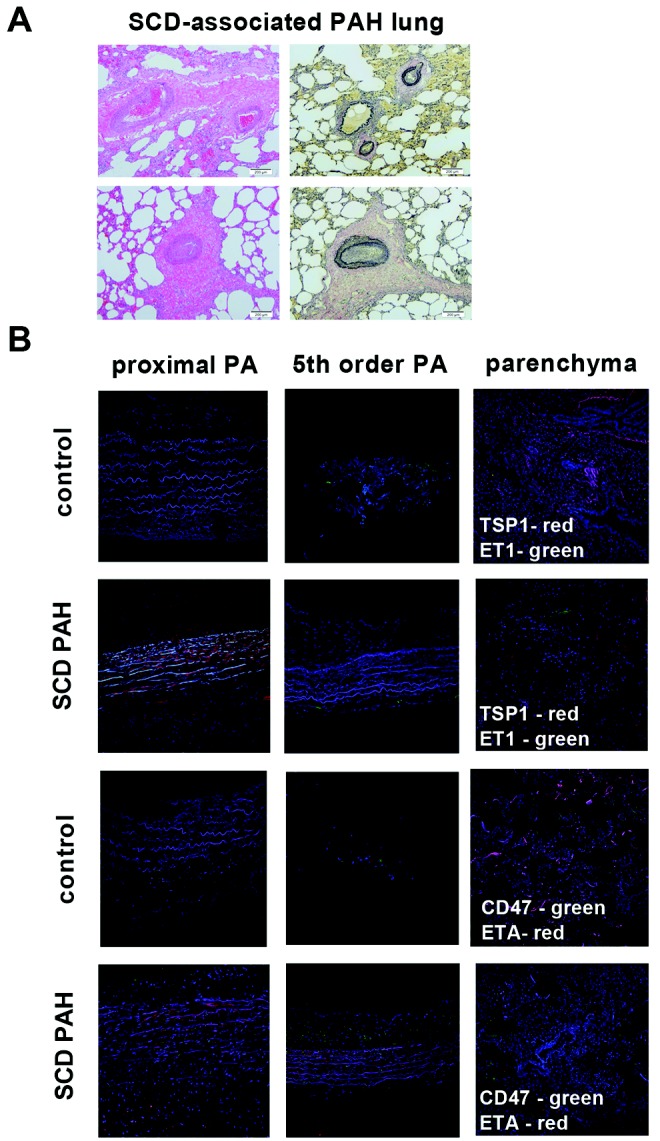
Sickle cell disease (SCD)–associated pulmonary arterial hypertension (PAH) lungs display general vasculopathy and upregulation of markers of PAH. A, Light microscopic examination of hematoxylin-eosin (right) and Verhoeff’s (left) stain of tissue sections of SCD-associated PAH whole lung demonstrated marked arterial thickening, inflammation, and hypertrophic collagen. In sections stained with Verhoeff’s, elastic fibers are black, collagen fibers are pink/red, and cell cytoplasm is yellow. Magnification, ×10; scale bars = 200 μM. B, Representative immunofluorescent cross sections of proximal pulmonary arteries, fifth-order pulmonary arteries, and peripheral parenchyma from control non-PAH and SCD-associated PAH lungs. The top two rows display thrombospondin 1 (TSP1; red) and endothelin 1 (ET-1; green), and the bottom two rows display CD47 (green) and endothelin receptor A (ETA; red). Magnification, ×20. PA: pulmonary arteries.
PAH-promoting pathways are dysregulated in both proximal and distal pulmonary arterial vessels from SCD-associated PAH lungs
Experiments employing control and SCD-associated PASMCs demonstrated induction of several PAH-promoting signaling axes. Using immunofluorescent histology, we investigated the anatomic expression patterns of these signaling axes in proximal and distal pulmonary arteries and parenchyma from SCD-associated PAH and control lungs. All anatomic sites, including proximal and distal pulmonary arteries and parenchyma from SCD-associated PAH lungs, displayed greater immunoreactive TSP1, ET-1, and ETA, whereas these proteins were less detectable in tissue sections from control lungs (Fig. 2B). Immunoreactive CD47 was expressed in control and diseased tissue sections. Especially interesting was the marked expression of TSP1 in the vascular smooth muscle cell layer of both proximal (main) pulmonary arterial segments and distal fifth-order pulmonary arteries. These results are prescient, as we and others have shown that TSP1 is a stress-secreted protein37 and that TSP1−/− mice are protected from hypoxia-induced PAH.25,38 CD47 is expressed widely,39 and control tissue sections demonstrated a modest level of immunoreactive protein. However, paralleling TSP1 overexpression, CD47 was increased in SCD pulmonary arterial vessels (Fig. 2B).
SCD PAH is associated with abnormalities of the pulmonary arterial extracellular matrix
Changes in the quality and quantity of the extracellular matrix of the pulmonary vasculature, particularly in the pulmonary arteries, are reported in PAH.40 It is unknown whether such alterations occur in the vasculature of SCD-associated PAH lungs. Immunofluorescent staining of cross sections of proximal pulmonary arteries from control non-PAH (Fig. 3A, right) and end-stage SCD-associated PAH (Fig. 3A, left) lungs demonstrated substantial elastin clipping (a maker of fiber degradation; Fig. 3A, circle) and abundant ectopically located collagen (in orange) in both the intimal and the adventitial layers of the diseased vessel (Fig. 3A, arrows). Also appreciated is the increased thickness of all layers (intima, media, and adventitia), especially the neointima of the SCD-associated pulmonary arteries.
Figure 3.
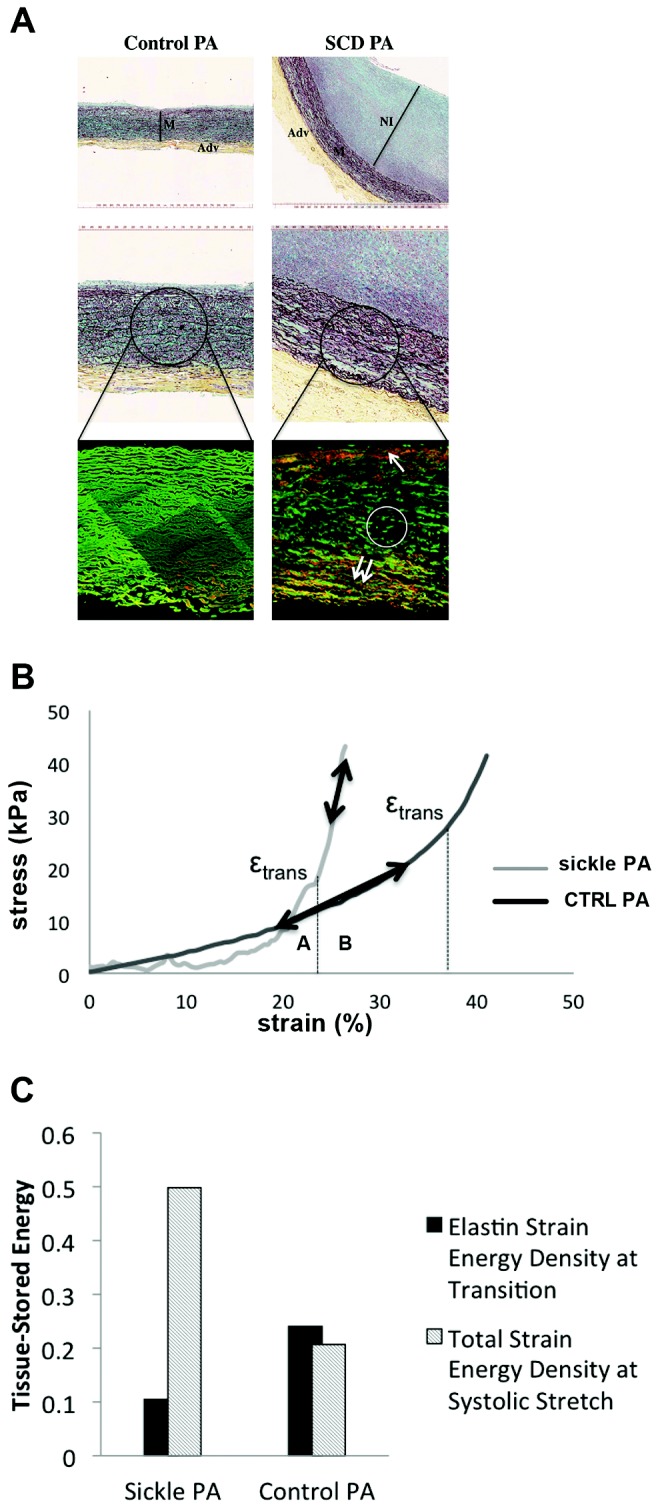
Vascular remodeling in sickle cell disease (SCD)–associated pulmonary arterial hypertension (PAH) contributes to abnormal biomechanics. A, Upper panels of control and SCD-associated pulmonary arterial tissue samples after Movat’s pentachrome staining demonstrating increased neointimal formation. Representative sections are presented. M: vascular media; Adv: vascular adventitia; NI: neointima. Lower panels show control and SCD-associated pulmonary arterial samples after 2-photon second-harmonic-generation microscopy, wherein elastin is shown in green and collagen is shown in red. The white circle depicts clipped medial elastin; the single arrow points to inner media collagen deposition, and the double-arrow points to outer media collagen deposition and highlights clipping and fragmentation of media elastin and accumulation of collagen in the inner and outer regions of the vascular media of the SCD sample. B, Stress-versus-strain behavior of fresh SCD-associated pulmonary arterial compared with non-PAH control (CTRL) pulmonary arterial segment. εtrans is the strain of transition from the elastin-dominant region (A) to the collagen-dominant region (B), associated with increased collagen engagement. C, Tissue-stored energy of fresh SCD-associated PAH pulmonary arterial segments compared with non-PAH control pulmonary arterial segments. PA: pulmonary arteries.
SCD-associated PAH vessels display abnormal mechanical properties
Studies suggest that histologic changes in the PAH vasculature correlate with changes in mechanical properties.27,41 We assessed the biaxial stress-strain profile of proximal pulmonary arterial segments from the diseased SCD-associated PAH lungs and non-PAH control lungs. It can be seen that the elastin-dominant region (region A to the left of the transition strain) shows almost no capability of handling load in the SCD pulmonary arteries. It can also be seen that in physiological conditions (region B) the SCD pulmonary arterial tissue operated strictly in the collagen-dominant region (Fig. 3B). Loss of elastin load-carrying capacity, early engagement of collagen, and collagen-mediated stiffening were appreciated in the SCD pulmonary arterial vessels. Additionally, very early collagen engagement in the circumferential direction was demonstrated in diseased SCD pulmonary arterial segments (λtrans ≈ 1.15 vs. 1.5 in diseased SCD pulmonary arteries vs. healthy vessels). This correlated with significantly greater stored energy in the SCD pulmonary arterial segment compared with the control pulmonary arteries (Fig. 3C).
SCD-associated PAH PASMCs produce more superoxide (O2•−) basally and in response to hypoxia
PAH, SCD associated or otherwise, is characterized by decreased bioavailability of nitric oxide (NO).42 NO interacts with its primary intracellular target, soluble guanylyl cyclase, to stimulate vascular smooth muscle cell relaxation and arterial vasodilation.43 NO also rapidly interacts with O2•− to generate the reactive nitrogen species peroxynitrite, limiting the bioavailability of NO and promoting vasoconstriction.44 Superoxide is produced by several enzymatic sources45-47 and contributes to SCD vasculopathy48,49 and to SCD PAH.19 SCD-associated PAH PASMCs produced significantly greater O2•−, as determined by cytochrome c assay under normoxia (21% FiO2) and after 3 hours of hypoxia (1% FiO2; Fig. 4A). Peroxynitrite can adversely alter protein function through nitration of key amino acid residues.50 Consistent with increased O2•− production by SCD PASMCs, levels of 3NT, a marker of protein nitration and reactive nitrogen species, were increased in SCD-associated PAH PASMCs compared with that in control cells (Fig. 4B). Interestingly, treating SCD PASMCs with a CD47 blocking antibody (clone B6H12, 1 μg/mL) inhibited hypoxia-mediated increases in 3NT.
Figure 4.

Sickle cell disease (SCD)–associated pulmonary arterial hypertension (PAH) pulmonary artery smooth muscle cells (PASMCs) produce more superoxide (O2•−) and display enhanced proliferation and increased matrix protein gene expression. A, PASMCs isolated from healthy control (CTRL) and SCD proximal pulmonary arteries were serum starved and treated in growth factor– and serum-free medium containing 0.1% bovine serum albumin (BSA) incubated in normoxia (Nx) or hypoxia (1% O2) for 3 hours. Superoxide production was measured in the 28,000 g membrane fraction and calculated from the initial linear rate of SOD-inhibitable cytochrome c reduction. Data represent the mean ratio of superoxide to total protein ± standard deviation (SD). Asterisks indicate a statistically significant difference (P < 0.05) compared with normoxic CTRL cells. B, PASMCs isolated from healthy CTRL and SCD proximal pulmonary arteries were serum starved and treated in growth factor– and serum-free medium containing 0.1% BSA with thrombospondin 1 (TSP1; 2.2 nmol/L) or hypoxia (1% O2) with or without CD47 blocking antibody (clone B6H12, 1 μg/mL) for 24 hours. Cell lysates were prepared and Western immunoblots performed. Representative data from 3 different experiments are shown. Densitometry is presented as the mean ratio of total protein to β-actin ± SD. Asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL, and the pound sign indicates a statistically significant difference (P < 0.05) compared with SCD-normoxia (Nx), SCD-TSP1, and SCD-hypoxia (Hx). C, PASMCs isolated from CTRL and SCD pulmonary arteries were plated at subconfluent density and grown in standard smooth muscle cell medium (Lonza) or under serum-free conditions, and proliferation was measured at 48 hours. Data are presented as mean ± SD (from 3 experiments). For starved, the single asterisk indicates a statistically significant difference (P < 0.05) compared with control 1, and the double asterisk indicates a statistically significant difference (P < 0.05) compared with control 1 and control 2; for replete, the triple asterisk indicates a statistically significant difference (P < 0.05) compared with control 1 and control 2. D, Cells from SCD or CTRL pulmonary arterial segments were treated as in A and collected in lysis buffer, RNA was extracted and reverse transcribed to complementary DNA, and reverse-transcription polymerase chain reaction was performed for the indicated collagen genes. Results were normalized to the housekeeping gene hypoxanthine phosphoribosyltransferase 1 and are presented as mean  ± SD (from 3 experiments). For collagen Iα1 (Col Iα1) messenger RNA (mRNA), single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, and double asterisk indicates a statistically significant difference (P < 0.05) compared with CTRL-TSP1; for collagen IIIα1 (Col IIIα1) mRNA, the single asterisk indicates a statistically significant difference (P < 0.05) compared with CTRL-Nx and CTRL-TSP1, the pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-Hx, and double asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, CTRL-TSP1, and CTRL-Hx; and for collagen IVα1 (Col IVα1) mRNA, single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx and CTRL-TSP1, the pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-Hx, and the double pound sign indicates a statistically significant difference (P < 0.05) compared with SCD-Nx and SCD-TSP1.
± SD (from 3 experiments). For collagen Iα1 (Col Iα1) messenger RNA (mRNA), single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, and double asterisk indicates a statistically significant difference (P < 0.05) compared with CTRL-TSP1; for collagen IIIα1 (Col IIIα1) mRNA, the single asterisk indicates a statistically significant difference (P < 0.05) compared with CTRL-Nx and CTRL-TSP1, the pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-Hx, and double asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx, CTRL-TSP1, and CTRL-Hx; and for collagen IVα1 (Col IVα1) mRNA, single asterisks indicate a statistically significant difference (P < 0.05) compared with CTRL-Nx and CTRL-TSP1, the pound sign indicates a statistically significant difference (P < 0.05) compared with CTRL-Hx, and the double pound sign indicates a statistically significant difference (P < 0.05) compared with SCD-Nx and SCD-TSP1.
SCD-associated PAH PASMCs display serum-independent proliferation
Overgrowth of PASMCs is found in PAH and promotes increased vascular resistance.51 In full growth medium, SCD-associated PASMCs proliferated faster than control PASMCs (Fig. 4C). Unexpectedly, the proliferative advantage of SCD PASMCs over control cells was accentuated when cells were cultured in the absence of serum.
Matrix gene expression is increased in SCD-associated PAH PASMCs
Immunohistologic analysis of pulmonary arterial segments from SCD lungs demonstrated overabundant and ectopic collagen deposition. We hypothesized that this may be secondary to upregulation of matrix genes. RT-PCR analysis of control and SCD-associated PAH PASMCs demonstrated increased mRNA levels of a number of matrix genes, including collagen Iα1, collagen IIIα1, and collagen IVα1, compared with normal PASMCs under normoxic and hypoxic conditions (Fig. 4D). Treatment with a CD47 blocking antibody (clone B6H12, 1 μg/mL) suppressed transcript levels of several matrix genes in control but not in SCD-associated PASMCs (Fig. 4D).
SCD-associated PAH vessels have abnormal vasoactive responses
Enhanced vasoconstriction and loss of vasodilation occurs in preclinical models of PAH and human disease.52,53 However, functional vasoactive responses of the distal pulmonary vasculature, in PAH in general and in SCD-associated PAH in particular, have not been previously investigated. Distal fifth-order pulmonary arterial vessels from normal and SCD-associated PAH lungs were gently harvested under microscopic control and immediately assayed using standard myographic technique. SCD-derived distal pulmonary arterial vessels showed increased vasoconstriction to the selective α1-adrenergic receptor agonist PE compared with normal pulmonary arterial vessels (Fig. 5A). Conversely distal pulmonary arterial segments from normal lungs displayed a normal dose-dependent sensitivity to the endothelial activating vasodilator Ach (Fig. 5B) and to the smooth muscle activator SNP (Fig. 5C). In contrast, distal pulmonary arterial vessels from SCD-associated PAH lungs were highly resistant to both Ach- and SNP-mediated vasodilation (Fig. 5B, 5C). In response to Ach, a classic activator of endothelial nitric oxide synthase, distal SCD-associated PAH vessels demonstrated vasoconstriction rather than vasodilation, suggesting a loss of endothelial cell capacity.
Figure 5.
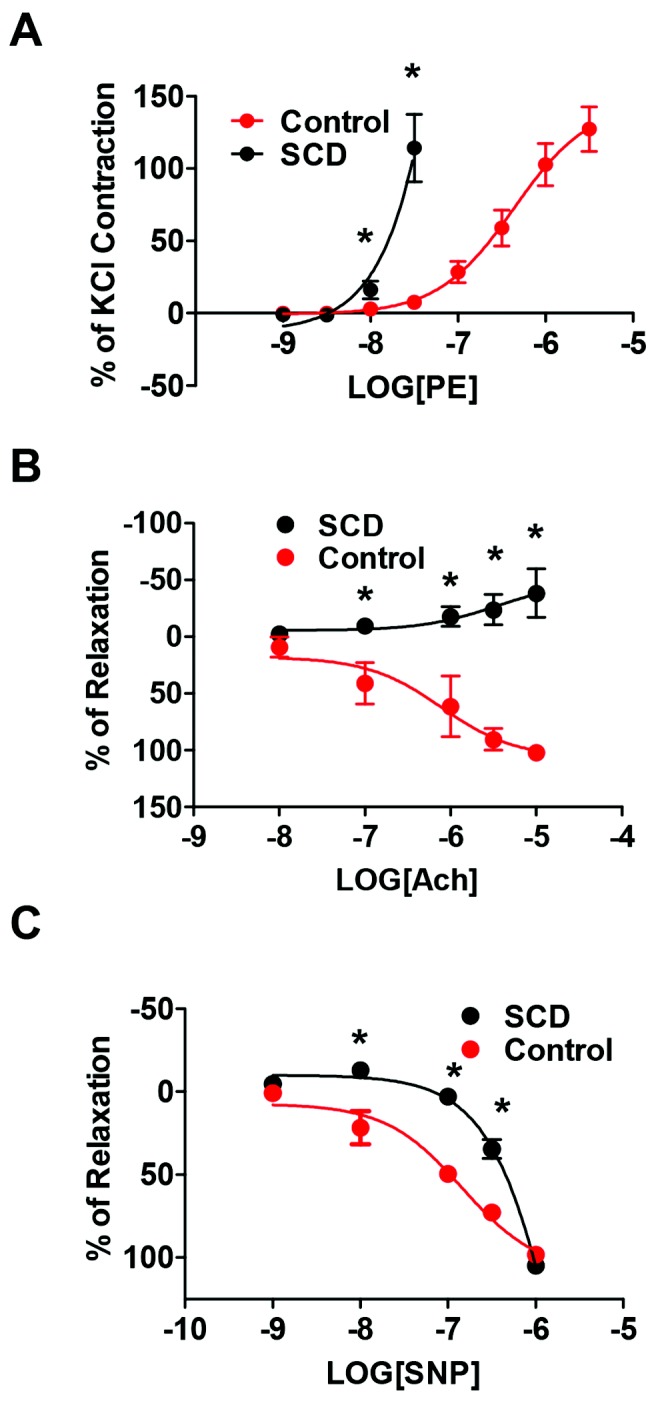
Distal fifth-order pulmonary arteries from end-stage sickle cell disease (SCD)–associated pulmonary arterial hypertension (PAH) lungs display increased vasoconstriction and loss of endothelial-dependent and endothelial-independent vasodilation. A, Vasoconstriction of control and SCD lung distal fifth-order pulmonary arteries was determined to a log dose of phenylephrine (PE) with results normalized to KCl response. Data are presented as mean ± standard deviation (SD; from 4 rings per treatment point). Asterisks indicate a statistically significant difference (P < 0.05) compared with control vessel. Control and SCD lung fifth-order pulmonary arteries were treated with a PE dose producing 80% maximum contraction (EC80) and then challenged with log dose concentrations of acetylcholine (Ach; B; 10−8 to 10−5 M; Sigma-Aldrich) or sodium nitroprusside (SNP; C; 10−9 to 10−6 M; Sigma-Aldrich), and vasodilation was determined. Data are presented as mean ± SD (from 4 rings at each treatment point). Asterisks indicate a statistically significant difference (P < 0.05) compared with control vessel.
SCD-associated PAH lung parenchyma shows gene expression changes relative to normal control tissue
Fold changes in SCD lung tissue (n = 1) relative to normal controls (n = 10) were calculated, and the top 10 upregulated and top 10 downregulated genes are reported in Tables 1 and 2, respectively. To confirm the technical validity of the microarray data, we selected 4 genes for RT-PCR quantification in technical replicates of the SCD lung tissue and normal control lung tissues, CLEC1B, DEFA4, GJA3, and SULT6B1. RT-PCR quantification of all 4 genes replicated the direction and magnitude of expression changes observed in the microarray analysis (Fig. 6A, 6B).
Table 1.
Upregulated genes in sickle cell disease (SCD)–associated pulmonary arterial hypertension (PAH) lungs versus control non-PAH lungs
| Gene | Description | Fold change (SCD vs. control) |
|---|---|---|
| SULT6B1 | Sulfotranferase family, cystolic, 6B, member 1 | 4.50 |
| OR5AN1 | Olfactory receptor, family 5, subfamily AN, member 1 | 3.90 |
| RBFOX1 | RNA binding protein, fox 1 homolog 1 | 3.23 |
| CXCL11 | Chemokine (C-X-C motif) ligand 11 | 3.04 |
| HAS2 | Hyaluronan synthase 2 | 3.02 |
| CT45A1 | Cancer/testis antigen family 45, member A1 | 3.02 |
| G6PC | Glucose-6-phosphate, catalytic subunit | 2.99 |
| ATXN3L | Ataxin 3–like | 2.95 |
| GJA3 | Gap junction protein, alpha 3 | 2.76 |
| AWAT2 | Acyl-CoA wax alcohol acyltranferase 2 | 2.61 |
Table 2.
Downregulated genes in sickle cell disease (SCD)–associated pulmonary arterial hypertension (PAH) lungs versus control non-PAH lungs
| Gene | Description | Fold change (SCD vs. control) |
|---|---|---|
| HRCT1 | Histidine-rich carboxyl terminus 1 | 0.31 |
| DEFA4 | Defensin, alpha 4, corticostatin | 0.32 |
| ADORA3 | Adenosine A3 receptor | 0.33 |
| ARPC5 | Actin-related protein 2/3 complex, subunit 5, 16 kDa | 0.36 |
| NTM | Neurotrimin | 0.36 |
| CHRM1 | Cholinergic receptor, muscarninic 1 | 0.37 |
| CLEC1B | C-type lectin domain family 1, member B | 0.39 |
| OLFM4 | Olfactomedin 4 | 0.41 |
| TNFRSF17 | Tumor necrosis factor receptor superfamily, member 17 | 0.42 |
| ZNF365 | Zinc finger protein 365 | 0.42 |
Figure 6.
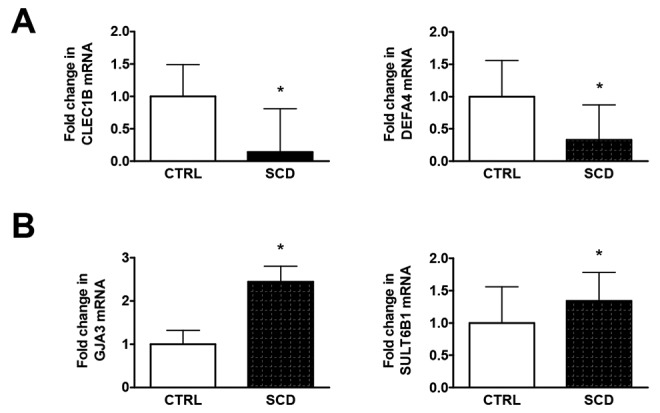
Reverse-transcription polymerase chain reaction validation of microarray data of changes in RNA expression in sickle cell disease (SCD) lung tissue of selected genes. Data represent the mean ± standard deviation of 5 technical replicates of the SCD lung tissue and 5 technical replicates each of 4 normal control lung tissue specimens. Asterisks indicate a statistically significant difference (P < 0.01) compared with control (CTRL).
Discussion
Characterization of plasma proteins54-56 and red blood cells57 in patients with SCD with and without PAH has found markers of inflammation, lipoproteins, and adhesion proteins increased and progenitor cells decreased. Several studies have documented the cardiopulmonary changes of this cohort.58-60 However, there is an absence of cellular- and tissue-specific data from SCD-associated PAH lungs. Our team has reported bilateral lung transplantation for the treatment of end-stage SCD-associated PAH.61 We present herein what is, to the best of our knowledge, the first multifaceted analysis of molecular, cellular, tissue, and vascular changes in SCD-associated PAH lungs.
Cell culture experiments with primary PASMCs harvested from proximal pulmonary arterial segments from SCD-associated PAH lungs found upregulation of genes known to promote PAH. In contrast to the known phenotypic drift induced by cell culture, passage of SCD PASMCs did not resolve the enhanced PAH pathway activation in these cells and suggests fixed autonomous changes. Endothelin markers have been linked to preclinical murine models of SCD and to human sickle cell red blood cells,62 and changes in plasma ET-1 levels are associated with clinical changes in disease.63,64 It was not known whether upregulation of endothelin in the blood extended to human SCD-associated PAH lungs and vascular cells from the same. Analysis of parenchyma, proximal and distal pulmonary arteries, and PASMCs from SCD-associated PAH lungs revealed significant upregulation of the ET-1–ETA signaling axis at multiple anatomic locations. The secreted protein TSP1 and its high-affinity receptor CD47 were also upregulated in PASMCs, vessels, and parenchyma from SCD lungs. Particularly impressive was the TSP1-CD47 induction in the mural compartment of proximal and distal fifth-order pulmonary arterial vessels. These data are novel and for the first time provide evidence of overexpression of the TSP1-CD47 signaling axis in the distal pulmonary vasculature in human PAH. These findings complement recent reports of increased lung parenchymal25 and plasma65 TSP1 expression in the setting of human PAH.
Extending immunofluorescent data, 2-photon microscopy confirmed extensive remodeling of the proximal pulmonary artery from SCD-associated PAH lungs with significant loss of elastin fiber integrity that, on mechanical testing, was associated with increased stiffness and complete loss of elastic capability. Specifically, at transition, the SCD pulmonary arterial segments had much less stored energy density in elastin because of fiber fracturing (nicking on 2-photon microscopy). Comparing the available energy storage to the control pulmonary arterial segments, the strain energy density of these tissues at peak systole was still primarily in elastin. Because the stiffer sickle pulmonary arterial segments stored a great deal more energy, it can be expected to require significantly more right ventricular work to deform the SCD vessels through the cardiac cycle. This, in turn, would be associated with higher stiffness afterload. Finally, the findings of collagen-based mechanics in the SCD pulmonary arteries correlated with results in human SCD PAMSC cultures that found gene induction of multiple collagen subtypes and extend reports of matrix induction in preclinical hypoxia-mediated PAH.66,67
Pathologic ROS, including O2•−, can increase vasoconstriction and limit vasodilation through several mechanisms.68 Control PASMCs demonstrated hypoxia-mediated increases in O2•− production. Conversely, SCD-associated PAH PASMCs displayed increased O2•− production under both normoxic and hypoxic conditions. Hyperactive O2•− production in SCD PASMCs may account, in part, for the decrease in both endogenous and exogenous NO response and the increase in PE sensitivity noted in SCD-associated distal pulmonary arteries. Together these data are intriguing given the identified roles TSP1 has in (1) limiting NO signaling in vascular cells,23,37 (2) directly activating NADPH oxidase to stimulate increased O2•− production in vascular smooth muscle cells,69 and (3) acutely inhibiting vasodilation70 to limit blood flow.71
As noted, cell autonomous upregulation of ET-1–ETA and TSP1-CD47 in SCD PASMCs did not resolve on extended cell passage. However, treating SCD PASMCs with a CD47 blocking antibody that prevents receptor activation by TSP1 minimized abnormal protein modifications (3NT protein levels were decreased) and corrected collagen IVα1 mRNA levels toward control while blocking the hypoxia-mediated increase in collagen IIIα1. These data in human SCD PASMCs are relevant in view of our finding that both TSP1−/− and CD47−/− mice show constitutive downregulation of pulmonary ET-1–ETA70 and that blocking CD47 activation in animals abrogates PAH.25 Though not directly tested, it is interesting to speculate whether the TSP1-CD47 axis promotes PAH, in part, through regulation of endothelin signaling.
Although caution must be exercised in interpreting the microarray expression data derived from a single SCD lung tissue, we were able to validate by RT-PCR the direction and magnitude of changes in expression of all 4 genes selected for individual quantification. In general, the genes expressing the greatest expression changes (Table 1) appear to be involved in cellular proliferation, extracellular matrix remodeling, metabolism, and signaling.
Limitations of the present study arise from the fact that some of the data are derived from a single source of lung tissue and are expected as this analysis was of tissue from the first ever lung transplant in a patient with SCD-associated PAH. Cognizant of this, care was exercised to reproduce results multiple times. Additionally, the results may be confounded by the use of PAH therapy in the patient prior to transplant. Finally, the patient did not have pure World Health Organization group 1 PAH in that, despite the presence of arterial remodeling consistent with idiopathic PAH, there was also evidence of pulmonary veno-oclusive disease and chronic thromboembolic disease. These limitations do not negate the interesting results of the study but rather temper generalization of the findings to PAH as a whole.
This work describes a detailed, comprehensive characterization of end-stage SCD-associated PAH lungs at the tissue, cellular, and genetic levels combined with unique ex vivo physiological and functional analyses of pulmonary arteries. Cellular, tissue, vascular, mechanical, and gene data confirmed global abnormalities, including (1) upregulated PAH markers, promoters, and genes; (2) aberrant and excessive matrix production; (3) pathologic ROS production; and (4) blunted vasodilation. Continued application of lung transplantation to patients with SCD-associated PAH will allow for validation of these results.
Source of Support: This work was supported by National Institutes of health (NIH) grant R01 HL-108954 and American Heart Association grant 11BGIA7210001 (to JSI); NIH grants P01 HL103455, U01 HL108642, and 3UL1RR024153 (to HCC); NIH grants U01 HL108642-01 and R03 HL095401 and Pittsburgh Foundation grant M2010-0055 (to FA); Pulmonary Hypertension Association Proof of Concept Grant (to FA, JSI); Parker B. Francis Foundation (to MPG); T32 Vascular Training Fellowship (to MSS); and AHA Postdoctoral Award 13POST14520003 (to NMR). This work was also supported by the Institute for Transfusion Medicine, the Hemophilia Center of Western Pennsylvania, and the Vascular Medicine Institute (to MPG, JSI, HCC, MTG).
Conflict of Interest: JSI is chair of the Scientific Advisory Boards of Vasculox (St. Louis, MO) and Radiation Control Technologies, Inc. (Rockville, MD).
References
- 1.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010;376:2018–2031. [DOI] [PubMed]
- 2.Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med 2000;342:1855–1865. [DOI] [PubMed]
- 3.Abbott KC, Hypolite IO, Agodoa LY. Sickle cell nephropathy at end-stage renal disease in the United States: patient characteristics and survival. Clin Nephrol 2002;58:9–15. [DOI] [PubMed]
- 4.Aguilar C, Vichinsky E, Neumayr L. Bone and joint disease in sickle cell disease. Hematol Oncol Clin North Am 2005;19:929–941, viii. [DOI] [PubMed]
- 5.Mehta SH, Adams RJ. Treatment and prevention of stroke in children with sickle cell disease. Curr Treat Options Neurol 2006;8:503–512. [DOI] [PubMed]
- 6.Graham JK, Mosunjac M, Hanzlick RL, Mosunjac M. Sickle cell lung disease and sudden death: a retrospective/prospective study of 21 autopsy cases and literature review. Am J Forensic Med Pathol 2007;28:168–172. [DOI] [PubMed]
- 7.Haque AK, Gokhale S, Rampy BA, Adegboyega P, Duarte A, Saldana MJ. Pulmonary hypertension in sickle cell hemoglobinopathy: a clinicopathologic study of 20 cases. Hum Pathol 2002;33:1037–1043. [DOI] [PubMed]
- 8.Fonseca GH, Souza R, Salemi VM, Jardim CV, Gualandro SF. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J 2012;39:112–118. [DOI] [PubMed]
- 9.Mehari A, Alam S, Tian X, et al. Hemodynamic predictors of mortality in adults with sickle cell disease. Am J Respir Crit Care Med 2013;187:840–847. [DOI] [PMC free article] [PubMed]
- 10.Mehari A, Gladwin MT, Tian X, Machado RF, Kato GJ. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA 2012;307:1254–1256. [DOI] [PMC free article] [PubMed]
- 11.Parent F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med 2011;365:44–53. [DOI] [PubMed]
- 12.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350:886–895. [DOI] [PubMed]
- 13.Ataga KI, Moore CG, Jones S, et al. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. Br J Haematol 2006;134:109–115. [DOI] [PubMed]
- 14.Machado RF, Barst RJ, Yovetich NA, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 2011;118:855–864. [DOI] [PMC free article] [PubMed]
- 15.Nouraie M, Lee JS, Zhang Y, et al. The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica 2013;98:464–472. [DOI] [PMC free article] [PubMed]
- 16.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med 2002;8:1383–1389. [DOI] [PubMed]
- 17.Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 2005;294:81–90. [DOI] [PMC free article] [PubMed]
- 18.Aslan M, Ryan TM, Adler B, et al. Oxygen radical inhibition of nitric oxide–dependent vascular function in sickle cell disease. Proc Natl Acad Sci USA 2001;98:15215–15220. [DOI] [PMC free article] [PubMed]
- 19.Hsu LL, Champion HC, Campbell-Lee SA, et al. Hemolysis in sickle cell mice causes pulmonary hypertension due to global impairment in nitric oxide bioavailability. Blood 2007;109:3088–3098. [DOI] [PMC free article] [PubMed]
- 20.De Franceschi L, Platt OS, Malpeli G, et al. Protective effects of phosphodiesterase-4 (PDE-4) inhibition in the early phase of pulmonary arterial hypertension in transgenic sickle cell mice. FASEB J 2008;22:1849–1860. [DOI] [PubMed]
- 21.Kaul DK, Nagel RL, Chen D, Tsai HM. Sickle erythrocyte-endothelial interactions in microcirculation: the role of von Willebrand factor and implications for vasoocclusion. Blood 1993;81:2429–2438. [PubMed]
- 22.Aslan M, Ryan TM, Townes TM, et al. Nitric oxide–dependent generation of reactive species in sickle cell disease: actin tyrosine induces defective cytoskeletal polymerization. J Biol Chem 2003;278:4194–4204. [DOI] [PubMed]
- 23.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci USA 2005;102:13141–13146. [DOI] [PMC free article] [PubMed]
- 24.Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD. CD47 is necessary for inhibition of nitric oxide–stimulated vascular cell responses by thrombospondin-1. J Biol Chem 2006;281:26069–26080. [DOI] [PubMed]
- 25.Bauer PM, Bauer EM, Rogers NM, et al. Activated CD47 promotes pulmonary arterial hypertension through targeting caveolin-1. Cardiovasc Res 2012;93:682–693. [DOI] [PMC free article] [PubMed]
- 26.Novelli EM, Kato GJ, Ragni MV, et al. Plasma thrombospondin-1 is increased during acute sickle cell vaso-occlusive events and associated with acute chest syndrome, hydroxyurea therapy, and lower hemolytic rates. Am J Hematol 2012;87:326–330. [DOI] [PMC free article] [PubMed]
- 27.Lammers SR, Kao PH, Qi HJ, et al. Changes in the structure-function relationship of elastin and its impact on the proximal pulmonary arterial mechanics of hypertensive calves. Am J Physiol Heart Circ Physiol 2008;295:H1451–H1459. [DOI] [PMC free article] [PubMed]
- 28.Rajkumar R, Konishi K, Richards TJ, et al. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol 2010;298:H1235–H1248. [DOI] [PMC free article] [PubMed]
- 29.Papierniak ES, Lowenthal DT, Mubarak K. Pulmonary arterial hypertension: classification and therapy with a focus on prostaglandin analogs. Am J Ther 2012;19:300–314. [DOI] [PubMed]
- 30.Galiè N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res 2004;61:227–237. [DOI] [PubMed]
- 31.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 2012;122:4306–4313. [DOI] [PMC free article] [PubMed]
- 32.Yu J, Taylor L, Wilson J, Comhair S, Erzurum S, Polgar P. Altered expression and signal transduction of endothelin-1 receptors in heritable and idiopathic pulmonary arterial hypertension. J Cell Physiol 2013;228:322–329. [DOI] [PMC free article] [PubMed]
- 33.Humbert M, Montani D, Perros F, Dorfmüller P, Adnot S, Eddahibi S. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vascul Pharmacol 2008;49:113–118. [DOI] [PubMed]
- 34.Isenberg JS, Annis DS, Pendrak ML, et al. Differential interactions of thrombospondin-1, -2, and -4 with CD47 and effects on cGMP signaling and ischemic injury responses. J Biol Chem 2009;284:1116–1125. [DOI] [PMC free article] [PubMed]
- 35.Dorfmüller P, Humbert M, Capron F, Muller KM. Pathology and aspects of pathogenesis in pulmonary arterial hypertension. Sarcoidosis Vasc Diffuse Lung Dis 2003;20:9–19. [PubMed]
- 36.Griffith SL, Rhoades RA, Packer CS. Pulmonary arterial smooth muscle contractility in hypoxia-induced pulmonary hypertension. J Appl Physiol 1994;77:406–414. [DOI] [PubMed]
- 37.Isenberg JS, Frazier WA, Roberts DD. Thrombospondin-1: a physiological regulator of nitric oxide signaling. Cell Mol Life Sci 2008;65:728–742. [DOI] [PMC free article] [PubMed]
- 38.Ochoa CD, Yu L, Al-Ansari E, Hales CA, Quinn DA. Thrombospondin-1 null mice are resistant to hypoxia-induced pulmonary hypertension. J Cardiothorac Surg 2010;5:32. [DOI] [PMC free article] [PubMed]
- 39.Soto-Pantoja DR, Stein EV, Rogers NM, Sharifi-Sanjani M, Isenberg JS, Roberts DD. Therapeutic opportunities for targeting the ubiquitous cell surface receptor CD47. Expert Opin Ther Targets 2013;17:89–103. [DOI] [PMC free article] [PubMed]
- 40.Medhora M, Bousamra M 2nd, Zhu D, Somberg L, Jacobs ER. Upregulation of collagens detected by gene array in a model of flow-induced pulmonary vascular remodeling. Am J Physiol Heart Circ Physiol 2002;282:H414–H422. [DOI] [PubMed]
- 41.Coflesky JT, Jones RC, Reid LM, Evans JN. Mechanical properties and structure of isolated pulmonary arteries remodeled by chronic hyperoxia. Am Rev Respir Dis 1987;136:388–394. [DOI] [PubMed]
- 42.Zuckerbraun BS, George P, Gladwin MT. Nitrite in pulmonary arterial hypertension: therapeutic avenues in the setting of dysregulated arginine/nitric oxide synthase signalling. Cardiovasc Res 2011;89:542–552. [DOI] [PMC free article] [PubMed]
- 43.Ignarro LJ. Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J Physiol Pharmacol 2002;53:503–514. [PubMed]
- 44.Squadrito GL, Pryor WA. The formation of peroxynitrite in vivo from nitric oxide and superoxide. Chem Biol Interact 1995;96:203–206. [DOI] [PubMed]
- 45.Drose S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol 2012;748:145–169. [DOI] [PubMed]
- 46.Katsuyama M, Matsuno K, Yabe-Nishimura C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J Clin Biochem Nutr 2012;50:9–22. [DOI] [PMC free article] [PubMed]
- 47.Vasquez-Vivar J, Kalyanaraman B, Martasek P. The role of tetrahydrobiopterin in superoxide generation from eNOS: enzymology and physiological implications. Free Radic Res 2003;37:121–127. [DOI] [PubMed]
- 48.Dias-Da-Motta P, Arruda VR, Muscara MN, et al. The release of nitric oxide and superoxide anion by neutrophils and mononuclear cells from patients with sickle cell anaemia. Br J Haematol 1996;93:333–340. [DOI] [PubMed]
- 49.Almeida CB, Franco-Penteado C, Saad ST, Costa FF, Conran N. Sickle cell disease serum induces NADPH enzyme subunit expression and oxidant production in leukocytes. Hematology 2010;15:422–429. [DOI] [PubMed]
- 50.Greenacre SA, Ischiropoulos H. Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic Res 2001;34:541–581. [DOI] [PubMed]
- 51.Morrell NW, Adnot S, Archer SL, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 2009;54:S20–S31. [DOI] [PMC free article] [PubMed]
- 52.Crosswhite P, Sun Z. Nitric oxide, oxidative stress and inflammation in pulmonary arterial hypertension. J Hypertens 2010;28:201–212. [DOI] [PMC free article] [PubMed]
- 53.Grunig E, Dehnert C, Mereles D, et al. Enhanced hypoxic pulmonary vasoconstriction in families of adults or children with idiopathic pulmonary arterial hypertension. Chest 2005;128:630S–633S. [DOI] [PubMed]
- 54.Niu X, Nouraie M, Campbell A, et al. Angiogenic and inflammatory markers of cardiopulmonary changes in children and adolescents with sickle cell disease. PloS ONE 2009;4:e7956. [DOI] [PMC free article] [PubMed]
- 55.Mestre B, Pitie M, Loup C, Claparols C, Pratviel G, Meunier B. Influence of the nature of the porphyrin ligand on the nuclease activity of metalloporphyrin-oligonucleotide conjugates designed with cationic, hydrophobic or anionic metalloporphyrins. Nucleic Acids Res 1997;25:1022–1027. [DOI] [PMC free article] [PubMed]
- 56.Klings ES, Anton Bland D, Rosenman D, et al. Pulmonary arterial hypertension and left-sided heart disease in sickle cell disease: clinical characteristics and association with soluble adhesion molecule expression. Am J Hematol 2008;83:547–553. [DOI] [PubMed]
- 57.Anjum F, Lazar J, Zein J, Jamaleddine G, Demetis S, Wadgaonkar R. Characterization of altered patterns of endothelial progenitor cells in sickle cell disease related pulmonary arterial hypertension. Pulm Circ 2012;2:54–60. [DOI] [PMC free article] [PubMed]
- 58.Linguraru MG, Pura JA, Van Uitert RL, et al. Segmentation and quantification of pulmonary artery for noninvasive CT assessment of sickle cell secondary pulmonary hypertension. Med Phys 2010;37:1522–1532. [DOI] [PMC free article] [PubMed]
- 59.Sachdev V, Kato GJ, Gibbs JS, et al. Echocardiographic markers of elevated pulmonary pressure and left ventricular diastolic dysfunction are associated with exercise intolerance in adults and adolescents with homozygous sickle cell anemia in the United States and United Kingdom. Circulation 2011;124:1452–1460. [DOI] [PMC free article] [PubMed]
- 60.Anthi A, Machado RF, Jison ML, et al. Hemodynamic and functional assessment of patients with sickle cell disease and pulmonary hypertension. Am J Respir Crit Care Med 2007;175:1272–1279. [DOI] [PMC free article] [PubMed]
- 61.George MP, Novelli EM, Shigemura N, et al. First successful lung transplantation for sickle cell disease with severe pulmonary arterial hypertension and pulmonary veno-occlusive disease. Pulm Circ 2013;3:952–958 (in this issue). [DOI] [PMC free article] [PubMed]
- 62.Phelan M, Perrine SP, Brauer M, Faller DV. Sickle erythrocytes, after sickling, regulate the expression of the endothelin-1 gene and protein in human endothelial cells in culture. J Clin Invest 1995;96:1145–1151. [DOI] [PMC free article] [PubMed]
- 63.Rybicki AC, Benjamin LJ. Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood 1998;92:2594–2596. [PubMed]
- 64.Ergul S, Brunson CY, Hutchinson J, et al. Vasoactive factors in sickle cell disease: in vitro evidence for endothelin-1-mediated vasoconstriction. Am J Hematol 2004;76:245–251. [DOI] [PubMed]
- 65.Frantz C. Role of thrombospondin-1 in pulmonary hypertension: a proof of concept [dissertation]. Universität des Saarlandes, 2011.
- 66.Estrada KD, Chesler NC. Collagen-related gene and protein expression changes in the lung in response to chronic hypoxia. Biomech Model Mechanobiol 2009;8:263–272. [DOI] [PMC free article] [PubMed]
- 67.Berg JT, Breen EC, Fu Z, Mathieu-Costello O, West JB. Alveolar hypoxia increases gene expression of extracellular matrix proteins and platelet-derived growth factor–B in lung parenchyma. Am J Respir Crit Care Med 1998;158:1920–1928. [DOI] [PubMed]
- 68.Fuchs B, Sommer N, Dietrich A, et al. Redox signaling and reactive oxygen species in hypoxic pulmonary vasoconstriction. Respir Physiol Neurobiol 2010;174:282–291. [DOI] [PubMed]
- 69.Csanyi G, Yao M, Rodriguez AI, et al. Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1. Arterioscler Thromb Vasc Biol 2012;32:2966–2973. [DOI] [PMC free article] [PubMed]
- 70.Bauer EM, Qin Y, Miller TW, et al. Thrombospondin-1 supports blood pressure by limiting eNOS activation and endothelial-dependent vasorelaxation. Cardiovasc Res 2010;88:471–481. [DOI] [PMC free article] [PubMed]
- 71.Isenberg JS, Hyodo F, Matsumoto K, et al. Thrombospondin-1 limits ischemic tissue survival by inhibiting nitric oxide–mediated vascular smooth muscle relaxation. Blood 2007;109:1945–1952. [DOI] [PMC free article] [PubMed]