

Abstract
Olfactory tumors, especially olfactory neuroblastomas (ON) and carcinomas with neuroendocrine differentiation (CND), are extremely rare, and little descriptive epidemiologic information is available. The objective of this study was to more fully describe selected olfactory tumors using a large population-based cancer incidence database. The Surveillance, Epidemiology and End Results (SEER) 9 registries limited-use data were reviewed from 1973 to 2006 for selected nasal cavity (C30.0) and accessory sinus (C31.0–31.9) tumors. Frequencies, incidence rates, and relative survival rates were estimated using SEER*Stat, v6.5.2. The majority of cases were squamous cell carcinoma (SCC), while the incidence of ON was greater than CND. For ON, the incidence was highest in the 60–79 year age group, while for SCC, the incidence was highest in the 80+ year age group. For CND, the incidence leveled off in the oldest age groups. Survival rates were highest for ON (>70% alive at 5 years after diagnosis) and poorest for CND (44% alive at 5 years). Adjuvant radiation therapy did not improve survival over surgery alone in ON. In SCC, survival was worse in patients who received adjuvant radiation compared to patients who had surgery alone. Our analysis confirms some previously published information, and adds new information about the incidence and demographics of ON and CND. In addition, our analysis documents the lack of benefit of adjuvant radiation in ON. It is not feasible to conduct prospective trials in patients with these rare diseases, and the importance of registry data in learning about olfactory tumors is emphasized.
Keywords: Epidemiology, Incidence, Survival, Olfactory, Neuroendocrine tumor, Esthioneuroblastoma
Introduction
Site classification of neoplasms of the olfactory area primarily places these tumors in the nasal cavity, but tumors are also seen in the adjacent paranasal sinuses: frontal, sphenoid, maxillary, and the ethmoid complex [1]. The olfactory area has direct extension to the CNS through the thin-walled cribiform plate. The area is bordered by rigid structures such as the hard palate and maxillary ostia, but also contains delicate structures such as the nasal turbinates and the aforementioned cribiform plate.
Olfactory area tumors represent a small fraction of head and neck malignancies, 0.2–0.8% [2], but a challenging diversity of rare histologies arises in this compact region. These include squamous cell carcinoma (encompassing nasopharyngeal carcinoma), neuroendocrine carcinoma, lymphoma, and olfactory neuroblastoma (i.e. esthioneuroblastoma) [1, 3, 4]. Olfactory area tumors also include other, less common, tumors such as those of the soft tissue, bone and cartilage, germ cell, and neuroectodermal (e.g. Ewing sarcoma and primitive neuroectodermal tumor [PNET]) tumors [1, 5].
The clinical presentation of olfactory area tumors is variable, ranging from sinonasal obstructive symptoms to vision impairment and CNS manifestations such as seizures [1, 4, 5]. These uncommon tumors often require a specialized surgical team (otolaryngologist, neurosurgeon, oral maxillofacial surgeon, and ophthalmologist), a tumor specialized medical or neuro-oncology team, and a radiation oncologist for optimal treatment [3, 4, 6–8].
Given the rarity of these tumors, there have been limited descriptive epidemiologic investigations. Much of the epidemiological information in the literature is based on single institution experiences or surgical cases [3, 9–11]. Only one population-based study on survival and prognostic factors for those with olfactory neuroblastoma has recently been published [12]. We used the SEER 9 registries limited-use data to describe population-based incidence and survival patterns for two selected neural based olfactory tumors: olfactory neuroblastoma (ON) and carcinomas with neuroendocrine differentiation (CND) and used the more common squamous cell carcinoma (SCC) as a basis for comparison.
Methods
The study population included data on all primary malignant tumors of the nasal cavity (ICD-O-3 site code C30.0) [13] and accessory sinuses (ICD-O-3 site codes C31.0– 31.9) from the Surveillance, Epidemiology, and End Results (SEER 9 registries limited-use dataset, April 2009) for cases diagnosed from 1973 to 2006. The SEER program is sponsored by the National Cancer Institute and includes, beginning in 1973, population-based incidence and survival data on all primary malignant cancers, and beginning in 2004, data on all primary brain tumors, uncertain and benign behavior. Five states (CT, HI, IA, NM, UT) and four population-based metropolitan areas (Atlanta, Detroit, San Francisco––Oakland, Seattle––Puget Sound) are included, representing approximately 10% of the U.S. population. Cases diagnosed at autopsy were excluded from the analyses.
The identified cases were grouped into three histologic sub-categories: ON (ICD-O-3 histology codes: 9364, 9473, 9500, 9520–9523), CND (ICD-O-3 histology codes: 8041, 8240, 8246), and SCC (ICD-O-3 histology codes: 8070– 8072). Age groups were defined to examine incidence patterns in the following way: children (0–19 years), and adults in 20 year age groupings (20–39, 40–59, 60–79, 80+ years). Relative survival rate estimates were evaluated for adults aged 20–64 and 65+ years. Cases with histologies not included in one of the three defined histologic sub-categories were excluded from the analyses (n = 2,925).
Frequencies, incidence rates, and relative survival rates were estimated using the SEER*Stat 6.5.2 software (Surveillance Research Program, 2008). Incidence rates were expressed per 100,000 and were age-adjusted to the 2000 US standard population. Frequencies and incidence rates were suppressed for cells with less than five cases. Relative survival was defined as the observed probability of survival adjusted for the expected survival rate of the US population for that age, sex, and race group. Survival time was calculated from the date of diagnosis to the date of death or last contact. Cases determined to be alive were censored at the date of last contact. Survival rates were not estimated for categories with fewer than 10 cases. SEER*Stat version 6.5.2 was utilized to obtain relative survival rates in 1-month intervals for a period of up to 10 years using the life-table method to properly account for right censoring. The Kaplan–Meier product-limit method was used to estimate observed survival in 1-month intervals for a period of up to 10 years. Survival curve comparisons were made using the Z-test procedure in SEER*Stat version 6.5.2.
Information about initial treatment was available only for surgery and radiation therapy. Chemotherapy, hormonal and immunotherapy treatment variables were not available for analyses. The variable for radiation therapy was categorized as “yes” (those patients who received any form of radiation therapy) or “no” (patients who received no radiation therapy). The variable for surgery was defined as “yes” (patients receiving any type of definitive or cancer-directed surgery) or “no” (patients who did not undergo definitive or cancer-directed surgery or who received exploratory or biopsy surgery only).
In an attempt to approximate the original Kadish staging system16, olfactory “stage” was created based on SEER Historic Stage classifications modified using the following extent of disease (EOD) variables: extension, lymph nodes, and metastases. Because of the number of changes the EOD classification system has undergone, we restricted the analyses to cases diagnosed between 1988 and 2006, thereby limiting analyses to the latest two EOD versions. In our study, Stage A included tumors categorized as “localized” using SEER Historic Stage. Stage B included SEER Historic Stage “regional” tumors without lymph node involvement or metastases that also had disease extension to adjacent connective tissue within the nasoethomoidal complex (code; 40). Stage C included tumors without lymph node involvement or metastases categorized as “regional” with disease extension codes not equal to 40. Because of the small number of cases with ON in Stage C, Stages B and C were combined. Stage D included all tumors categorized as “distant” using SEER Historic Stage or tumors that had lymph node involvement or metastases, regardless of the SEER Historic Stage classification. Unstaged tumors were those classified as unstaged using the SEER Historic Stage variable and were not included in the analyses. Due to the small number of subjects with CND, survival by stage could not be assessed.
Results
In the SEER 9 registries limited-use data set from 1973 to 2006, a total of 5,974 tumors were identified that were located in the nasal cavity or accessory sinuses. Of these tumors, 2,498 (41.8%) were classified as SCC, 353 (5.9%) as ON, and 78 (1.3%) as CND. The average annual age-adjusted incidence rate of CND (0.010; 95% CI: 0.008–0.013) was significantly lower than the rate for ON (0.046; 95% CI: 0.042–0.051), and the incidence rates of CND and ON were both significantly lower than that of SCC (0.340; 95% CI: 0.327–0.354; Table 1). Greater numbers of males than females were diagnosed within each of the three histology subtypes. Although the average annual age-adjusted rates were also observed to be higher for males than females, the gender difference was only statistically significant for SCC. Individuals categorized as other than white or black race were observed to have the highest average annual age-adjusted incidence rate although the rate comparisons with white and black race were not statistically significant.
Table 1.
Distribution and average annual age-adjusted incidencea of selected malignant olfactory tumors [olfactory neuroblastoma (ON), carcinoma with neuroendocrine differentiation (CND), and squamous cell carcinoma (SCC)] by gender and race; Surveillance, Epidemiology, and End Results (SEER) Program, 1973–2006b
| ON N (%) | AAR (CI) | CND N (%) | AAR (CI) | SCC N (%) | AAR (CI) | |
|---|---|---|---|---|---|---|
| Total | 353 (100) | 0.046 (0.042–0.051) | 78 (100) | 0.010 (0.008–0.013) | 2,498 (100) | 0.340 (0.327–0.354) |
| Gender | ||||||
| Male | 191 (54.1) | 0.054 (0.046–0.062) | 48 (61.5) | 0.014 (0.010–0.019) | 1,583 (63.4) | 0.491 (0.467–0.516) |
| Female | 162 (45.9) | 0.040 (0.034–0.047) | 30 (38.5) | 0.007 (0.005–0.011) | 915 (36.6) | 0.223 (0.208–0.238) |
| Race | ||||||
| White | 293 (83.0) | 0.047 (0.042–0.052) | 61 (78.2) | 0.010 (0.008–0.013) | 2,049 (82.0) | 0.330 (0.316–0.345) |
| Black | 20 (5.7) | 0.027 (0.016–0.042) | 8 (10.3) | 0.012 (0.005–0.023) | 215 (8.6) | 0.369 (0.320–0.423) |
| Other | 38 (10.8) | 0.058 (0.041–0.080) | 9 (11.5) | 0.014 (0.006–0.026) | 220 (8.8) | 0.391 (0.340–0.448) |
Average annual age-adjusted rates (AAR) are per 100,000 and are age-adjusted to the 2000 U.S. standard million population. CI––95% Confidence Interval
Source: Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence SEER 9 Regs Limited-Use, Nov 2008 Sub (1973–2006) <Katrina/Rita Population Adjustment>––Linked to County Attributes––Total US, 1969–2006 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Cancer Statistics Branch, released April 2009, based on the November 2008 submission
The incidence rates for ON tumors peaked in the 60–79 year age group, while the incidence for SCC continued to increase through the oldest age group (Fig. 1). The incidence of CND tumors leveled off in the oldest age groups. For both the CND and SCC histologies, very few children (0–19 years) were diagnosed. Median age at diagnosis was 51 years for ON, 56 years for CND, and 66 years for SCC.
Fig. 1.

Average annual age-specific incidence rates of selected malignant olfactory tumors; SEER 9 registries data 1973-2006. a olfactory neuroblastoma (ON), and carcinoma with neuroendocrine differentiation (CND), b squamous cell carcinoma (SCC) [* For SCC and CND, the numbers of cases in the 00–19 year age group were too small to report.] Rates (AAR) are per 100,000 and are age-adjusted to the 2000 US standard million population
Among the three olfactory histology groupings, survival was observed to be best for cases diagnosed with ON (Fig. 2). Two-, five-, and ten-year relative survival estimates for ON were 82% (95% CI: 76.6–86.1%), 72% (95% CI: 65.8–77.8%), and 62% (95% CI: 54.3–69.5%), respectively. For CND, five- and ten-year relative survival estimates were lowest at 44% (95% CI: 29.8–57.9%), and 39% (23.3–54.0%), respectively, while five- and ten-year relative survival estimates for SCC were intermediate (53% (95% CI: 50.0–55.3%), and 45% (95% CI: 42.1–48.3%), respectively). Females with ON were observed to have slightly better survival than males (Fig. 2a) although these differences were not statistically significant (P = 0.15). Males with SCC had higher relative survival than females (P < 0.05). Patients diagnosed between the ages of 20–64 had much better survival 10 years after diagnosis than those aged 65 years and older for those with ON (P; 0.059) and SCC (P < 0.0001) (Fig. 2b).
Fig. 2.

Relative survival of selected malignant olfactory tumors [olfactory neuroblastoma (ON), carcinoma with neuroendocrine differentiation (CND), and squamous cell carcinoma (SCC)] by a gender and b age at diagnosis groups; SEER 9 Registries data 1973–2006
Information on initial treatment with surgery and radiation was available for ON and SCC. Numbers were too small to report by subgroup for CND. Those who received surgery had significantly better survival than those who did not have surgery or had a biopsy only for both ON (P < 0.001) and SCC (P < 0.0001) (Fig. 3a). For patients with ON, there were no survival differences between those who did or did not have radiation as part of initial therapy (P = 0.62) (Fig. 3b), and the use of adjuvant radiation following surgery did not provide any additional survival advantage (P = 0.79) (Fig. 3c). In contrast, patients with SCC who underwent radiation therapy following surgery had significantly worse survival than those who had surgery alone (P < 0.0001). However, cases undergoing radiation therapy following surgery were predominantly stage C (43%) or D (20%). In comparison, the majority of cases having surgery alone were stage A (63%).
Fig. 3.

Relative survival of selected malignant olfactory tumors [olfactory neuroblastoma (ON) and squamous cell carcinoma (SCC)] by a site-directed surgery status, b radiation therapy (RT) status and c for SCC and ON tumors with site-directed surgery, relative survival for radiation therapy (RT) status; SEER 9 Registries data 1973–2006
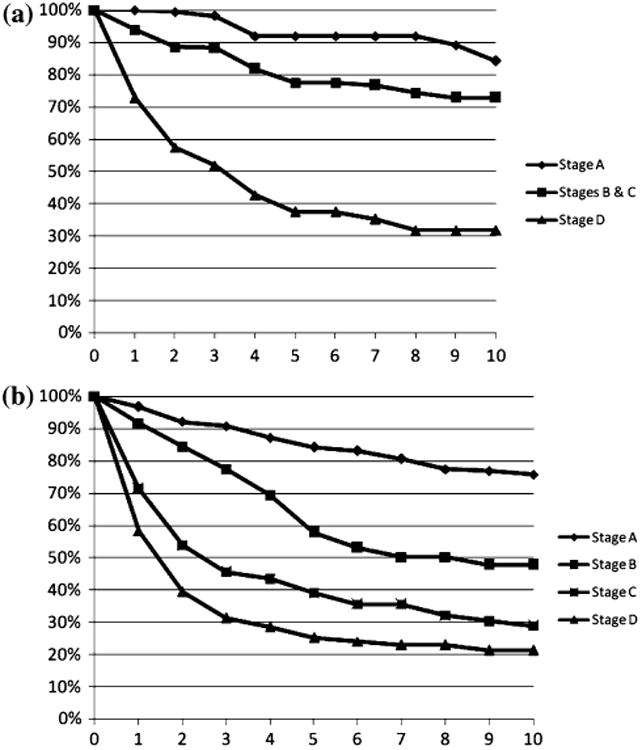
Two hundred thirty-six cases with ON and 1,153 cases with SCC were diagnosed between 1988 and 2006 and were available for staging. Eight cases with ON and 98 cases with SCC were unstaged and were excluded from the analyses. For ON cases, those with Stage D had significantly poorer survival than those with Stage A (P < 0.001), or Stages B and C (P < 0.001) (Fig. 4a). Those with Stage A tumors had survival rates that were marginally better than the survival rates observed for cases with Stage B and C tumors (P = 0.06). For SCC cases, those with Stage A had significantly better survival compared to those with stages B (P < 0001), C (P < 0001), and D (P < 0001) (Fig. 4b). Those with Stage D had significantly poorer survival than those with Stage B tumors (P < 0001) and also when compared to survival for those with Stage C tumors (P < 0.01).
Fig. 4. Relative survival a olfactory neuroblastoma (ON) and b squamous cell carcinoma (SCC) by modified SEER Historic Stage classification; SEER 9 Registries data 1988–2006.
Discussion
We provide a population-based evaluation of the epidemiology and outcome data for selected olfactory area tumors. Our analysis confirms that the majority of cases in the nasal cavity area are SCC. This tumor has been postulated to be highly influenced by environmental risk factors (such as tobacco and alcohol use) [14]. As expected, males had a higher incidence rate of SCC, and the incidence rate of SCC continued to rise through the oldest age groups. The race group defined as ‘other than white or black’ had a higher incidence of SCC than white or black populations in the US, although this was not statistically significant. A subtype of SCC, nasopharyngeal carcinoma, has a higher incidence rate in Asian and in North African countries than in the US [15, 16]. These various factors support an environmental role in SCC tumorigenesis.
The incidence of ON was greater than that of CND. Increased incidence rates of ON and CND were present in males as compared to females, but the differences in incidence rates were not statistically significant or as prominent as the difference in SCC. Similar to SCC, a higher incidence in the other than white or black racial group was observed for ON and CND, although the difference was not statistically significant. We do not know of a similar racial or geographical association for ON and CND as has been found for SCC. The median age at diagnosis in ON and CND is nearly a decade younger than that in SCC. In addition, the incidence rate plateaus or declines with advancing age for ON and CND as compared to SCC where the incidence continues to rise through the oldest ages.
As expected, survival rates are the highest in ON, and poorest for CND, relative to SCC. Both ON and SCC are associated with a greater than 50% 5 year survival. This may reflect inherent tumor susceptibility to treatment, less aggressive behavior, or the fact that these diseases have restrictive pathologic diagnosis criteria, including primarily well behaved or very differentiated tumors [5]. On the other hand, carcinomas with neuroendocrine differentiation can have varying degrees of differentiation, including a poorly differentiated subtype that is likely to be treatment resistant [17, 18].
Most notable in our study is the apparent lack of any additional benefit of radiation following surgery in patients with ON. There is general agreement about the role of surgery as initial treatment for this tumor. While adjuvant radiation is often used, its role seems less well established. Chamberlain described the use of chemotherapy in six patients with ON having CNS metastases [19]. All patients had originally been treated with curative intent with surgery followed by radiation. Similarly, in a meta-analysis of 26 studies including 390 patients, Dulguerov et al., reported that the most frequently used (44% of patients) treatment strategy was the combination of surgery and radiation, and that the combination was associated with statistically significantly better survival than with radiation alone, although survival in the combination group was not significantly different than in those receiving surgery alone [4]. In an analysis of the SEER data, Jethanamest et al. reported that among 274 cases of ON for whom treatment data were available 62% received combination therapy, 26.7% received surgery alone and 9.5% received radiation alone [12]. The group receiving combination therapy had the longest mean survival; however, the difference in mean survival was only significant for those receiving combination therapy compared to those receiving radiation alone. Dulguerov et al. reviewed 220 cases of carcinoma of the nasal cavity and paranasal sinuses at two institutions over a 20-year period and found that survival was better in patients treated with surgery and radiation or surgery alone, compared to those treated with radiation alone [3]. The analysis did not include the 42 cases of ON that were identified. Porter et al. recently described twelve patients from a single institution who underwent surgery for advanced (Stage C) ON [11]. Although the median survival of patients who received adjuvant chemotherapy was 83 months, and for patients not receiving chemotherapy 78 months, the small number of patients precludes drawing any conclusions. Regardless, the authors suggested that adjuvant radiation prolongs the time to relapse and that chemotherapy (cisplatin and etoposide) may add an additional benefit. Treatment with chemotherapy was not available for analysis in our review, and as noted above, we did not find any improvement in survival with radiation over surgery alone.
Another unexpected finding is that patients with SCC fared worse with receiving radiation therapy. Our analysis demonstrates that more patients with advanced stages were included in this group, and therefore it is likely that RT does provide a benefit in this poor prognostic group. The alternative is RT provides no benefit or causes harm, but this goes against the established benefit of RT in squamous histology of head and neck neoplasms [20, 21]. In a recent SEER study of advanced stage head and neck SCC treated with RT as primary local treatment modality, which had over 6,000 patients, 5-year overall survival curves were similar to the RT treated SCC overall survival curves in this study [21].
There are limitations to our study, foremost of which is the lack of detailed clinical data (such as other co-morbid conditions and more detailed treatment and extent of disease information) that may impact survival estimates. It is possible that there may be variation in diagnosis, coding, and reporting practices among the different regions in the US. Classification of histology can be problematic, especially for tumors in the olfactory area. The effect of numerous histologic categories and a lack of consensus regarding diagnostic criteria for certain subtypes have also been noted and may result in misclassification [5, 17, 22, 23].
Despite these limitations, registry data remain a very useful mechanism for studying rare tumors such as ON and CND. Combined, these tumors accounted for only about 7% of tumors of the nasal cavity or accessory sinuses identified in the database for a period of 30+ years. Case series at single institutions underscore the fact that these diseases are very uncommon, and that prospective studies to evaluate treatment are not feasible [3, 10]. The case series do provide more detailed information about treatment, including treatment for recurrent disease, and treatment with chemotherapy. However, the numbers of cases included are too small to draw any conclusions. The SEER data are population-based, and therefore, represent all cases of olfactory tumors regardless of the manner of treatment. The Jethanamest et al. SEER analysis was restricted to 274 cases of ON for whom treatment data were available [12] while our analysis of the SEER data includes data on 353 cases of ON.
In summary, our analysis supports previously published information, suggests a lack of benefit of adjuvant radiation in ON, and adds new information about the incidence and demographics of ON and CND. In addition, our analysis highlights the importance of registry data for learning about these rare diseases.
Acknowledgments
Sources of Support: This study was conducted under contract to the Central Brain Tumor Registry of the United States, which received funding from American Brain Tumor Association, National Brain Tumor Foundation, Pediatric Brain Tumor Foundation and the Division of Cancer Control and Population Sciences, National Cancer Institute, National Institutes of Health, Department of Health and Human Services, under contract #HHSN26100800766P.
Abbreviations
- CND
Carcinomas with neuroendocrine differentiation
- CNS
Central nervous system
- ICD-O-3
International Classification of Diseases for Ocology, Third Edition
- ON
Olfactory neuroblastoma
- SCC
Squamous cell carcinoma
- SEER
Surveillance, Epidemiology, and End Results Program
Contributor Information
J. Lee Villano, Department of Medicine, The University of Illinois at Chicago, Chicago, IL, USA.
Linda Bressler, Department of Pharmacy Practice, The University of Illinois at Chicago, Chicago, IL, USA.
Jennifer M. Propp, Department of Epidemiology/Biostatistics, School of Public Health, University of Illinois at Chicago, 1603 W. Taylor St. (M/C 923), Chicago, IL 60612, USA; Central Brain Tumor Registry of the United States, Chicago, IL, USA
Tibor Valyi-Nagy, Department of Pathology, The University of Illinois at Chicago, Chicago, IL, USA.
Iman K. Martin, Department of Epidemiology/Biostatistics, School of Public Health, University of Illinois at Chicago, 1603 W. Taylor St. (M/C 923), Chicago, IL 60612, USA
Therese A. Dolecek, Department of Epidemiology/Biostatistics, School of Public Health, University of Illinois at Chicago, 1603 W. Taylor St. (M/C 923), Chicago, IL 60612, USA; Central Brain Tumor Registry of the United States, Chicago, IL, USA
Bridget J. McCarthy, Email: bjm3@uic.edu, Department of Epidemiology/Biostatistics, School of Public Health, University of Illinois at Chicago, 1603 W. Taylor St. (M/C 923), Chicago, IL 60612, USA; Central Brain Tumor Registry of the United States, Chicago, IL, USA.
References
- 1.Barnes L. Pathology and genetics of head and neck tumours. IARC Press; Lyon: 2005. International Academy of Pathology, World Health Organization, International Agency for Research on Cancer. [Google Scholar]
- 2.Davies L, Welch HG. Epidemiology of head and neck cancer in the United States. Otolaryngol Head Neck Surg. 2006;135:451–457. doi: 10.1016/j.otohns.2006.01.029. [DOI] [PubMed] [Google Scholar]
- 3.Dulguerov P, Jacobsen MS, Allal AS, Lehmann W, Calcaterra T. Nasal and paranasal sinus carcinoma: are we making progress? A series of 220 patients and a systematic review. Cancer. 2001;92:3012–3029. doi: 10.1002/1097-0142(20011215)92:12<3012::aid-cncr10131>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 4.Dulguerov P, Allal AS, Calcaterra TC. Esthesioneuroblastoma: a meta-analysis and review. Lancet Oncol. 2001;2:683–690. doi: 10.1016/S1470-2045(01)00558-7. [DOI] [PubMed] [Google Scholar]
- 5.Ejaz A, Wenig BM. Sinonasal undifferentiated carcinoma: clinical and pathologic features and a discussion on classification, cellular differentiation, and differential diagnosis. Adv Anat Pathol. 2005;12:134–143. doi: 10.1097/01.pap.0000163958.29032.56. [DOI] [PubMed] [Google Scholar]
- 6.Fitzek MM, Thornton AF, Varvares M, Ancukiewicz M, McIntyre J, Adams J, Rosenthal S, Joseph M, Amrein P. Neuroendocrine tumors of the sinonasal tract. Results of a prospective study incorporating chemotherapy, surgery, and combined proton-photon radiotherapy. Cancer. 2002;94:2623–2634. doi: 10.1002/cncr.10537. [DOI] [PubMed] [Google Scholar]
- 7.Musy PY, Reibel JF, Levine PA. Sinonasal undifferentiated carcinoma: the search for a better outcome. Laryngoscope. 2002;112:1450–1455. doi: 10.1097/00005537-200208000-00023. [DOI] [PubMed] [Google Scholar]
- 8.Rischin D, Porceddu S, Peters L, Martin J, Corry J, Weih L. Promising results with chemoradiation in patients with sinonasal undifferentiated carcinoma. Head Neck. 2004;26:435–441. doi: 10.1002/hed.10396. [DOI] [PubMed] [Google Scholar]
- 9.Zafereo ME, Fakhri S, Prayson R, Batra PS, Lee J, Lanza DC, Citardi MJ. Esthesioneuroblastoma: 25-year experience at a single institution. Otolaryngol Head Neck Surg. 2008;138:452–458. doi: 10.1016/j.otohns.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 10.Diaz EM, Jr, Johnigan RH, 3rd, Pero C, El-Naggar AK, Roberts DB, Barker JL, DeMonte F. Olfactory neuroblastoma: the 22-year experience at one comprehensive cancer center. Head Neck. 2005;27:138–149. doi: 10.1002/hed.20127. [DOI] [PubMed] [Google Scholar]
- 11.Porter AB, Bernold DM, Giannini C, Foote RL, Link MJ, Olsen KD, Moynihan TJ, Buckner JC. Retrospective review of adjuvant chemotherapy for esthesioneuroblastoma. J Neurooncol. 2008;90:201–204. doi: 10.1007/s11060-008-9645-y. [DOI] [PubMed] [Google Scholar]
- 12.Jethanamest D, Morris LG, Sikora AG, Kutler DI. Esthesioneuroblastoma: a population-based analysis of survival and prognostic factors. Arch Otolaryngol Head Neck Surg. 2007;133:276–280. doi: 10.1001/archotol.133.3.276. [DOI] [PubMed] [Google Scholar]
- 13.Fritz AG World Health Organization. International classification of diseases for oncology: ICD-O. World Health Organization; Geneva: 2000. [Google Scholar]
- 14.Blot WJ, McLaughlin JK, Winn DM, Austin DF, Greenberg RS, Preston-Martin S, Bernstein L, Schoenberg JB, Stemhagen A, Fraumeni JF., Jr Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988;48:3282–3287. [PubMed] [Google Scholar]
- 15.Stiller CA. International patterns of cancer incidence in adolescents. Cancer Treat Rev. 2007;33:631–645. doi: 10.1016/j.ctrv.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Yu MC, Yuan JM. Epidemiology of nasopharyngeal carcinoma. Semin Cancer Biol. 2002;12:421–429. doi: 10.1016/s1044579x02000858. [DOI] [PubMed] [Google Scholar]
- 17.Rosenthal DI, Barker JL, Jr, El-Naggar AK, Glisson BS, Kies MS, Diaz EM, Jr, Clayman GL, Demonte F, Selek U, Morrison WH, Ang KK, Chao KS, Garden AS. Sinonasal malignancies with neuroendocrine differentiation: patterns of failure according to histologic phenotype. Cancer. 2004;101:2567–2573. doi: 10.1002/cncr.20693. [DOI] [PubMed] [Google Scholar]
- 18.Smith SR, Som P, Fahmy A, Lawson W, Sacks S, Brandwein M. A clinicopathological study of sinonasal neuroendocrine carcinoma and sinonasal undifferentiated carcinoma. Laryngoscope. 2000;110:1617–1622. doi: 10.1097/00005537-200010000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Chamberlain MC. Treatment of intracranial metastatic esthesioneuroblastoma. Cancer. 2002;95:243–248. doi: 10.1002/cncr.10679. [DOI] [PubMed] [Google Scholar]
- 20.Cooper JS, Porter K, Mallin K, Hoffman HT, Weber RS, Ang KK, Gay EG, Langer CJ. National cancer database report on cancer of the head and neck: 10-year update. Head Neck. 2009;31:748–758. doi: 10.1002/hed.21022. [DOI] [PubMed] [Google Scholar]
- 21.Rusthoven KE, Raben D, Chen C. Improved survival in patients with Stage III-IV Head and neck cancer treated with radiotherapy as primary local treatment modality. Int J Radiat Oncol Biol Phys. 2008;72:343–350. doi: 10.1016/j.ijrobp.2007.12.046. [DOI] [PubMed] [Google Scholar]
- 22.Iezzoni JC, Mills SE. “Undifferentiated” small round cell tumors of the sinonasal tract: differential diagnosis update. Am J Clin Pathol. 2005;124(Suppl):S110–S121. doi: 10.1309/59RBT2RK6LQE4YHB. [DOI] [PubMed] [Google Scholar]
- 23.Silva EG, Butler JJ, Mackay B, Goepfert H. Neuroblastomas and neuroendocrine carcinomas of the nasal cavity: a proposed new classification. Cancer. 1982;50:2388–2405. doi: 10.1002/1097-0142(19821201)50:11<2388::aid-cncr2820501126>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]