

ABSTRACT
Kisspeptin stimulates gonadotropin-releasing hormone (GnRH) neurons via the kisspeptin receptor, Kiss1r. In rodents, estrogen-responsive kisspeptin neurons in the rostral hypothalamus have been postulated to mediate estrogen-induced positive feedback induction of the preovulatory luteinizing hormone (LH) surge. However, conflicting evidence exists regarding the ability of mice lacking Kiss1r to display LH surges in response to exogenous hormones. Whether the discrepancy reflects different mouse strains used and/or utilization of different surge-induction paradigms is unknown. Here, we tested multiple hormonal paradigms in one Kiss1r knockout (KO) model to see which paradigms, if any, could generate circadian-timed LH surges. Kiss1r KO and wild-type (WT) females were ovariectomized, given sex steroids in various modes, and assessed several days later for LH levels in the morning or evening (when surges occur). Serum LH levels were very low in all morning animals, regardless of genotype or hormonal paradigm. In each paradigm, virtually all WT females displayed clear LH surges in the evening, whereas none of the KO females demonstrated LH surges. The lack of LH surges in KO mice reflects a lack of GnRH secretion rather than diminished pituitary responsiveness from a lifetime lack of GnRH exposure because KO mice responded to GnRH priming with robust LH secretion. Moreover, high cfos-GnRH coexpression was detected in WT females in the evening, whereas low cfos-GnRH coexpression was present in KO females at all time points. Our findings conclusively demonstrate that WT females consistently display LH surges under multiple hormonal paradigms, whereas Kiss1r KO mice do not, indicating that kisspeptin-Kiss1r signaling is mandatory for GnRH/LH surge induction.
Keywords: estradiol, GnRH, GPR54, Kiss1, Kiss1r, kisspeptin, LH surge, positive feedback
Kiss1r knockout mice fail to generate any resemblance of a luteinizing hormone (LH) surge or gonadotropin-releasing hormone (GnRH) neuronal activation, even after GnRH priming, indicating that kisspeptin-Kiss1r signaling is mandatory for the GnRH/LH surge.
INTRODUCTION
In mammals, the preovulatory luteinizing hormone (LH) surge, which triggers ovulation, is governed by neurons in the forebrain and preoptic area that release gonadotropin-releasing hormone (GnRH). Throughout the estrous cycle, low levels of ovarian-derived estradiol (E2) inhibit GnRH secretion via negative feedback until proestrus, when increasing levels of E2 exert positive feedback on hypothalamic neural circuits to induce a preovulatory GnRH/LH surge [1, 2]. In rodents, the E2-induced LH surge is timed by a circadian clock such that the surge occurs exclusively in the late afternoon/early evening of proestrus, around the time of lights off [3, 4]. The stimulatory effects of E2 on GnRH and LH secretion have been shown to be dependent on estrogen receptor α (ERα), as female mice with neuronal deletions in ERα but not ERβ fail to exhibit E2-induced LH surges [5]. However, these ERα-mediated effects on GnRH do not occur directly in GnRH neurons since these cells lack ERα [6, 7]. In addition, progesterone and its receptor (PR) have also been shown to be important contributors to the LH surge, as PR KO mice are unable to produce an E2-induced LH surge [8, 9].
The neuropeptide kisspeptin, encoded by the Kiss1 gene, and its receptor, Kiss1r (formerly termed GPR54), are critical for puberty and fertility in mammals [10, 11]. Kisspeptin potently stimulates GnRH secretion and hence LH secretion via direct binding of Kiss1r in GnRH cells [12, 13]. Two separate populations of kisspeptin-synthesizing neurons have been characterized in the mammalian hypothalamus, one in the arcuate nucleus (ARC; infundibular nucleus in primates) and one more rostrally in the preoptic region. In rodents, the preoptic kisspeptin population lies specifically in the continuum comprising the anterior ventral periventricular nucleus and neighboring periventricular nucleus (AVPV/PeN) [14–16]. Sex steroids, such as E2, suppress the levels of Kiss1 mRNA in the ARC but robustly elevate Kiss1 expression in the AVPV/PeN [16–18]. Since kisspeptin can directly stimulate GnRH neuron electrical activity and GnRH secretion [19, 20] and virtually all Kiss1 neurons in the AVPV/PeN express ERα [18], it is believed that E2's positive feedback effects on GnRH/LH secretion are mediated by AVPV/PeN Kiss1 neurons [21]. Indeed, on the afternoon of proestrus in rats, there is an increase in the number of cfos-expressing Kiss1 neurons in the AVPV/PeN that coincides with cfos induction in GnRH neurons [17, 22]. Similarly, cfos is induced in Kiss1 neurons in the AVPV/PeN of mice exclusively at the time of the LH surge but not at other times [23]. Thus, Kiss1 neuronal activation prior to or at the time of the LH surge may play a critical role in driving the surge. Supporting this, kisspeptin neurons in the AVPV/PeN are sexually dimorphic in cell number and Kiss1 expression (both greater in females than males), correlating with the sexually dimorphic nature of the LH surge in rodents (occurs only in females) [16, 24].
Conflicting evidence currently exists concerning the vitality of kisspeptin-Kiss1r signaling in the initiation of an E2-induced LH surge. Dungan et al. [25] surprisingly reported that Kiss1r is not essential for generating an E2-induced LH surge, with adult Kiss1r KO females capable of displaying LH surges in response to exogenous E2 treatment. However, in stark contrast, using both a different Kiss1r KO mouse model and a different E2 paradigm for inducing LH surges, Clarkson et al. [26] reported that kisspeptin-Kiss1r signaling is necessary for GnRH neuronal activation and the LH surge. These latter results were more consistent with the presumption that E2 exerts its positive feedback effects on GnRH/LH secretion via ERα in AVPV/PeN Kiss1 neurons. The contradictory findings might be attributed to differences in the transgenic mouse lines and/or the estrogen paradigms employed by the two groups, though this issue has not been addressed, leaving the issue unresolved. The controversy was heightened by a more recent finding that selective ablation of kisspeptin cells in transgenic mice did not prevent females from sexually maturing or being fertile in adulthood [27].
In order to further define the roles of kisspeptin-Kiss1r signaling in the preovulatory LH surge, we sought to resolve the controversy regarding the ability of Kiss1r KO mice to exhibit LH surges. Using the Kiss1r KO mouse strain that was reported to display LH surges, our overall goal was 2-fold: 1) to determine whether Kiss1r KO mice can in fact display GnRH neuronal activation and an LH surge under various surge-inducing paradigms, including the different paradigms used by each of the previous two studies as well as additional paradigms not previously tested in Kiss1r KO mice and 2) if Kiss1r KO females do in fact surge, begin to identify novel neural LH surge circuits underlying this kisspeptin-independent process. Indeed, several other brain factors have been suggested to perhaps play a role in the LH surge mechanism [3, 28, 29], but whether they are sufficient to drive the surge without kisspeptin signaling is unclear. Despite this latter initial goal, our cumulative results conclusively indicate that Kiss1r is in fact essential for producing an LH surge under each and every hormonal paradigm examined.
MATERIALS AND METHODS
Animals
Experiments were conducted on adult (10–12 wk old) female Kiss1r KO and wild-type (WT) littermates generated from Kiss1r heterozygous breeders from a Kiss1r KO mouse line originally created by Omeros, Inc. (Seattle, WA), and kindly provided to us by Dr. Robert Steiner (University of Washington, Seattle, WA). This Kiss1r KO mouse line was previously shown by us to be completely hypogonadal and infertile and to have undetectable reproductive hormone levels [25, 30, 31]. All females were genotyped after weaning via PCR analysis of tail DNA. Mice were provided food and water ad libitum and housed in groups of two to three under a 12L:12D cycle (lights off at 1800 h). All experiments were performed in agreement with the National Institutes of Health Animal Care and Use Guidelines and with authorization from the Animal Care and Use Committee of the University of California, San Diego. For all surgeries, mice received Buprenex analgesic (1.5 μg, s.c.) following surgery.
Blood and Brain Collection and Hormone Assays
At specific circadian times (see below), mice were anesthetized with isoflurane and blood drawn via retro-orbital bleeding just prior to killing via rapid decapitation. Serum was isolated from the blood samples and stored at −20°C. Serum LH levels were measured by the University of Virginia's Ligand Assay Core (Charlottesville, VA) by a sensitive sandwich RIA assay. Specifically, serum LH is measured by a sensitive two-site sandwich immunoassay using monoclonal antibodies against bovine LH (no. 581B7) and the human LH-beta subunit (no. 5303), as described previously [32–34]. Mouse LH reference prep (AFP5306A; provided by Dr. A.F. Parlow and the National Hormone and Peptide Program) is used as standard. The limit of detectability for the mouse LH assay is 0.04 ng/ml. Circulating serum E2 levels were also measured by University of Virginia's Ligand Assay Core, using Calbiotech ELISA. This mouse E2 assay has a sensitivity of 3 pg/ml. For animals in experiment 1, brains were collected at killing and immediately frozen on dry ice before being stored at −80°C. Frozen brains were sectioned on a cryostat into five sets of 20-μm sections and thaw-mounted on Superfrost-plus slides that were stored at −80°C until assaying via in situ hybridization.
Double-Label In Situ Hybridization
For double-label in situ hybridization (ISH) of cfos in GnRH neurons (experiment 5), slide-mounted brain sections encompassing the entire forebrain and preoptic area were fixed in 4% paraformaldehyde, pretreated with acetic anhydride, rinsed in 2× SSC (sodium citrate, sodium chloride), delipidated in chloroform, dehydrated in ethanol (70%, 95%, 100%), and air-dried. Radio-labeled (33P) antisense cfos (0.04 pmol/ml) and digoxigenin-labeled Gnrh (1:500) riboprobes were combined with tRNA, denatured, dissolved together in hybridization buffer, and applied to each slide (100 μl/slide). Slides were coverslipped and placed in a humidity chamber at 55°C for 16 h. Following hybridization, slides were washed in 4× SSC and then placed into RNAse (37mg/ml RNAse A in 0.15 M sodium chloride, 10 mM Tris, 1 mM EDTA, pH 8.0) for 30 min at 37°C, then in RNAse buffer without RNase at 37°C. After a wash in 2× SSC at room temperature, slides were washed in 0.1× SSC at 62°C for 1 h and then incubated in 2× SSC with 0.05% Triton X-100 containing 2% sheep serum (NSS) for 1 h at room temperature. The slides were then washed in buffer 1 (100 mM Tris-HCl pH 7.5, 150 mM NaCl) and incubated overnight at room temperature with anti-DIG antibody conjugated to alkaline phosphatase (Roche) diluted 1:500 in buffer 1 containing 1% NSS and 0.3% Triton X-100. The following day, slides were washed with buffer 1 and incubated with Vector Red alkaline phosphatase substrate (Vector Labs, Burlingame, CA) for 1 h at room temperature. Slides were then dried, dipped in emulsion, stored at 4°C, and developed and coverslipped 9 days later.
ISH slides were analyzed with an automated grains imaging processing system (Dr. Don Clifton, University of Washington) by a person unaware of the treatment group of each slide. Red fluorescent DIG-containing (GnRH) cells were identified under fluorescence microscopy and the grain-counting software used to quantify the number of silver grains (cfos mRNA) overlying each cell. Signal-to-background ratios for individual cells were calculated by the program, and a cell was considered double labeled if its ratio was >3.
Hormonal Paradigm 1: LH Surge Induction by Constant Elevated Estradiol
Mice were anesthetized with isoflurane and then ovariectomized (OVX) and given E2 implants. Silastic brand (Dow Corning, Midland, MI; inner diameter = 0.20 cm, outer diameter = 0.318 cm) capsules containing 0.625 μg of 17-β E2 dissolved in sesame oil were subcutaneously implanted. This hormone paradigm typically produces constantly elevated serum E2 levels of ∼20–30 pg/ml, resembling mouse proestrus levels [35]. Under this hormonal milieu, female mice will normally produce a daily circadian-timed LH surge, occurring each evening exclusively around the time of lights off (1800 h) [23, 35]. Adult WT and Kiss1r KO females treated with this E2 regimen were killed 2 days after surgery either in the morning (between 1000 and 1100 h) or in the evening just after lights off (1810–1830 h), and their blood was collected for LH and E2 analysis (n = 7–8/group).
Hormonal Paradigm 2: LH Surge Induction by Rising Estradiol Levels
Mice were anesthetized with isoflurane and OVX and subcutaneously given low-dose E2 implants (Silastic tubing; inner diameter 0.10 cm, outer diameter 0.21 cm) containing crystalline 17-β E2 dissolved in Silastic medical adhesive at 1 μg/20 g body weight (BW) [26, 36, 37]. Five days after this low-estrogen treatment, mice were subcutaneously injected in the morning with 1 μg/20 g BW estradiol benzoate (EB) in sesame oil (100 μl) to produce elevated proestrus-like E2 levels on the following day [36, 37]. This hormone paradigm produces low diestrous levels of serum E2 for several days and then increases serum E2 to proestrus levels over the last 2 days, mirroring the natural cyclical rise in E2 during a female rodent's typical estrous cycle. Under this hormonal regimen, female mice will normally produce a robust circadian-timed LH surge on the day after EB injection, occurring around the time of lights off [36, 37]. Adult WT and Kiss1r KO females treated with this E2+EB paradigm were killed the day after the EB injection either in the morning (between 1000 and 1100 h) or in the evening just after lights off (1810–1830 h), and their blood was collected for LH and E2 analyses (n = 7–10/group).
Hormonal Paradigm 3: LH Surge Induction by a Combination of Estradiol and Progesterone
Anesthetized mice were OVX and subcutaneously given low-dose E2 implants (Silastic tubing; inner diameter 0.04 inch, outer diameter 0.85 inch) containing crystalline 17-β E2 dissolved in Silastic medical adhesive at 1 μg/20 g BW, as with the previous hormonal paradigm above. Five days after this low-estradiol treatment, mice were subcutaneously injected in the morning with 1 μg/20 g BW EB in sesame oil (100 μl). On the next day, mice were injected subcutaneously with progesterone (P) dissolved in sesame oil and given at a dose of 300 μg/20 g BW [36, 37]. This hormone paradigm produces low diestrous levels of serum E2 for several days, followed by elevated proestrus-like levels of E2 and elevated serum P, resembling a female rodent's typical estrous cycle. As with the other hormonal regimens, female mice treated with this E2+P paradigm will normally produce a circadian-timed LH surge on the day of the P injection, occurring in the evening around the time of lights off. Adult WT and Kiss1r KO females treated with this E2+P paradigm were killed the day of the P injection, either in the morning (between 1130 and 1200 h) or in the evening just after lights off (1810–1830 h), and their blood was collected for LH analysis (n = 7–8/group).
Hormonal Paradigm 4: GnRH Priming Prior to LH Surge Induction by Elevated E2
This regimen combined GnRH priming with the hormonal paradigm 1 above. In Kiss1r KO mice, an inability of females to produce an LH surge might be attributed to a chronic lack of gonadotrope exposure (and hence responsiveness of the pituitary) to GnRH. Thus, adults were first primed with exogenous GnRH to stimulate the pituitary. Mice received a single daily subcutaneous GnRH injection (200 ng) every other day for a total of three injections. On the last day of priming, blood was drawn 15 min after the GnRH injection to assess pituitary's ability to secrete LH in response to GnRH. Two days later, all mice were OVX and implanted subcutaneously with a Silastic E2 implant producing elevated proestrus levels of E2, as in hormonal paradigm 1. Two days later, blood was collected prior to killing at either 1100 or 1830 h, and the serum was analyzed for LH levels (n = 7–11/group).
LH Surge Criteria
There is no universally accepted criterion to define an LH surge; in experimentally E2-treated female mice, we typically define an LH surge as being at least 0.60 ng/ml or greater (most LH surges are above 1.00 ng/ml). However, for the present study, wherever possible, we used the criterion delineated by Dungan et al. [25] in their Kiss1r KO paper, so that our data could better be compared with theirs. That study used a value of the mean LH for all morning mice plus two times the SD as a threshold for identifying an LH surge. This criterion is experiment specific (since morning values may differ between experiments); in their own experiment, it equated to an LH surge being greater than 0.53 ng/ml. Of note, in experiment 1 of the present study, all morning mice had identical LH values (all below the limit of detection), and, as such, no SD could be calculated. Thus, for experiment 1, we used our typical LH surge criteria of 0.60 ng/ml. For experiments 2, 3, and 4, the Dungan et al. [25] criteria could also be used and were 0.45, 0.53, and 0.16 ng/ml, respectively. Regardless, we note that in all these experiments, the number of mice classified as displaying an LH surge was identical using either of the two criteria (0.60 ng/ml or morning mean + 2 SD).
Statistical Analyses
All data are expressed as the mean ± SEM for each group. In all experiments, differences were analyzed by analysis of variance, followed by post hoc comparisons via Fisher (protected) LSD test. For all comparisons, statistical significance was set at P < 0.05. All analyses were performed in Statview 5.0.1 (SAS Institute, Cary, NC).
RESULTS
Experiment 1: LH Surge Induction by Constant Elevated E2 in Kiss1r KO and WT Mice
A previous report [25] suggested that Kiss1r KO females (same strain as our mice) could display LH surges in response to constant elevated proestrus levels of E2 (hormonal paradigm 1). This experiment tested if, in fact, adult Kiss1r KO female mice were capable of displaying a circadian-timed LH surge in response to elevated levels of E2. After treatment with hormonal paradigm 1, all WT females displayed robust LH surges at the evening time point (range: 0.60–9.10 ng/ml), whereas LH levels were extremely low in WT females in the morning (∼0.08 ng/ml; Fig. 1), consistent with a circadian induction of the LH surge in the evening. In contrast, no Kiss1r KO females exhibited any hint of LH surges in the evening (range: 0.08–0.11 ng/ml; Fig. 1). Indeed, LH levels of all Kiss1r KO mice were extremely low at both time points (at or below the limit of detectability; Fig. 1), significantly lower than those of evening WT females (P < 0.001) and similar to levels of morning WT females.
FIG. 1.
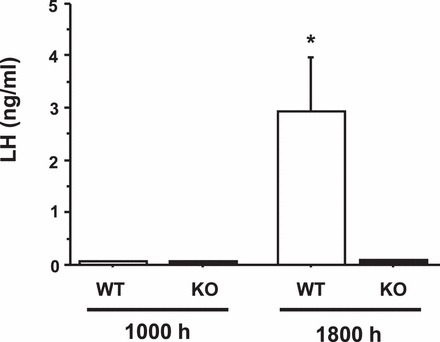
Mean serum LH levels in adult female WT and Kiss1r KO mice exposed to a positive feedback regimen of constant elevated E2. Two days after E2 implantation, blood was collected from mice at either a morning (approx. 1000 h) or an evening (lights off; 1800 h) time point. *Significantly different from all other groups (P < 0.01). All data are expressed as the mean ± SEM for each group.
Experiment 2: LH Surge Induction by Rising E2 Levels in Kiss1r KO and WT Females
This experiment tested if adult Kiss1r KO female mice could display an LH surge in response to rising levels of E2 that mimic those of a normal estrous cycle (hormonal paradigm 2). This E2+EB paradigm failed to elicit LH surges in a different Kiss1r KO line [26] but was not previously tested in our Kiss1r KO mouse line (which had been reported by Dungan et al. [25] to be capable of displaying surges with a different paradigm). We found that all WT females exposed to this E2+EB paradigm displayed robust LH surges at the evening time point (range: 2.03–8.30 ng/ml), whereas LH levels in the morning were extremely low in WT females (Fig. 2). Conversely, no Kiss1r KO females exhibited LH surges in the evening (range: 0.04–0.09 ng/ml; Fig. 2). As in experiment 1, LH levels of Kiss1r KO mice were extremely low at both morning and evening time points, significantly lower than those of evening WT females (P < 0.001) and similar to levels of morning WT females.
FIG. 2.
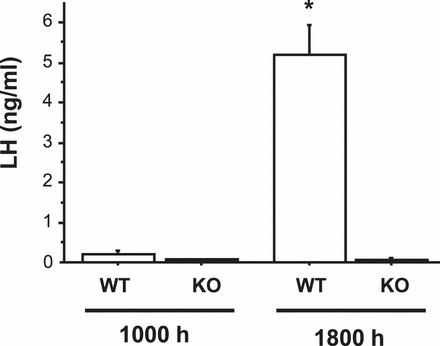
Mean serum LH levels in female WT and Kiss1r KO mice exposed to a positive feedback regimen of low E2 followed by an injection of EB to induce rising E2. The day after EB injection, blood was collected from mice at either a morning (1000 h) or an evening (lights off; 1800 h) time point. *Significantly different from all other groups (P < 0.01). All data are expressed as the mean ± SEM for each group.
Experiment 3: LH Surge Induction by a Combination of E2 and P in Kiss1r KO and WT Females
In gonadal-intact female rodents, the LH surge is induced by rising E2 on proestrus that is accompanied by rising P levels. P has been proposed to modulate the surge onset, duration, and/or magnitude. Here, we used an E2+EB+P paradigm (hormonal paradigm 3) to test whether P may be required for Kiss1r KO female mice to produce an LH surge. We found that under this E2+EB+P paradigm, most (75%) WT females displayed an LH surge at the evening time point (range of surging females: 0.70–1.48 ng/ml; Fig. 3). LH levels were very low in all WT females killed in the morning. As before, and in contrast to the WTs, Kiss1r KO females treated with E2+EB+P did not generate any semblance of an LH surge at the evening time point (Fig. 3). All evening values of LH were extremely low in KO females (being at the limit of detection in all mice) and were significantly different from WT evening levels (P < 0.001) and similar to morning levels of both genotypes.
FIG. 3.
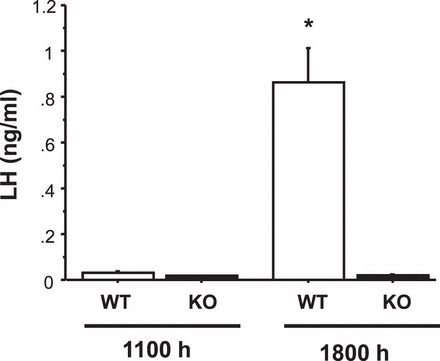
Mean serum LH levels in female WT and Kiss1r KO mice exposed to a positive feedback paradigm of low E2 followed by morning injections of EB and then, 1 day later, P. The day of the P injection, blood was collected from mice at either a morning (approx. 1100 h) or an evening (lights off; 1800 h) time point. *Significantly different from all other groups (P < 0.01). All data are expressed as the mean ± SEM for each group.
Experiment 4: GnRH Priming of the Pituitary Prior to LH Surge Induction by E2
The inability of Kiss1r KO females to produce an LH surge under the three different hormonal paradigms above might possibly be attributed to a lifetime lack of pituitary exposure to sufficient GnRH and hence reduced gonadotrope responsiveness to a GnRH surge signal. If so, a positive feedback paradigm might trigger a GnRH surge but not an accompanying LH surge. To assess this possibility, adult females of both genotypes were first primed with GnRH injections over the course of several days before being subjected to an LH surge-induction protocol. After the final GnRH priming injection, blood levels were analyzed for LH levels. Unlike control mice not given GnRH, both WT and Kiss1r KO mice displayed large elevations in serum LH 15 min after GnRH treatment (Fig. 4A; P < 0.01 relative to controls not receiving GnRH), indicating that the pituitaries of both genotypes were highly responsive to GnRH stimulation. All GnRH-primed mice were then treated with constant E2 to see if an LH surge could now be induced in Kiss1r KO mice. As in the previous experiments, WT mice showed robust LH surges in the evening (range 0.96–1.65 ng/ml), whereas none of the previously GnRH-primed Kiss1r KO mice displayed an LH surge in response to elevated E2 (Fig. 4B). LH levels of GnRH-primed Kiss1r KO mice were extremely low (below limit of detectability) at both time points, similar to morning WT levels and significantly lower than WT evening levels (P < 0.01).
FIG. 4.
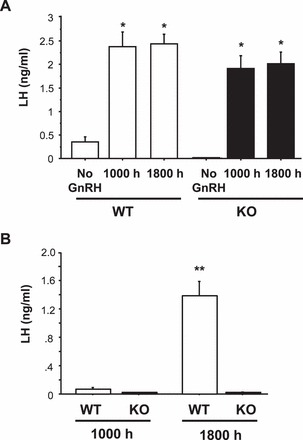
Mean serum LH levels in female WT and Kiss1r KO mice exposed to a positive feedback regimen of constant elevated E2 after several days of GnRH priming. A) LH levels in mice 15 min after the final GnRH priming injection. GnRH stimulated robust LH secretion in both genotypes, indicating a responsive pituitary. *Significantly different from non-GnRH treated group (P < 0.01). B) Two days after the last GnRH priming treatment, mice were OVX and given E2, and blood was collected 2 days later at a morning (1000 h) or an evening (lights off; 1800 h) time point. **Significantly different from all other groups. All data are expressed as the mean ± SEM for each group.
Experiment 5: Positive Feedback Induction of cfos in GnRH Neurons in WT but Not Kiss1r KO Females
The lack of LH surges in Kiss1r KO mice in the previous experiments suggested an absence of a preceding GnRH surge. To assess whether positive feedback can or cannot activate GnRH neurons in Kiss1r KO mice, we measured cfos induction in GnRH neurons of WT and Kiss1r KO females that had been treated with hormonal paradigm 1. GnRH neurons were analyzed in three regions of the GnRH neural network: the medial septum, the OVLT, and the anterior POA. There were no statistical differences between genotypes or groups in the number of GnRH neurons counted in each area or overall (∼100 total cells/animal on average; not shown). Mirroring the LH results in experiment 1, WT females had high cfos-GnRH coexpression in the evening but not in the morning (Figs. 5, 6), indicating that their GnRH neurons were activated in the evening, in line with a GnRH/LH surge. In WT evening females, elevated cfos induction was detected in GnRH neurons in all three brain regions, with the highest level of cfos-GnRH coexpression in the OVLT and POA. Conversely, Kiss1r KO females did not have elevated cfos-GnRH coexpression in the evening in any area of the GnRH neuronal network (Figs. 5, 6). The level of cfos induction in GnRH neurons of KO mice was low (<10%) in both the morning and the evening, similar to WT morning levels. This outcome suggests that the lack of LH surges in Kiss1r KO mice is because the GnRH system is not activated rather than the pituitary being unresponsive to a GnRH surge signal.
FIG. 5.
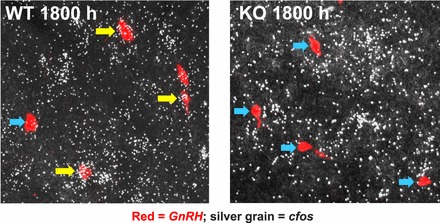
Representative photomicrographs of cfos mRNA coexpression in GnRH neurons in WT and Kiss1r KO female mice exposed to an LH surge paradigm of constant elevated E2 and sacrificed in the evening. Yellow arrows denote examples of GnRH cells (red fluorescence) with significant cfos (silver grains) coexpression. Blue arrows designate example GnRH neurons that did not have cfos induction.
FIG. 6.
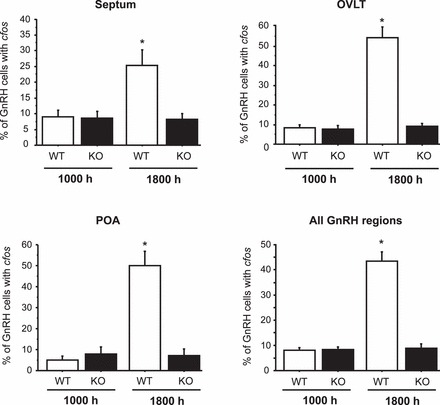
Mean percent of GnRH neurons coexpressing cfos in female mice exposed to an LH surge paradigm of constant elevated E2. Two days after E2 implantation, brains were collected from mice at either a morning (1000 h) or an evening (1800 h) time point. Brains were assayed for cfos induction in GnRH cells using double-label ISH. Mean levels of cfos-GnRH coexpression in mice from different time-points were quantified in three brain regions within the GnRH neural system as well as the entire GnRH population. *Significantly different from all other groups (P < 0.01). All data are expressed as the mean ± SEM for each group.
E2 Levels in the Different Paradigms
Circulating serum E2 levels were measured in mice of both genotypes from each of the experiments. There were no significant differences in E2 levels between WT and KO mice at either morning or evening time points in any of the hormonal paradigms examined (Table 1). In addition, Table 1 summarizes the percent of animals in each group that displayed an LH surge under each hormonal paradigm.
TABLE 1.
Serum estradiol (E2) levels and percentage of mice surging in WT and Kiss1r KO mice treated with different hormonal paradigms to induce evening LH surges.
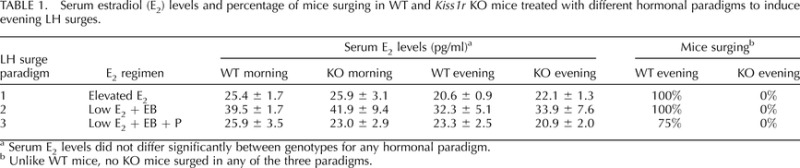
Serum E2 levels did not differ significantly between genotypes for any hormonal paradigm.
Unlike WT mice, no KO mice surged in any of the three paradigms.
DISCUSSION
A wealth of evidence accumulated over the past decade has implicated the kisspeptin system in directly mediating the positive feedback effects of E2 on the GnRH and LH surges that drive ovulation [3]. However, despite this, several prominent reports have cast doubt on whether this kisspeptin positive feedback model is in fact correct and whether other neural circuits may instead be involved. Indeed, the first published analysis of the ability of Kiss1r KO females to display LH surges in response to exogenous E2 surprisingly reported that these females can in fact show LH surges after E2 supplementation, suggesting that kisspeptin-Kiss1r signaling is not necessary for mice to generate LH surges [25]. However, soon thereafter, another report argued the opposite: that Kiss1r KO mice cannot display LH surges when given E2, supporting the developing dogma that kisspeptin-Kiss1r signaling is essential for positive feedback [26]. The reasons for these opposite and discrepant findings were not addressed, though it could have been due to technical differences, as the two studies used both different Kiss1r KO strains and different E2 paradigms. The mouse model that surged was not tested with the E2 paradigm that failed to elicit surges, and the E2 paradigm that did elicit surges was never tested in the mouse model failing to surge with the other protocol. Thus, direct comparison between the two studies was limited and lacking. Here, we sought to directly address this controversy to establish which E2 paradigms, if any, could in fact elicit LH surges in Kiss1r KO females. We convincingly demonstrate that Kiss1r KO females are incapable of generating LH surges under any hormonal paradigm tested, including even after GnRH priming.
Unlike WT mice, which consistently displayed LH surges in the evening, Kiss1r KO females did not produce a single steroid-induced LH surge under any of the three hormonal paradigms tested. Indeed, Kiss1r KO females did not even demonstrate a semblance of an LH surge or any elevation of LH of any kind: in all cases, LH levels in Kiss1r KO mice were extremely low and typically undetectable, regardless of time of day. Combining all experiments, the overall percent of evening WT females displaying an LH surge was 94% (30 of 32 mice), whereas the overall percent of evening KO females that surged was 0% (0 of 33 mice). The complete lack of surges in the KO mice could not be attributed to deficits in E2, as serum E2 levels were similar between genotypes in all experiments. These data indicate quite clearly that mice cannot generate LH surges in the absence of kisspeptin signaling.
In the absence of any other data, the lack of detectable LH surges in the Kiss1 KO mice could, in theory, be attributable to defects at several levels: the inability of GnRH neurons to be activated and release a surge of GnRH and/or an inability of the pituitary to respond to an incoming GnRH signal and secrete a surge of LH. The latter possibility could stem from a lifetime lack of gonadotrope exposure to GnRH, resulting in a severely reduced responsiveness to the first GnRH signal. Indeed, in normal rodents, the LH surge magnitude increases over the end of pubertal maturation, eventually producing full magnitude surges in adulthood [1]. Given that Kiss1r KO mice are essentially prepubertal, even at adult ages, their gonadotrope system may, in theory, resemble that of a prepubertal animal, perhaps explaining the lack of a robust LH surge on first-time exposure to GnRH in an experimental positive feedback paradigm. We tested this possibility by priming the pituitaries of adult Kiss1r KO mice with GnRH injections; after the third GnRH injection, LH was robustly elevated in Kiss1r KO and WT mice, indicating a responsive pituitary. However, when subsequently challenged with an E2 surge paradigm, these GnRH-primed Kiss1r KO mice, unlike WTs, failed to produce any semblance of a LH surge. Thus, the lack of an LH surge likely reflects lack of a GnRH surge rather than diminished pituitary responsiveness to incoming GnRH. This conclusion was supported by our finding of high cfos-GnRH coexpression in WT females at the evening time point but low cfos-GnRH coexpression in evening Kiss1r KO mice, indicating that GnRH neurons are not properly activated by E2 positive feedback paradigms in Kiss1r KO mice.
Our findings support those of Clarkson et al. [26], who found that another line of Kiss1r KO mice do not surge after E2+EB treatment. Moreover, we extend those findings to show that several additional positive feedback paradigms not tested in that study, including one involving constant elevated E2 and another involving P supplementation, also do not elicit LH surges in Kiss1r KO mice. Moreover, this is the first study to document the inability of Kiss1r KO mice to surge even after GnRH priming. Thus, our conclusions that Kiss1r is necessary for GnRH activation and the LH surge are in agreement with those of the Clarkson et al. [26] study. However, the reason for the discrepancy between our present findings and the previous finding by Dungan et al. [25] regarding the ability of elevated E2 (hormonal paradigm 1) to induce LH surges and GnRH neuronal activation in the identical Kiss1r KO strain as ours is unknown. Our mice were the same age, housed in the same conditions (though at different universities), exposed to the same E2 paradigm, and had similar E2 levels as in the Dungan study. Thus, there is unlikely to be any major technical differences between our two studies. We do note that our Kiss1r KO mouse line was originally on a mixed 129/BL6 background; our lab has further backcrossed our colony several generations into C57Bl6, though it still remains a mixed 129/BL6 strain. We therefore cannot rule out a subtle “strain” difference between our mice and those used in the Dungan study, but we do not believe this would be enough to fully induce or prevent GnRH activation or an LH surge, as there is no indication of gross disparities in the ability of strains to generate LH surges. We do note that the mean LH surge levels for the Kiss1r KO mice reported in the Dungan study were of lower magnitude than in our study, and not all of those mice surpassed the designated threshold level to qualify as an LH surge, indicating that a decent proportion of those Kiss1r KO mice may not have actually “surged.” Moreover, the morning values for GnRH neuronal activation of both WT and KO were uncharacteristically high in the Dungan study (25–30%), nearly reaching typical evening “surge” levels (∼40–50%). In our mice, morning levels of GnRH neuron activation were below 9% in both genotypes, which is more consistent with known low levels of activation at nonsurge times. We do not have a clear reason for this discrepancy between studies but note that our study assessed cfos and GnRH mRNA using ISH, whereas Dungan et al. [25] assessed Fos and GnRH protein using IHC.
Our study utilized three different hormonal paradigms to induce LH surges in female mice, providing a context to compare the efficacy of different experimental models of positive feedback. Overall, all three paradigms were efficient at inducing LH surges in WT females. The percent of WT females surging and the magnitude of the surge were not different between constant elevated E2 (paradigm 1) and low E2+EB (paradigm 2), though the latter method produced slightly higher circulating E2 levels than the former. If anything, this may suggest that the lower E2 levels achieved in paradigm 1, having full efficacy for surge induction, may be slightly more physiological, though this is likely not a huge difference. Interestingly, the paradigm including P (paradigm 3) yielded slightly lower mean LH surge levels and had two WT animals that failed to reach the surge criteria. This was surprising, as in rats, P is thought to increase the magnitude of the LH surge. However, P is also known to advance and/or shorten duration of the surge in rats [2]. Thus, it is likely that in our mice treated with P, the LH surge either began earlier or had a shorter duration than in the other non-P hormonal paradigms. If so, we may have been sampling toward the end of the surge event right after lights off and catching the latter part of the surge rather than its peak. Alternatively, the LH surge system in mice is quite easily perturbed by stress, and it is possible (though probably unlikely) that handling and injections of P earlier that day increased stress levels, which reduced the LH surge later that evening. Regardless of the observed slightly lower surge magnitude, the fact remains that a surge was still present. Indeed, our data clearly and convincingly show that the majority (75%) of WT mice show an LH surge under this P paradigm, whereas none of the KO mice displayed any evidence of LH elevation at all, demonstrating the necessity of Kiss1r for this surge process.
In conclusion, our results suggest that kisspeptin-Kiss1r signaling is essential for the sex steroid-induced activation of GnRH neurons and LH surge in mice. The inability of Kiss1r KO mice to surge is likely to inactivation of GnRH neurons, presumably by kisspeptin signaling arising from the anterior hypothalamus. Although one of our initial goals when embarking on this project was to potentially identify novel neural circuits that may be able to generate an LH surge in the absence of kisspeptin-Kiss1r signaling, our findings rather indicate that kisspeptin-Kiss1r signaling is a necessary and essential component of the positive feedback induction of the LH surge and hence ovulation. While several other brain factors, such as GABA/glutamate and RFRP-3, have been suggested to perhaps play a role in the LH surge mechanism [3, 28, 29], these or other factors do not appear to be sufficient to drive the surge without kisspeptin signaling and may therefore be modulatory to the surge rather than essential. Indeed, our findings suggest that there are not additional parallel pathways independent of kisspeptin that are sufficient for the LH surge process, at least in rodents.
ACKNOWLEDGMENT
The authors thank Sangeeta Dhamija, Joshua Kim, and Alison Lawrence for their technical and animal support.
Footnotes
Supported by National Science Foundation grant IOS-1025893. Additional support was provided by the Eunice Kennedy Shriver NICHD/NIH (SCCPIR) grants U54-HD012303 (U.C. San Diego) and U54 HD-28934 (University of Virginia Ligand Assay and Analysis Core). J.F. was supported by a summer research award from Pomona College (Claremont, CA).
REFERENCES
- Ojeda SR, Skinner MK. Puberty in the rat. Neill JD. (ed.), Knobil and Niell's Physiology of Reproduction, 3rd ed. San Diego: Elsevier; 2006: 2061 2126. [Google Scholar]
- Freeman ME. Neuroendocrine control of the ovarian cycle in the rat. : Neill JD. (ed.), Physiology of Reproduction, 3rd ed. San Diego: Elsevier; 2006: 2283 2326. [Google Scholar]
- Khan AR, Kauffman AS. The role of kisspeptin and RFamide-related peptide-3 neurones in the circadian-timed preovulatory luteinising hormone surge. J Neuroendocrinol 2012; 24: 131 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolson KP, Chappell PE. The changes they are a-timed: metabolism, endogenous clocks, and the timing of puberty. Front Endocrinol (Lausanne) 2012; 3: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Grone HJ, Todman MG, Korach KS, Greiner E, Perez CA, Schutz G, Herbison AE. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 2006; 52: 271 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabovszky E, Shughrue PJ, Merchenthaler I, Hajszan T, Carpenter CD, Liposits Z, Petersen SL. Detection of estrogen receptor-beta messenger ribonucleic acid and 125I-estrogen binding sites in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology 2000; 141: 3506 3509. [DOI] [PubMed] [Google Scholar]
- Hrabovszky E, Steinhauser A, Barabas K, Shughrue PJ, Petersen SL, Merchenthaler I, Liposits Z. Estrogen receptor-beta immunoreactivity in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology 2001; 142: 3261 3264. [DOI] [PubMed] [Google Scholar]
- Chappell PE, Lydon JP, Conneely OM, O'Malley BW, Levine JE. Endocrine defects in mice carrying a null mutation for the progesterone receptor gene. Endocrinology 1997; 138: 4147 4152. [DOI] [PubMed] [Google Scholar]
- Chappell PE, Schneider JS, Kim P, Xu M, Lydon JP, O'Malley BW, Levine JE. Absence of gonadotropin surges and gonadotropin-releasing hormone self-priming in ovariectomized (OVX), estrogen (E2)-treated, progesterone receptor knockout (PRKO) mice. Endocrinology 1999; 140: 3653 3658. [DOI] [PubMed] [Google Scholar]
- de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A 2003; 100: 10972 10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr,, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, et al. The GPR54 gene as a regulator of puberty. N Engl J Med 2003; 349: 1614 1627. [DOI] [PubMed] [Google Scholar]
- Kauffman AS. Coming of age in the kisspeptin era: sex differences, development, and puberty. Mol Cell Endocrinol 2010; 324: 51 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley AE, Clifton DK, Steiner RA. Kisspeptin signaling in the brain. Endocr Rev 2009; 30: 713 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson J, Herbison AE. Postnatal development of kisspeptin neurons in mouse hypothalamus: sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology 2006; 147: 5817 5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottsch ML, Cunningham MJ, Smith JT, Popa SM, Acohido BV, Crowley WF, Seminara S, Clifton DK, Steiner RA. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology 2004; 145: 4073 4077. [DOI] [PubMed] [Google Scholar]
- Kauffman AS, Gottsch ML, Roa J, Byquist AC, Crown A, Clifton DK, Hoffman GE, Steiner RA, Tena-Sempere M. Sexual differentiation of Kiss1 gene expression in the brain of the rat. Endocrinology 2007; 148: 1774 1783. [DOI] [PubMed] [Google Scholar]
- Adachi S, Yamada S, Takatsu Y, Matsui H, Kinoshita M, Takase K, Sugiura H, Ohtaki T, Matsumoto H, Uenoyama Y, Tsukamura H, Inoue K, et al. Involvement of anteroventral periventricular metastin/kisspeptin neurons in estrogen positive feedback action on luteinizing hormone release in female rats. J Reprod Dev 2007; 53: 367 378. [DOI] [PubMed] [Google Scholar]
- Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology 2005; 146: 3686 3692. [DOI] [PubMed] [Google Scholar]
- Han SK, Gottsch ML, Lee KJ, Popa SM, Smith JT, Jakawich SK, Clifton DK, Steiner RA, Herbison AE. Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci 2005; 25: 11349 11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messager S, Chatzidaki EE, Ma D, Hendrick AG, Zahn D, Dixon J, Thresher RR, Malinge I, Lomet D, Carlton MB, Colledge WH, Caraty A, et al. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc Natl Acad Sci U S A 2005; 102: 1761 1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison AE. Estrogen positive feedback to gonadotropin-releasing hormone (GnRH) neurons in the rodent: the case for the rostral periventricular area of the third ventricle (RP3V). Brain Res Rev 2008; 57: 277 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JT, Popa SM, Clifton DK, Hoffman GE, Steiner RA. Kiss1 neurons in the forebrain as central processors for generating the preovulatory luteinizing hormone surge. J Neurosci 2006; 26: 6687 6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JL, Clifton DK, de la Iglesia HO, Steiner RA, Kauffman AS. Circadian regulation of Kiss1 neurons: implications for timing the preovulatory GnRH/LH surge. Endocrinology 2009; 150: 3664 3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma T, Sakakibara M, Yamada S, Kinoshita M, Iwata K, Tomikawa J, Kanazawa T, Matsui H, Takatsu Y, Ohtaki T, Matsumoto H, Uenoyama Y, et al. Significance of neonatal testicular sex steroids to defeminize anteroventral periventricular kisspeptin neurons and the GnRH/LH surge system in male rats. Biol Reprod 2009; 81: 1216 1225. [DOI] [PubMed] [Google Scholar]
- Dungan HM, Gottsch ML, Zeng H, Gragerov A, Bergmann JE, Vassilatis DK, Clifton DK, Steiner RA. The role of kisspeptin-GPR54 signaling in the tonic regulation and surge release of gonadotropin-releasing hormone/luteinizing hormone. J Neurosci 2007; 27: 12088 12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson J. d'Anglemont de Tassigny X, Moreno AS, Colledge WH, Herbison AE. Kisspeptin-GPR54 signaling is essential for preovulatory gonadotropin-releasing hormone neuron activation and the luteinizing hormone surge. J Neurosci 2008; 28: 8691 8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Boehm U. Female reproductive maturation in the absence of kisspeptin/GPR54 signaling. Nat Neurosci 2011; 14: 704 710. [DOI] [PubMed] [Google Scholar]
- Christian CA, Moenter SM. The neurobiology of preovulatory and estradiol-induced gonadotropin-releasing hormone surges. Endocr Rev 2010; 31: 544 577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottem EN, Godwin JG, Krishnan S, Petersen SL. Dual-phenotype GABA/glutamate neurons in adult preoptic area: sexual dimorphism and function. J Neurosci 2004; 24: 8097 8105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman AS, Park JH, McPhie-Lalmansingh AA, Gottsch ML, Bodo C, Hohmann JG, Pavlova MN, Rohde AD, Clifton DK, Steiner RA, Rissman EF. The kisspeptin receptor GPR54 is required for sexual differentiation of the brain and behavior. J Neurosci 2007; 27: 8826 8835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poling MC, Kauffman AS. Sexually dimorphic testosterone secretion in prenatal and neonatal mice is independent of kisspeptin-Kiss1r and GnRH signaling. Endocrinology 2012; 153: 782 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallest PC, Trader GL, Darrow JM, Shupnik MA. Regulation of rat luteinizing hormone beta gene expression in transgenic mice by steroids and a gonadotropin-releasing hormone antagonist. Biol Reprod 1995; 53: 103 109. [DOI] [PubMed] [Google Scholar]
- Haavisto AM, Pettersson K, Bergendahl M, Perheentupa A, Roser JF, Huhtaniemi I. A supersensitive immunofluorometric assay for rat luteinizing hormone. Endocrinology 1993; 132: 1687 1691. [DOI] [PubMed] [Google Scholar]
- Matteri RL, Roser JF, Baldwin DM, Lipovetsky V, Papkoff H. Characterization of a monoclonal antibody which detects luteinizing hormone from diverse mammalian species. Domest Anim Endocrinol 1987; 4: 157 165. [DOI] [PubMed] [Google Scholar]
- Christian CA, Mobley JL, Moenter SM. Diurnal and estradiol-dependent changes in gonadotropin-releasing hormone neuron firing activity. Proc Natl Acad Sci U S A 2005; 102: 15682 15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronson FH. The regulation of luteinizing hormone secretion by estrogen: relationships among negative feedback, surge potential, and male stimulation in juvenile, peripubertal, and adult female mice. Endocrinology 1981; 108: 506 516. [DOI] [PubMed] [Google Scholar]
- Bronson FH. Vom Saal FS. Control of the preovulatory release of luteinizing hormone by steroids in the mouse. Endocrinology 1979; 104: 1247 1255. [DOI] [PubMed] [Google Scholar]