

Abstract
Heparan sulfate (HS) proteoglycans (HSPGs) participate in several aspects of inflammation because of their ability to bind to growth factors, chemokines, interleukins and extracellular matrix proteins as well as promote inflammatory cell trafficking and migration. We investigated whether HSPGs play a role in the development of airway remodeling during chronic allergic asthma using mice deficient in endothelial- and leukocyte-expressed N-deacetylase/N-sulfotransferase-1 (Ndst1), an enzyme involved in modification reactions during HS biosynthesis. Ndst1-deficient and wild-type (WT) mice exposed to repetitive allergen (ovalbumin [OVA]) challenge were evaluated for the development of airway remodeling. Chronic OVA-challenged WT mice exhibited increased HS expression in the lungs along with airway eosinophilia, mucus hypersecretion, peribronchial fibrosis, increased airway epithelial thickness and smooth muscle mass. In OVA-challenged Ndst1-deficient mice, lung eosinophil and macrophage infiltration as well as airway mucus accumulation, peribronchial fibrosis and airway epithelial thickness were significantly lower than in allergen-challenged WT mice along with a trend toward decreased airway smooth muscle mass. Leukocyte and endothelial Ndst 1 deficiency also resulted in significantly decreased expression of IL-13 as well as remodeling-associated mediators such as VEGF, FGF-2 and TGF-β1 in the lung tissue. At a cellular level, exposure to eotaxin-1 failed to induce TGF-β1 expression by Ndst1-deficient eosinophils relative to WT eosinophils. These studies suggest that leukocyte and endothelial Ndst1-modified HS contribute to the development of allergen-induced airway remodeling by promoting recruitment of inflammatory cells as well as regulating expression of pro-remodeling factors such as IL-13, VEGF, TGF-β1 and FGF-2 in the lung.
Keywords: allergen-induced airway remodeling, FGF-2, heparan sulfates, N-Deacetylase/N-Sulfotransferase-1, TGF-β
Introduction
Heparan sulfates (HS) are linear glycosaminoglycans (GAGs) covalently attached to a core protein occurring as HS proteoglycans (HSPGs). HSPGs are the most abundant sulfated GAGs in the lung parenchyma and are expressed by all cells in the body (Papakonstantinou and Karakiulakis 2009). Because of their ability to bind to multiple growth factors and their receptor tyrosine kinases as well as chemokines, interleukins, enzymes, enzyme inhibitors, lipases and apolipoproteins, extracellular matrix and plasma proteins (Bishop et al. 2007), HSPGs play a critical role in various physiological aspects including normal development and growth control, cell signaling and morphogenesis, cellular crosstalk, organization of basement membrane barriers, nutritional metabolism, injury and wound repair (Bishop et al. 2007), inflammatory responses (Parish 2006) and even microbial adhesion to host cells (Rostand and Esko 1997). The role of sulfotransferases in generating critical binding sites or niches for proteins to interact with GAGs is well recognized (Kusche-Gullberg and Kjellen 2003). The modification reaction involving N-sulfation which is unique to HS and does not occur in other sulfated GAGs is initiated with N-deacetylation followed by N-sulfation of selected N-acetylglucosamine residues and is carried out by a family of four bifunctional glucosaminyl N-deacetylase/N-sulfotransferase isoenzymes (Ndst1–4) (Kusche-Gullberg and Kjellen 2003).
The extent of sulfation and distribution of the sulfate substitutions on the HS backbone appear to play a vital role in regulating normal physiology. A systemic deletion of Ndst1 is lethal for mice due to forebrain defects (Ringvall et al. 2000) and various other defects are associated with targeted disruption of the Ndst1 gene (Ringvall and Kjellen 2010) which include skeletal malformation (Grobe et al. 2005; Pallerla et al. 2007), anophthalmia (Pan et al. 2006), lung hypoplasia with respiratory distress (Fan et al. 2000) and abnormal functioning of bone morphogenetic protein (Hu et al. 2009). In addition, HSPGs play an important role during conditions of inflammation. Endothelial HSPGs were found to promote neutrophilic inflammation in several acute inflammatory models (Wang et al. 2005). This is further supported by studies demonstrating that endothelial HSPGs promote recruitment of leukocytes to inflammatory sites by acting as ligands for adhesion molecules such as L-selectin as well as for chemokines and thus sequestering chemotactic gradients (Celie et al. 2007; Massena et al. 2010). Recent studies have shown that inactivation of the enzyme uronyl 2-O-sulfotransferase downstream from Ndst1 in endothelial cells resulting in a change in HS structure that is caused by increased N-sulfation and 6-O-sulfation of glucosamine units in response to the decreased 2-O-sulfation of uronic acid residues enhances neutrophil infiltration during acute inflammation in mice (Axelsson et al. 2012). This underscores the importance of the frequency and distribution of sulfate residues to the differential role of HSPGs during inflammation.
Using mice deficient in endothelial and leukocyte Ndst1 (Ndst1f/fTekCre+), we have previously demonstrated that endothelial but not leukocyte HSPGs play an important role in the development of airway inflammation including airway hyperresponsiveness (AHR) by supporting recruitment of inflammatory cells, specifically eosinophils, to the airways in a murine model of acute allergic airway inflammation (Zuberi et al. 2009). Allergic asthma, on the other hand, is a chronic disease caused by repetitive allergen exposure and is distinguished by several structural changes in the airways, including airway epithelial alterations, subepithelial fibrosis, increased airway smooth muscle mass and increased airway vascularization that are collectively termed as airway remodeling (Hamid and Tulic 2009). Given the previously identified role for Ndst1-modified HSPGs in supporting recruitment of inflammatory cells to allergic airways (Zuberi et al. 2009) and the ability of mediators such as cytokines, chemokines and growth factors released by these cells to drive airway remodeling (Al-Muhsen et al. 2011), we have examined whether these HSPGs contribute to the development of airway inflammation with features of remodeling in a murine model of chronic allergen-induced airway inflammation.
Results
HS expression in lungs of allergen-challenged WT and Ndst1f/fTekCre+ mice
Previous studies have demonstrated altered metabolism of proteoglycans, including small HSPGs, by bronchial fibroblasts from asthmatics that may result in increased proteoglycan deposition in the bronchial mucosa and contribute to AHR as well as airway remodeling (Westergren-Thorsson et al. 2002). Increased HSPG expression has also been shown in the airways of allergen-exposed rats as well as of asthmatic subjects (Venkatesan et al. 2012). We examined HSPG expression in the lungs of chronic allergen-challenged mice using a previously characterized phage display anti-HS antibody (Kurup et al. 2007) that interacts with saccharides containing N-, 2-O- and 6-O-sulfate as well as internal 2-O-sulfate groups. An increase in these sulfated proteoglycans was noted in the lungs of allergen-challenged WT mice relative to control mice. While HSPG expression was noted by endothelial cells and airway epithelial cells in the saline and allergen-exposed mice, inflammatory cells recruited to the airways also stained positive for HSPG in the allergen-exposed group (Figure 1A, left panels and B, center panel). In the lungs of allergen-exposed Ndst1f/fTekCre+ mice, HSPG expression was observed largely by airway epithelial cells and there was an overall reduction in HSPG expression consistent with the phenotype of these mice, i.e. endothelial and leukocyte-specific deficiency of Ndst1 (Figure 1A, right panels and B, bottom panel). Sub-epithelial basement membrane and smooth muscle cells were positive for expression of these sulfated proteoglycans in WT and Ndst1f/fTekCre+ mice.
Fig. 1.

Lung HS expression after allergen exposure. (A) Expression of sulfated PG in lungs of control and repetitively OVA-challenged WT and Ndst1f/fTekCre+ mice examined by immunofluorescence staining with VSV-tagged phage display-derived single chain antibody AO4B08 that interacts with the N-, 2-O-, and 6-O-sulfated saccharide motif as well as an internal 2-O-sulfate group. A non-GAG binding antibody MPB49 was used as a control. White arrows indicate positive (bright green) staining in the endothelium (Endo) and leukocytes (Leu). Red arrows indicate positive staining in the airway epithelium (Epi). Staining with a control antibody MPB49 in OVA-challenged Ndst1f/fTekCre+ mice is shown. Scale bar represents 30 μm. (B) Expression of sulfated PG by leukocytes recruited to the lungs of OVA-challenged WT and Ndst1f/fTekCre+ mice by immunofluorescence staining with antibody AO4B08. Staining with antibody MPB49 is shown as the control. Scale bar represents 10 μm. Representative data of n = 3 mice/group is shown.
Ndst1f/fTekCre+ mice exhibit attenuated cellular infiltration of the airways after chronic allergen challenge
We next compared inflammatory cellular responses in WT and Ndst1f/fTekCre+ mice. After chronic allergen challenge, WT mice demonstrated a significantly larger number of inflammatory cells in the lung tissue and bronchoalveolar lavage fluid (BALF) in comparison with Ndst1f/fTekCre+ mice (Figure 2A and B). A marked increase was noted in the number of eosinophils, neutrophils and lymphocytes in the allergen-challenged WT mice relative to the saline-exposed (control) group with a tendency for macrophages also to be higher (Figure 2B and C). Ndst1f/fTekCre+ mice also showed a similar trend after allergen exposure. However, compared with allergen-exposed WT mice, the number of eosinophils and macrophages in the BALF of allergen-challenged Ndst1f/fTekCre+ was significantly reduced with a trend toward a decrease in the number of neutrophils and lymphocytes as well albeit not statistically significant. In addition, allergen-exposed WT mice exhibited significantly higher lung tissue infiltration by eosinophils and macrophages relative to corresponding Ndst1f/fTekCre+ mice as assessed by eosinophil-specific major basic protein (MBP) (Figure 3A and B) and macrophage-specific F4/80 (Figure 3C and D) immunostaining, respectively. The number of lung tissue eosinophils and macrophages in control WT and Ndst1f/fTekCre+ mice were negligible.
Fig. 2.
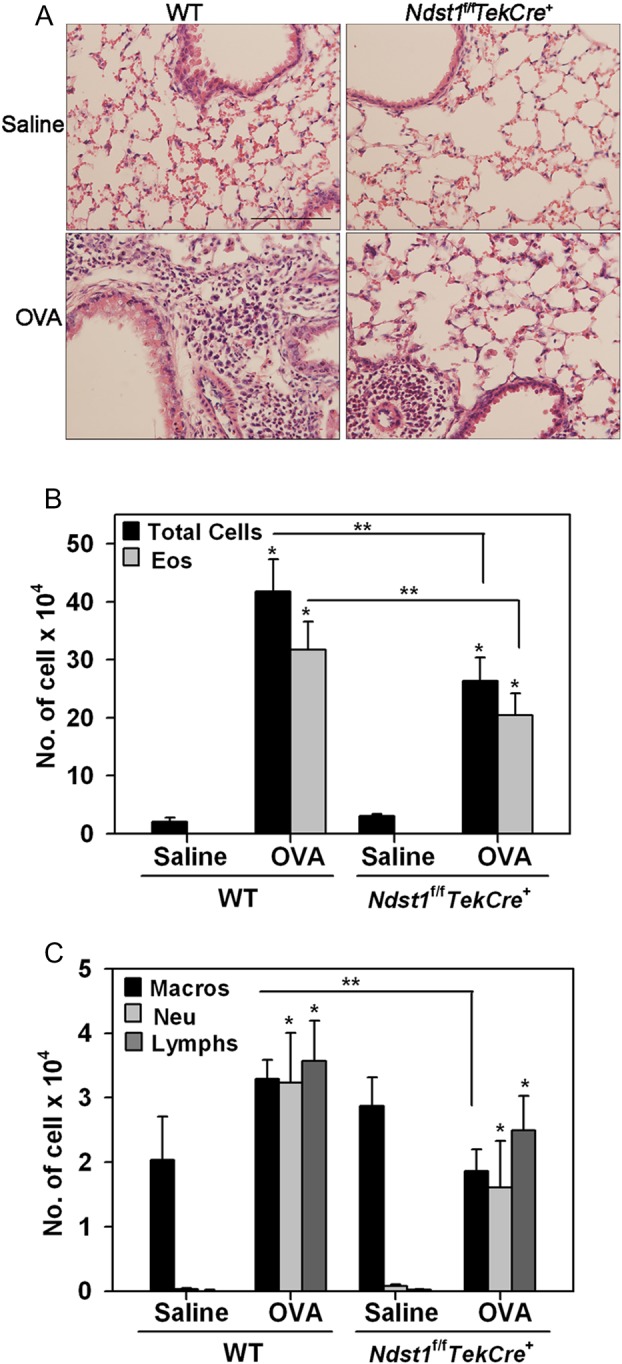
Reduced cellular infiltration of the airways in allergen-exposed Ndst1f/fTekCre+ mice. Lung tissue and BALF was collected from control and allergen-challenged WT and Ndst1f/fTekCre+ mice 24 h after the last challenge. (A) Cellular infiltration was evaluated by H&E staining of paraformaldehyde-fixed paraffin embedded lung tissue sections. Images representative of each group are shown. Scale bar represents 100 μm. (B and C) BALF was evaluated for total as well as differential cell counts by microscopic evaluation of cytocentrifuged slides based on morphological criteria after Hema 3 staining. Total and eosinophil [Eos] counts are shown in B; macrophage [Macros], neutrophil [Neu], and lymphocyte [Lymphs] counts are shown in C. Data represent mean ± SEM. Combined data of n = 5 mice for control groups and 7–8 mice for allergen-challenged groups is shown in B and C. *P < 0.01 in B and <0.04 in C for comparison of control vs. allergen-exposed mice. **P < 0.05 in B and C for comparison of allergen-challenged groups.
Fig. 3.

Reduced tissue eosinophils and macrophages in allergen-exposed Ndst1f/fTekCre+ mice. Lung tissue from control and allergen-exposed WT and Ndst1f/fTekCre+ mice was evaluated for eosinophils and macrophages by immunohistochemical staining. (A) Eosinophils were identified with eosinophil-specific MBP using rat mAb against murine MBP. A representative image of lung sections from each group is shown. (B) MBP-positive cells in randomly selected non-overlapping microscopic fields of lung tissue (5 fields/mouse for control group and 17 ± 2 [Mean ± STD] fields/mouse for allergen-exposed groups) were counted at a magnification of ×400. (C) Lung tissue macrophages were identified with rat anti-mouse F4/80. A representative image of lung sections from each group is shown. (D) The number of F4/80-positive cells in randomly selected non-overlapping microscopic fields (10 fields/mouse) of the alveolar tissue was counted at a magnification of ×400. Scale bar in A and C represents 100 μm. Combined data (Mean ± SEM) of n = 5–6 mice/group is shown in B and D. *P < 0.01 in for comparison of control vs. allergen-exposed mice and **P < 0.02 for comparison of allergen-challenged groups.
Allergen-challenged Ndst1f/fTekCre+ mice display lower levels of IFN-γ and IL-13
Chronic allergic inflammation including asthma is a complex disease involving many inflammatory cytokines that are central to almost every aspect of the immune response to an allergen. Th2 (IL-4, IL-5 and IL-13) and Th1 (IL-2 and IFN-γ) cytokine as well as TNF-α levels in the lung tissue of control and allergen-challenged WT and Ndst1f/fTekCre+ mice were determined (Table I). In addition, eotaxin-1 in the BALF of these mice was measured. In WT mice, repetitive allergen challenge induced a significant increase in levels of IL-5, IL-13, IFN-γ and eotaxin-1. Compared with WT mice, allergen-exposed Ndst1f/fTekCre+ mice showed significantly lower levels of lung IL-13 and IFN-γ. No change was noted in IL-4 levels in either group after allergen exposure. Allergen-induced IL-5 and eotaxin-1 expression in Ndst1f/fTekCre+ mice was similar to that observed in WT counterparts. IL-2 levels decreased to a similar extent in both groups after chronic allergen exposure while no significant differences were observed in TNF-α levels.
Table I.
Lung cytokine and eotaxin-1 levels in chronic allergen-exposed WT and Ndst1f/fTekCre+ mice
| Cytokine | WT |
Ndst1f/fTekCre+ |
||
|---|---|---|---|---|
| Saline | OVA | Saline | OVA | |
| IFNγa | 33.75 ± 5.02 | 99.84 ± 18.86* | 38.87 ± 5.24 | 25.61 ± 9.44** |
| IL-2a | 10.10 ± 2.5 | 6.3 ± 0.49 | 10.43 ± 1.73 | 5.81 ± 0.37 |
| IL-4a | 9.58 ± 0.66 | 11.67 ± 1.23 | 9.29 ± 0.4 | 8.45 ± 0.47 |
| IL-5a | 0.25 ± 0.84 | 84.11 ± 19.81* | 1.73 ± 1.12 | 81.73 ± 21.90* |
| IL-13a | 25.0 ± 4.07 | 39.41 ± 5.17* | 26.30 ± 2.66 | 27.05 ± 2.91** |
| TNF-αa | 106.57 ± 30.24 | 127.66 ± 38.85 | 106.65 ± 15.42 | 75.88 ± 11.71 |
| Eotaxin-1b | 12.9 ± 1.67 | 24.71 ± 5.47* | 13.46 ± 0.82 | 30.13 ± 8.6 |
apg/100 mg protein in lung tissue.
bpg/mL BALF.
*P < 0.05 for comparison of control vs. allergen-exposed mice and **P < 0.05 for comparison of allergen-challenged groups. n = 5–8 mice/group.
Allergen-challenged Ndst1f/fTekCre+ mice exhibit reduced airway remodeling
Since HSPGs are involved in biological functions such as activation of chemokines, enzymes and growth factors to modulate inflammatory responses, we examined the impact of altered endothelial and leukocyte HSPG expression caused by Ndst1 deficiency on airway remodeling, a distinctive feature of chronic airway inflammation associated with repetitive allergen exposure. As expected, WT mice demonstrated significantly increased airway mucus metaplasia, peribronchial fibrosis (based on tissue collagen), airway epithelial thickness, another feature characteristic of remodeled airways (Cohen et al. 2007) and airway smooth muscle mass after repetitive allergen exposure (Figures 4 and 5). While some degree of airway mucus metaplasia (Figure 4A) and peribronchial fibrosis (Figure 4C) was visible even in allergen-exposed Ndst1f/fTekCre+ mice relative to corresponding control mice, levels were significantly lower than in WT allergen-exposed mice (Figure 4B and D). Further, allergen-challenged Ndst1f/fTekCre+ mice exhibited significantly attenuated epithelial thickness compared with corresponding WT mice (Figure 5A and B). A similar trend was noted with respect to airway smooth muscle hypertrophy/hyperplasia, although the decrease in smooth muscle mass in allergen-exposed Ndst1f/fTekCre+ was not significantly lower than in WT mice (Figure 5C and D). Chronic airway inflammation is also associated with vascular remodeling that is largely mediated by vascular endothelial growth factor (VEGF) which is expressed and secreted by structural cells as well as inflammatory cells recruited to the airways (Meyer and Akdis 2013). We examined VEGF expression by inflammatory cells, predominantly macrophages (based on cell morphology), in the lung tissue of chronic allergen-challenged WT vs. Ndst1f/fTekCre+ mice by immunohistochemistry. Although an increased number of VEGF-positive inflammatory cells were present in the alveolar spaces of allergen-challenged WT as well as Ndst1f/fTekCre+ mice relative to respective control mice (Figure 5E and F), the number of VEGF-positive cells in the Ndst1f/fTekCre+ allergen-challenged group was significantly lower compared with corresponding WT counterparts.
Fig. 4.

Decreased airway mucus secretion and peribronchial fibrosis in allergen-exposed Ndst1f/fTekCre+ mice. (A and B) Airway mucus secretion in control and allergen-challenged WT and Ndst1f/fTekCre+ mice was examined by PAS staining of lung sections. Images representative of each group are shown. PAS-positive area in airways (5–8 airways/mouse) was quantitated by ImageJ analysis of captured images. (C and D) Peribronchial fibrosis around airways was examined by staining lung sections with Masson's trichrome stain for collagen deposition. Images representative of each group are shown. Fibrotic area was quantitated (2–13 airways/mouse with similar basement membrane length of 660 ± 20 μm) by image analysis using ImageJ. Scale bar in A and C represents 100 μm. Combined data (Mean ± SEM) of n = 5–6 mice/group is shown. *P < 0.01 for comparison of control vs. allergen-exposed mice in B and D and **P < 0.05 in B and <0.01 in D for comparison of allergen-challenged groups.
Fig. 5.

Epithelial thickness, smooth muscle mass and expression of VEGF is decreased in allergen-challenged Ndst1f/fTekCre+ mice. (A and B) Epithelial thickness in all intact airways was measured in H&E-stained lung sections from control (25–27 airways) and allergen-exposed (32–42 airways) WT and Ndst1f/fTekCre+ mice using ImageJ. Images representative of each group are shown. Arrows indicate the airway epithelium in OVA-challenged WT and Ndst1f/fTekCre+ mice. Scale bar represents 50 μm. (C and D) Airway smooth muscle mass in lung sections from control and allergen-exposed WT and Ndst1f/fTekCre+ mice was examined by immunohistochemical staining for α-SMA. Images representative of each group are shown. Peribronchial smooth muscle mass was quantitated (2–14 airways/mouse with similar basement membrane length of 714 ± 20 μm) by image analysis using ImageJ. Scale bar represents 100 μm. (E and F) VEGF expression by inflammatory cells in lung tissue from control and OVA-challenged WT and Ndst1f/fTekCre+ mice was detected by immunohistochemistry with polyclonal antibodies against VEGF. The number of VEGF-positive cells in randomly selected non-overlapping microscopic fields (5 fields/mouse) of the alveolar tissue in lung sections was counted at a magnification of ×400. Representative images are shown for each group. Scale bar represents 100 μm. Combined data (Mean ± SEM) of n = 5–6 mice/group is shown. *P < 0.01 for comparison of control vs. allergen-exposed mice in B, D and F, and **P < 0.01 for comparison of allergen-challenged groups in B and F.
Allergen-challenged Ndst1f/fTekCre+ mice exhibit decreased levels of lung TGF-β1 and FGF-2
Given that airway remodeling was attenuated in allergen-exposed Ndst1f/fTekCre+ mice relative to WT mice, we investigated the level of expression of TGF-β1 and FGF-2 in the lungs of these mice as their role in promoting events such as cell proliferation and differentiation that contribute to airway remodeling is well recognized (Bosse and Rola-Pleszczynski 2008; Halwani et al. 2011). Consistent with increased airway remodeling, allergen exposure significantly induced total TGF-β1 levels in BALF of WT mice. While BALF TGF-β1 levels in allergen-exposed Ndst1f/fTekCre+ mice were also higher than in corresponding control mice, they were significantly lower than in the allergen-exposed WT mice (Figure 6A). Further, evaluation of lung sections for total TGF-β1 expression by immunohistochemistry correlated positively with these findings (Figure 6B). An increased number of TGF-β1-positive inflammatory cells, predominantly macrophages (based on cell morphology), were present in the alveolar spaces of allergen-challenged WT mice relative to control mice (Figure 6B and C). In allergen-exposed Ndst1f/fTekCre+ mice, there were significantly fewer TGF-β1-positive inflammatory cells in the alveolar spaces compared with WT counterparts. Further, intensity of TGF-β1-positive staining of the inflammatory cells was lower in the allergen-exposed Ndst1f/fTekCre+ mice compared with WT mice (Figure 6B). In the case of TGF-β1 and also VEGF (described above), the number of cells positive for expression of these growth factors correlated with the reduced number of F4/80-positive macrophages present in the lung tissue of these mice after allergen challenge (Figure 3C and D).
Fig. 6.
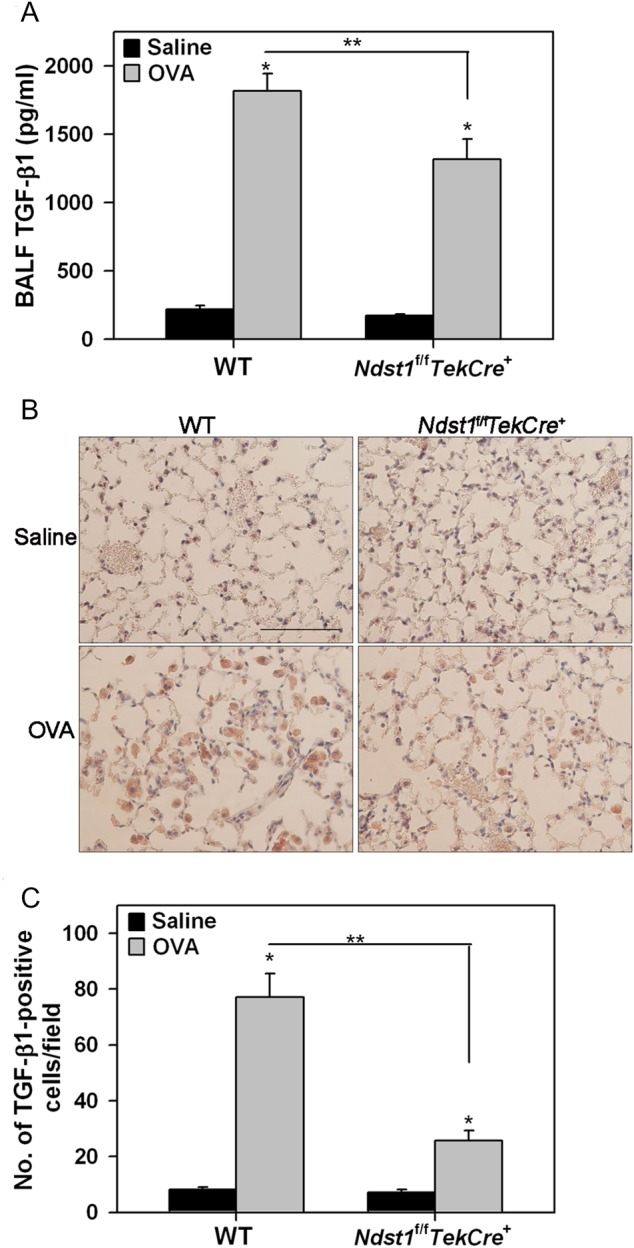
Lung TGF-β1 expression is reduced in allergen-challenged Ndst1f/fTekCre+ mice. (A) TGF-β1 in BALF from control (5–6 mice/group) and allergen-exposed (7–8 mice/group) WT and Ndst1f/fTekCre+ mice was determined by ELISA. (B) TGF-β1 expression in lung tissue of these mice was detected by immunohistochemistry with polyclonal antibodies against TGF-β1. Images representative of each group are shown. Scale bar represents 100 μm. (C) The number of TGF-β1-positive cells in randomly selected non-overlapping microscopic fields (5 fields/mouse) of the alveolar tissue in lung sections from control and allergen-exposed mice (n = 5–6 mice/group) were counted at a magnification of ×400. Data represents mean ± SEM. *P < 0.01 in A and C for comparison of control vs. allergen-exposed mice. **P < 0.02 in A and C for comparison of allergen-challenged groups.
Since TGF-β1 levels in the BALF was higher in allergen-challenged WT mice relative to Ndst1f/fTekCre+ mice and eosinophils constitute the major inflammatory cell type in allergic airways, expression of TGF-β1 at a cellular level was examined in bone marrow (BM)-cultured eosinophils from naïve (unstimulated) WT and Ndst1f/fTekCre+ mice by quantitative real-time RT–PCR (qPCR). In cells that were unstimulated but cultured in medium which contains IL-5 as part of the routine culture medium, expression of TGF-β1 mRNA was similar in WT and Ndst1f/fTekCre+ mice (Figure 7A). However, since activated eosinophils are a major source of TGF-β1 in asthmatic airways that can contribute to fibrotic changes in the lungs (Minshall et al. 1997), TGF-β1 expression by WT and Ndst1f/fTekCre+ eosinophils with and without exposure to eotaxin-1 was examined. Although exposure to eotaxin-1 did not have any effect on Ndst1 expression in WT eosinophils (Figure 7B), it induced a near 2-fold increase in TGF-β1 expression in WT eosinophils, but had no effect on Ndst1f/fTekCre+ eosinophils (Figure 7C), suggesting that Ndst1-modified HS may play a role in regulating TGF-β1 expression in activated eosinophils. Baseline expression of FGF-2 was similar in WT and Ndst1f/fTekCre+ mice and no effect was noted in WT eosinophils after exposure to eotaxin-1 (data not shown).
Fig. 7.

Expression of TGF-β1 by eosinophils from WT and Ndst1f/fTekCre+ mice. (A) Expression of TGF-β1 in unstimulated eosinophils cultured from BM of WT and Ndst1f/fTekCre+ mice by qPCR. (B) Expression of Ndst1 mRNA by eosinophils from WT mice with and without activation (100 nM eotaxin-1 for ∼16 h). (C) Expression of TGF-1 mRNA by eosinophils from WT and Ndst1f/fTekCre+ mice with and without activation with eotaxin-1. Combined data of three independent experiments with eosinophils from 2 different mice/experiment is shown (n = 6 mice/group). Results are expressed as fold change in expression relative to expression in WT cells in A and untreated cells in B and C that is set as 1. Data represent mean ± SEM. *P < 0.025 in C for comparison of untreated vs. eotaxin-1-treated eosinophils.
Evaluation of lung tissue FGF-2 expression by immunohistochemistry demonstrated that allergen-exposure results in a dramatic increase in FGF-2 expression mostly associated with macrophages (based on morphology) in the alveolar spaces of WT mice (Figure 8A and B). Negligible FGF-2 staining was noted in control mice of both groups. FGF-2 expression in allergen-exposed Ndst1f/fTekCre+ mice was only marginally higher than in corresponding control mice. Additionally, quantitative assessment of cellular FGF-2 expression based on intensity of staining demonstrated that FGF-2 expression by lung tissue macrophages in allergen-exposed Ndst1f/fTekCre+ mice is significantly lower compared with WT counterparts (Figure 8C).
Fig. 8.
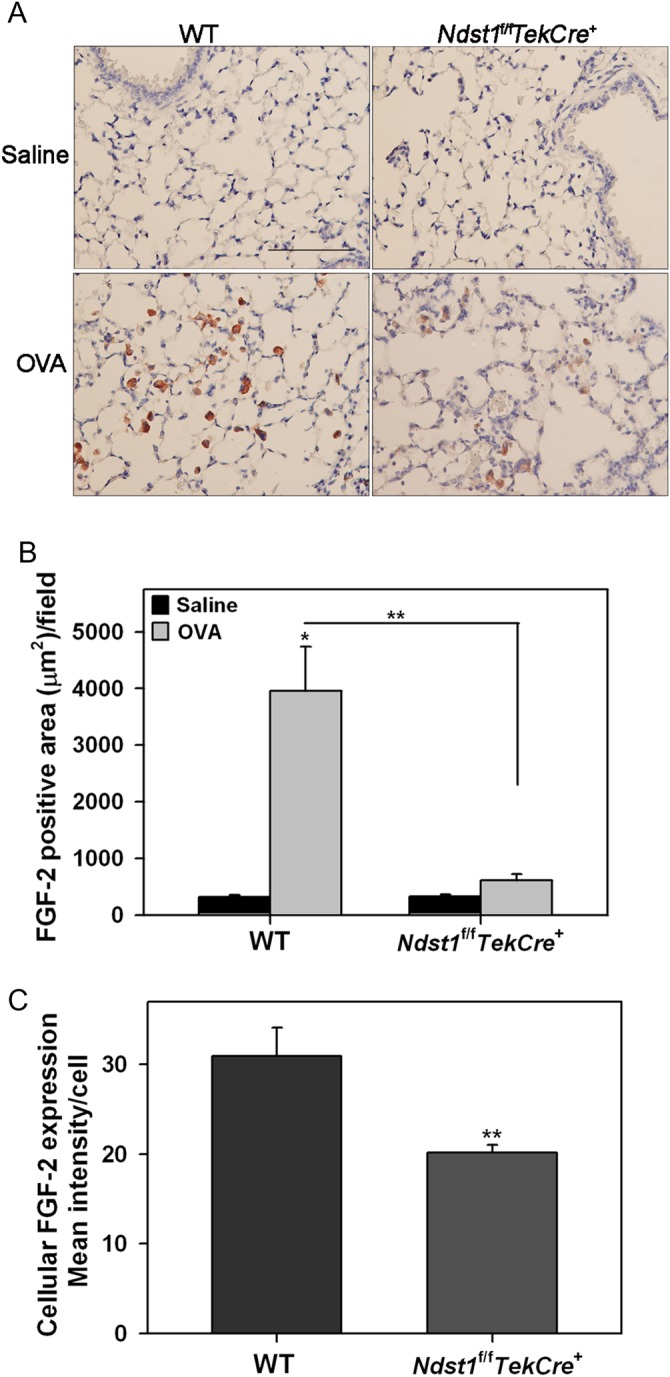
Allergen-challenged Ndst1f/fTekCre+ mice exhibit decreased lung FGF-2 expression. (A) FGF-2 expression in lung tissue of control and allergen-exposed WT and Ndst1f/fTekCre+ mice was detected by immunohistochemistry with polyclonal antibodies against FGF-2. Images representative of each group are shown. Scale bar represents 100 μm. (B) FGF-2 expression in the alveolar tissue was quantitated by image analysis using ImageJ. Five randomly selected non-overlapping fields were analyzed/mouse (n = 6 mice/group). (C) FGF-2 expression by individual inflammatory cells in the alveolar spaces of allergen-challenged WT and Ndst1f/fTekCre+ mice (n = 6 mice/group) was quantitated using ImageJ. Images of 5 non-overlapping microscopic fields were captured at a magnification of ×200 and the intensity of FGF-2 expression by 10 randomly selected cells (predominantly macrophages based on cell morphology) in each field was measured. Data represents mean ± SEM. *P < 0.01 in B for comparison of control vs. allergen-exposed WT mice. **P < 0.01 in B and C for comparison of allergen-challenged groups.
Discussion
HSPGs have multiple roles in inflammatory responses such as supporting leukocyte recruitment and transmigration as well as mediating cell proliferation and differentiation during establishment of acute and chronic inflammation (Parish 2006). We have previously demonstrated that endothelial HSPGs play an important role during the establishment of acute allergic airway inflammation by facilitating recruitment of inflammatory cells, particularly eosinophils, to the airways (Zuberi et al. 2009). Persistent exposure to allergens causes chronic airway inflammation including asthma that is characterized by lung tissue remodeling in addition to all the inflammatory responses seen with acute allergic inflammation (Hamid and Tulic 2009). Although pathological changes associated with airway remodeling are described extensively, mechanisms underlying chronic inflammation-associated airway remodeling are not fully identified. Here we have extended our previous observations with Ndst1-modified HSPGs and allergic airway inflammation to demonstrate that chronic allergen exposure results in increased HSPG expression in the lungs and that HSPGs contribute to the development of airway remodeling associated with sustained allergen exposure.
Inflammation caused by persistent recruitment of eosinophils, lymphocytes, macrophages, neutrophils and mast cells to airway tissues is assumed to be the initiating event for airway remodeling (Tagaya and Tamaoki 2007). In the present study, mice deficient in endothelial and leukocyte Ndst1 demonstrated significantly decreased recruitment of eosinophils and macrophages to the airways and lung tissue after exposure to chronic allergen challenge compared with corresponding WT mice. However, the reduction in neutrophil and lymphocyte numbers was only slightly lower than that in the WT mice. While the overall reduction in leukocyte recruitment to the airways of chronic allergen-exposed Ndst1f/fTekCre+ mice is consistent with our previous findings in acute allergen-exposed Ndst1f/fTekCre+ mice (Zuberi et al. 2009), the reduction in the number of each cell type recruited relative to allergen-exposed WT mice was not as striking as in the previous study. This could be a consequence of the repetitive allergen exposure resulting in sustained cellular recruitment and inflammation. Previous studies by us and others have revealed an important role for HSPGs in promoting leukocyte trafficking and recruitment in other models of acute inflammation as well such as neutrophilic inflammation (Wang et al. 2005), neuroinflammation (Floris et al. 2003; Zhang et al. 2012) and nephritis (Rops et al. 2008) by functioning as ligands for leukocyte-expressed receptors like L-selectin and Mac-1 as well as by facilitating chemokine gradients for transmigration (Diamond et al. 1995; Celie et al. 2005, 2007; Wang et al. 2005; Parish 2006). Further, our previous studies showed that trafficking of circulating leukocytes in lung microvessels of allergen-challenged Ndst1f/fTekCre+ mice was significantly lower than that observed in corresponding WT mice and that endothelial-expressed HSPGs, but not eosinophil-expressed HSPGs, are essential for efficient eosinophil rolling under conditions of flow (Zuberi et al. 2009). In the current study, eotaxin-1 levels in chronic allergen-challenged Ndst1f/fTekCre+ mice were similar to that in WT mice and are not likely to be a contributing factor for the reduced airway eosinophilia in these mice. However, cell surface HSPGs are also known to interact with and/or regulate activity of other eosinophil- and/or macrophage-selective chemokines which are important in the context of allergic asthma such as RANTES and MCP-1 (Zimmermann et al. 2003; Pease and Williams 2006) to assist cellular recruitment (Hoogewerf et al. 1997; Ali et al. 2002; Hardy et al. 2004). While levels of these cytokines were not measured in the current study, altered endothelial and leukocyte HSPG expression in the Ndst1f/fTekCre+ mice may impact their retention or biological activity at sites of inflammation resulting in decreased eosinophil and macrophage recruitment after allergen-exposure.
Several pro-inflammatory Th2 type cytokines (e.g. IL-5, IL-13) released by leukocytes recruited to the airways are key players contributing to airway remodeling in chronic asthma (Tagaya and Tamaoki 2007; Al-Muhsen et al. 2011). In the present study, levels of IL-4, -5 and -13 were measured as representative Th2 cytokines in the lungs of allergen-exposed Ndst1f/fTekCre+ and WT mice. Allergen-induced IL-13 levels were significantly lower in Ndst1f/fTekCre+ mice relative to corresponding WT mice while IL-5 levels remained similar to those in WT mice. T cells are the predominant producers of both these cytokines and the number of lymphocytes in allergen-challenged Ndst1f/fTekCre+ mice was not significantly lower than in WT counterparts to account for the decreased IL-13. Studies have shown that expression and release of IL-13 is induced by airway epithelial cells when they undergo injury and repair as in airway remodeling (Allahverdian et al. 2008) and can thus be an additional source of IL-13 in asthmatic airways. Since allergen-challenged Ndst1f/fTekCre+ mice demonstrate an overall reduction in airway remodeling, IL-13 expression and release by the airway epithelium in these mice may be lower than in corresponding WT mice and thus account for the lower IL-13 levels in the lung tissue. Human eosinophils (Spencer et al. 2009) and macrophages express IL-13 (Hancock et al. 1998) and decreased recruitment of the murine counterparts of these two cell types in the BALF as well as the lung tissue of chronic allergen-challenged Ndst1f/fTekCre+ mice in the current study could in part contribute to the reduced IL-13 levels. Another possibility is an indirect effect of eosinophil- and/or macrophage-released factor(s) that may regulate IL-13 expression and release by T cells. Reduced eosinophil and macrophage recruitment in allergen-exposed Ndst1f/fTekCre+ mice would thus result in decreased IL-13 release by recruited T cells. However, additional studies are needed to further examine this. Consistent with the reduced lung cellular inflammation and IL-13 levels, airway epithelial thickness, mucus hypersecretion and peribronchial fibrosis were significantly attenuated in allergen-exposed Ndst1f/fTekCre+ mice relative to corresponding WT mice. Likewise, airway smooth muscle mass in these mice was reduced to levels observed in saline-exposed control Ndst1f/fTekCre+ mice, although the reduction compared with allergen-exposed WT was not statistically significant.
Another allergen-induced cytokine that was significantly reduced in Ndst1f/fTekCre+ mice was the Th1 cytokine IFN-γ. IFN-γ binds to basement membrane as well as cell surface HSPGs which protects IFN-γ from proteolytic cleavage and promotes increased stability (Lortat-Jacob 2006). Altered expression of HSPG by the endothelium and leukocytes in Ndst1f/fTekCre+ mice may affect the binding and thus the stability of IFN-γ resulting in decreased levels of this cytokine in the lung. While allergic asthma is largely a Th2-driven inflammatory disease, IFN-γ has also been shown to play a proinflammatory role. Elevated serum IFN-γ in atopic asthma correlates with impaired lung function (ten Hacken et al. 1998) and a co-operative role for this cytokine with Th2 cytokines in allergen-induced AHR has been identified more so in a chronic setting (Kumar et al. 2006). More recently, a role for IFN-γ was shown in the development of allergen-induced AHR, eosinophilic inflammation and airway remodeling, specifically subepithelial fibrosis and hyperplasia of mucus-secreting goblet cells, via mast cell IFN-γ receptor (Yu et al. 2011). Consistent with these findings, diminished levels of lung IFN-γ could be an added explanation for the attenuated airway remodeling in allergen-challenged Ndst1f/fTekCre+ mice.
In addition to IL-13 and IFNγ, expression of VEGF, total TGF-β1 and FGF-2 in the lungs of allergen-challenged Ndst1f/fTekCre+ mice were significantly lower than in corresponding WT mice. HSPGs are known to bind to and regulate the biological activity of VEGF (Robinson et al. 2006; Xu et al. 2011), TGF-β1 (Lyon et al. 1997) and FGF-2 (Maccarana et al. 1993). VEGF, a known pro-angiogenic growth factor (Meyer and Akdis 2013), is elevated in asthmatic patients (Hoshino et al. 2001) as well as chronic allergen-exposed mice (Lee et al. 2006). Recent studies indicate that alveolar macrophage-derived VEGF is necessary for allergic airway inflammation in asthmatic mice (Song et al. 2012). IL-13, which is elevated in allergen-exposed mice, is known to induce release of specific isoforms of VEGF (Corne et al. 2000; Wen et al. 2003). In the current study, increased recruitment of inflammatory cells that express VEGF to the lungs along with elevated IL-13 levels and retention of secreted VEGF by binding to HSPG are all likely to contribute to vascular remodeling in the airways of allergen-challenged WT mice. In contrast, decreased cellular inflammation along with lower IL-13 levels and less binding and retention due to altered HSPGs expression caused by endothelial and leukocyte Ndst1 deficiency may lead to reduced vascular remodeling in the lungs of allergen-challenged Ndst1f/fTekCre+ mice. Recent studies have shown that HSPG isolated from endothelial cells treated with micro RNA to suppress Ndst1 have reduced affinity for VEGF and the treated cells are less responsive to VEGF (Kasza et al. 2013). FGF-2 and TGF-β1 promote bronchial smooth muscle cell and fibroblast proliferation and differentiation and thus contribute to airway remodeling (Bosse and Rola-Pleszczynski 2008; Halwani et al. 2011). While the effect of FGF-2 on differentiation of smooth muscle cells appears to be controversial (Bosse and Rola-Pleszczynski 2008; Schuliga et al. 2013), its proliferative effects on human (Hetzel et al. 2005) and mouse (Xiao et al. 2012) fibroblasts as well as endothelial cells (Sahni and Francis 2004) is well established. Besides endothelial cells, epithelial cells, fibroblasts and airway smooth muscle cells, eosinophils and macrophages are producers of FGF-2 (Cordon-Cardo et al. 1990; Kranenburg et al. 2002; Stenfeldt and Wennerås 2004; Yum et al. 2011) and TGF-β1 (Halwani et al. 2011) and expression of both these growth factors is induced after repetitive allergen exposure in mice, especially by macrophages (Yum et al. 2011). FGF-2 levels are known to be elevated in the BALF and sputum of patients with asthma (Redington et al. 2001; Bissonnette et al. 2014). Thus, elevated FGF-2 levels in chronic allergen-challenged airways can exert proinflammatory effects on various cell types in the lungs.
Decreased expression of VEGF, TGF-β1 and FGF-2 in the lungs of allergen-exposed Ndst1f/fTekCre+ mice, especially by macrophages in the alveolar tissue (based on cell morphology), could be attributed to a reduced number of tissue macrophages in these mice as well as a reduced expression of these growth factors at a cellular level. Binding of these growth factors to HSPGs allows creation of a local reservoir and protection from degradation. Altered expression of HSPGs by endothelial cells and inflammatory leukocytes in Ndst1f/fTekCre+ mice may lead to decreased binding and retention of these growth factors released by eosinophils and macrophages themselves or other cell types in response to sustained allergen exposure, thus resulting in reduced cell activation in these mice. Further, intracellular Ndst1-modified HSPG may also play a role in regulating expression of these growth factors. Intracellular HSPG in general have received less attention relative to those in the extracellular matrix or on the cell surface, although they have been reported to have biological functions in storage granules, the nucleus and other intracellular organelles (Kolset et al. 2004). Based on previous studies indicating that eotaxin does not interact with HS or a range of GAG (Ellyard et al. 2007), our data demonstrating induction of TGF-β1 expression by eotaxin-1 in WT but not Ndst1f/fTekCre+ eosinophils suggest a potential role for intracellular Ndst1-modified HSPG in regulating expression of TGF-β1 by these cells. Although no effect was noted on FGF-2 expression in eosinophils after eotaxin-1 exposure, it is possible that this chemokine may just not be an inducer of FGF-2 while other inflammatory mediators of chronic airway inflammation may have an alternate effect. Overall, combined with reduced recruitment of inflammatory cells, release of decreased levels of VEGF, TGF-β1 and FGF-2 during inflammatory conditions or in response to inflammatory mediators, as exemplified in the case of TGF-β1 by eosinophils, may result in attenuated airway remodeling in chronic allergen exposed Ndst1f/fTekCre+ mice.
In summary, our studies demonstrate a role for HSPGs in promoting airway remodeling associated with chronic allergic airway inflammation and suggest that targeting HSPG in the lungs to modulate inflammatory cell recruitment and pro-inflammatory activity of mediators may serve as a mechanism for potential pharmacological intervention to inhibit airway remodeling associated with chronic allergic asthma.
Materials and methods
Animal model of chronic allergic airway inflammation
Ndst1f/fTekCre+ mice on C57Bl/6 background and deficient in endothelial and leukocyte Ndst1 were generated as described previously (Fuster et al. 2007; Zuberi et al. 2009). Offspring were genotyped using genomic DNA isolated from tail clippings as described (Wang et al. 2005; Fuster et al. 2007). Wild-type (WT) C57Bl/6 and Ndst1f/fTekCre+ mice (8–12 weeks) were exposed to chronic allergen challenge with ovalbumin (OVA) (Grade V, Sigma Chemical Co., St Louis, MO) as described previously (Cho et al. 2010, 2011). Briefly, mice were sensitized with 50 µg OVA in 0.5 mg aluminum hydroxide by subcutaneous injection on days 0, 7, 14 and 21 and then challenged intranasally with OVA (20 µg/mouse) on days 23, 25, 28. This was followed by biweekly intranasal challenges with OVA for an additional 4 weeks. Age and gender matched WT and Ndst1f/fTekCre+ mice sensitized and challenged with saline instead of OVA were used as controls. All studies involving mice were performed following procedures and standards approved by the local Institutional Animal Care and Use Committee.
BALF and lung tissue collection
Mice were sacrificed 24 h after the last allergen challenge and lungs were lavaged with 1 mL of saline. Total cell counts in the BALF were determined using a hemocytometer. BALF was centrifuged and the supernatant was stored at −80°C for further evaluation. Differential cell counts were determined based on morphological criteria from cytospin slides of BALF cells stained with Hema 3 staining system (Fisher Diagnostics, Middletown, VA). Right lungs were snap-frozen and stored at −80°C while left lungs were perfused with 4% paraformaldehyde to preserve pulmonary structure, fixed in 4% paraformaldehyde for 48 h at 4°C and paraffin-embedded.
Measurement of lung Th1/Th2 cytokines
Protein concentration in supernatants of lung tissue homogenates prepared in lysis buffer (PBS containing 1% Triton X-100 and protease inhibitors) was measured (BCA Protein Assay Kit, Pierce Biotechnology, Rockford, IL). Th1 (IL-2 and IFN-γ)/Th2 (IL-4, IL-5, IL-13) cytokine and TNF-α levels in the lung tissue supernatants was determined using cytometric bead array Flex Set kits (BD Biosciences, San Diego, CA) according to the manufacturer with a FACScan flow cytometer equipped with CellQuest Pro™ Software (BD Biosciences) for data acquisition and FlowJo Software (Tree Star, Inc., Ashland, OR) for analysis or a FACSCanto II flow cytometer (for IL-13) with FACSDiva™ Software for data acquisition and analysis. Level of each cytokine was expressed as pg cytokine/100 mg protein.
Measurement of eotaxin-1 and TGF-β1 by ELISA
Eotaxin-1 and total TGF-β1 in the BALF was measured using ELISA kits (R & D Systems, Minneapolis, MN) according to the manufacturers' recommendations. Optical density of the color developed was measured at 450/570 nm with a microplate reader (FLUOstar OPTIMA, BMG LABTECH, Durham, NC) and the concentration of eotaxin-1 or TGF-β1 in the sample was determined against a standard curve generated with a detection limit of 15.6 pg/mL.
Lung histology
To examine HS expression in lung tissue, immunofluorescence studies were performed using a VSV-tagged phage display-derived single chain antibody AO4B08, that interacts with the N-, 2-O-, and 6-O-sulfated saccharide motif, as well as an internal 2-O-sulfate group (Kurup et al. 2007) and a non-GAG binding antibody MPB49 as the control (both antibodies kindly provided by Dr. T. van Kuppevelt, Radboud University Nijmegen Medical Center, The Netherlands). Bound antibodies were detected with monoclonal anti-VSV glycoprotein (Sigma-Aldrich, St. Louis, MO) followed by FITC-conjugated goat anti-mouse IgG. To examine cellular infiltration of lungs, tissue sections were stained with Harris Modified Hematoxylin and Shandon instant eosin (H&E, Thermo Fisher Scientific Co., Pittsburgh, PA). Lung tissue eosinophils and macrophages were detected by immunohistochemical staining of lung sections of mice from two independent allergen challenge studies for eosinophil-specific MBP with rat mAb against mouse MBP (1 μg/mL) or with rat anti-mouse F4/80 (10 μg/mL, Abcam, Cambridge, MA) (Zuberi et al 2009), respectively. MBP-positive or F4/80-positive cells in randomly selected non-overlapping microscopic fields were counted at a magnification of ×400 and results were expressed as average number of MBP-positive or F4/80-positive cells per field. For MBP, 5 fields for saline groups and an average of 17 ± 2 (mean ± STD) fields for allergen-challenged groups were counted per slide and for F4/80, 10 fields were counted per slide. For expression of VEGF, TGF-β1 and FGF-2, lung tissue sections were stained with polyclonal antibodies against VEGF (4 µg/mL), total TGF-β1 (1 μg/mL) or FGF-2 (0.2 μg/mL) (all from Santa Cruz Biotechnology, Dallas, TX). VEGF or TGF-β1-positive cells (predominantly macrophages based on cell morphology) were counted in 5 randomly selected non-overlapping fields of the alveolar tissue per slide at a magnification of ×400. FGF-2 expression was quantitated in images of 5 non-overlapping fields of the alveolar tissue per slide at a magnification of ×100 using ImageJ image analysis system (Abramoff et al. 2004). The results were presented as FGF-2 positive area (μm2) per field. FGF-2 expression by individual inflammatory cells in the alveolar spaces was also quantitated using ImageJ. Images of 5 non-overlapping microscopic fields were captured at a magnification of ×200 and the intensity of FGF-2 expression by 10 randomly selected cells (predominantly macrophages based on cell morphology) in each field was measured. Results were expressed as mean intensity/cell.
Airway remodeling analysis
Lungs of representative mice from two independent allergen challenge studies were analyzed for airway mucus accumulation, peribronchial fibrosis, epithelial thickness and peribronchial smooth muscle hypertrophy/hyperplasia. Both large and small airways were examined in all analyses. To determine airway mucus production, lung sections were stained with periodic acid-Schiff's (PAS) reagent (Sigma-Aldrich®, St. Louis, MO). PAS-positive area was quantitated using ImageJ and expressed as percent PAS-positive area per airway (Kang et al. 2012). Subepithelial fibrosis was assessed using Masson's trichrome stain (Sigma-Aldrich®) and quantitated by ImageJ. Results were expressed as area of fibrosis (μm2) per μm basement membrane length (Ge et al. 2010). Airway epithelial thickness in H&E stained lung sections was measured as described previously (Doherty et al. 2012). Thickness of the epithelium from the bottom of the basement membrane to the mucosal surface of the airway epithelium was measured using ImageJ. All intact airways (up to 10) were analyzed and four different areas were measured per airway. To determine airway smooth muscle hypertrophy/hyperplasia, expression of α-smooth muscle actin (SMA) was evaluated by immunostaining using mAb against murine SMA (0.4 μg/mL, Sigma-Aldrich®). Peribronchial smooth muscle mass was quantitated by ImageJ and expressed as SMA positive area (μm2) per mm basement membrane length (Ge et al. 2010).
Murine eosinophils
Eosinophils were cultured from BM of naïve WT and Ndst1f/fTekCre+ mice as described previously (Dyer et al. 2008) but with minor modifications as stated in our earlier studies (Bahaie et al. 2012). Cells between 12–14 days of culture differentiated based on Hema 3 staining and verified for expression of both MBP and Siglec-F (>95% eosinophils) (Bahaie et al. 2012) were used in studies.
qPCR
Eosinophils from WT and Ndst1f/fTekCre+ mice were activated by overnight culture (∼16 h) in medium containing eotaxin-1 (100 nM) as described in our previous study (Ha et al. 2013) and then evaluated for expression of TGF-β1 or FGF-2 by qPCR using previously published primers specific for each molecule (Teshima-Kondo et al. 2004; Sullivan et al. 2009). qPCR was carried out in a iQ™5 multicolor real-time PCR detection System (Bio-Rad, Hercules, CA) as described in our previous studies (Ha et al. 2013). The amount of TGF-β1 or FGF-2 mRNA in each sample was calculated based on its threshold cycle, Ct, suggested by the software (IQ™5 Optical System software) after subtraction of the Ct of the housekeeping gene β-actin. Results are expressed as fold change in expression relative to expression in untreated cells.
Statistical analysis
The results are expressed as the mean ± SE. Statistical significance was determined by unpaired Student's t-test. A P value of <0.05 was considered as significant.
Funding
This work was supported by National Institutes of Health [U19-AI70535 and AI35796 to P.S. and HL107150 to J.D.E.].
Conflict of interest
None declared.
Abbreviations
HS, heparan sulfate; GAG, glycosaminoglycan; HSPG, hepran sulfate proteoglycan; Ndst1, glucosaminyl N-deacetylase/N-sulfotransferase-1; AHR, airway hyperresponsiveness; BALF, bronchoalveolar lavage fluid; FGF-2, fibroblast growth factor-2; MBP, major basic protein; OVA, ovalbumin; PAS, periodic acid-Schiff; SMA, α-smooth muscle actin; TGF-β1, transforming growth factor- β1; VEGF, vascular endothelial growth factor.
References
- Abramoff MD, Magalhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J Allergy Clin Immunol. 2011;128:451–462. doi: 10.1016/j.jaci.2011.04.047. [DOI] [PubMed] [Google Scholar]
- Ali S, Fritchley SJ, Chaffey BT, Kirby JA. Contribution of the putative heparan sulfate-binding motif BBXB of RANTES to transendothelial migration. Glycobiology. 2002;12:535–543. doi: 10.1093/glycob/cwf069. [DOI] [PubMed] [Google Scholar]
- Allahverdian S, Harada N, Singhera GK, Knight DA, Dorscheid DR. Secretion of IL-13 by Airway Epithelial Cells Enhances Epithelial Repair via HB-EGF. Am J Respir Cell Mol Biol. 2008;38:153–160. doi: 10.1165/rcmb.2007-0173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson J, Xu D, Kang BN, Nussbacher JK, Handel TM, Ley K, Sriramarao P, Esko JD. Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood. 2012;120:1742–1751. doi: 10.1182/blood-2012-03-417139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahaie NS, Hosseinkhani RM, Ge XN, Kang BN, Ha SG, Blumenthal MN, Jessberger R, Rao SP, Sriramarao P. Regulation of eosinophil trafficking by SWAP-70 and its role in allergic airway inflammation. J Immunol. 2012;188:1479–1490. doi: 10.4049/jimmunol.1102253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Bissonnette EY, Madore AM, Chakir J, Laviolette M, Boulet LP, Hamid Q, Bergeron C, Maghni K, Laprise C. Fibroblast growth factor-2 is a sputum remodeling biomarker of severe asthma. J Asthma. 2014;51:119–126. doi: 10.3109/02770903.2013.860164. [DOI] [PubMed] [Google Scholar]
- Bosse Y, Rola-Pleszczynski M. FGF2 in asthmatic airway-smooth-muscle-cell hyperplasia. Trends Mol Med. 2008;14:3–11. doi: 10.1016/j.molmed.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Celie JW, Keuning ED, Beelen RHJ, Drager AM, Zweegman S, Kessler FL, Soininen R, van den Born J. Identification of L-selectin Binding Heparan Sulfates Attached to Collagen Type XVIII. J Biol Chem. 2005;280:26965–26973. doi: 10.1074/jbc.M502188200. [DOI] [PubMed] [Google Scholar]
- Celie JW, Rutjes NW, Keuning ED, Soininen R, Heljasvaara R, Pihlajaniemi T, Dräger AM, Zweegman S, Kessler FL, Beelen RH, et al. Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am J Pathol. 2007;170:1865–1878. doi: 10.2353/ajpath.2007.070061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JY, Pham A, Rosenthal P, Miller M, Doherty T, Broide DH. Chronic OVA allergen challenged TNF p55/p75 receptor deficient mice have reduced airway remodeling. Int Immunopharmacol. 2011;11:1038–1044. doi: 10.1016/j.intimp.2011.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JY, Song DJ, Pham A, Rosenthal P, Miller M, Dayan S, Doherty T, Varki A, Broide DH. Chronic OVA allergen challenged Siglec-F deficient mice have increased mucus, remodeling, and epithelial Siglec-F ligands which are up-regulated by IL-4 and IL-13. Respiratory Research. 2010;11:154. doi: 10.1186/1465-9921-11-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L, Xueping E, Tarsi J, Ramkumar T, Horiuchi TK, Cochran R, DeMartino S, Schechtman KB, Hussain I, Holtzman MJ, et al. Epithelial Cell Proliferation Contributes to Airway Remodeling in Severe Asthma. Am J Respir Crit Care Med. 2007;176:138–145. doi: 10.1164/rccm.200607-1062OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordon-Cardo C, Vlodavsky I, Haimovitz-Friedman A, Hicklin D, Fuks Z. Expression of basic fibroblast growth factor in normal human tissues. Lab Invest. 1990;63:832–840. [PubMed] [Google Scholar]
- Corne J, Chupp G, Lee CG, Homer RJ, Zhu Z, Chen Q, Ma B, Du Y, Roux F, McArdle J, et al. IL-13 stimulates vascular endothelial cell growth factor and protects against hyperoxic acute lung injury. J Clin Invest. 2000;106:783–791. doi: 10.1172/JCI9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Alon R, Parkos CA, Quinn MT, Springer TA. Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD1) J Cell Biol. 1995;130:1473–1482. doi: 10.1083/jcb.130.6.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty TA, Khorram N, Sugimoto K, Sheppard D, Rosenthal P, Cho JY, Pham A, Miller M, Croft M, Broide DH. Alternaria induces STAT6-dependent acute airway eosinophilia and epithelial FIZZ1 expression that promotes airway fibrosis and epithelial thickness. J Immunol. 2012;188:2622–2629. doi: 10.4049/jimmunol.1101632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer KD, Moser JM, Czapiga M, Siegel SJ, Percopo CM, Rosenberg HF. Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow. J Immunol. 2008;181:4004–4009. doi: 10.4049/jimmunol.181.6.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellyard JI, Simson L, Bezos A, Johnston K, Freeman C, Parish CR. Eotaxin Selectively Binds Heparin: An Interaction That Protects Eotaxin From Proteolysis And Potentiates Chemotactic Activity In Vivo. J Biol Chem. 2007;282:15238–15247. doi: 10.1074/jbc.M608046200. [DOI] [PubMed] [Google Scholar]
- Fan G, Xiao L, Cheng L, Wang X, Sun B, Hu G. Targeted disruption of NDST-1 gene leads to pulmonary hypoplasia and neonatal respiratory distress in mice. FEBS Lett. 2000;467:7–11. doi: 10.1016/s0014-5793(00)01111-x. [DOI] [PubMed] [Google Scholar]
- Floris S, van den Born J, van der Pol SMA, Dijkstra CD, De Vries HE. Heparan Sulfate Proteoglycans Modulate Monocyte Migration across Cerebral Endothelium. J Neuropathol Exp Neurol. 2003;62:780–790. doi: 10.1093/jnen/62.7.780. [DOI] [PubMed] [Google Scholar]
- Fuster MM, Wang L, Castagnola J, Sikora L, Reddi K, Lee PHA, Radek KA, Schuksz M, Bishop JR, Gallo RL, et al. Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J Cell Biol. 2007;177:539–549. doi: 10.1083/jcb.200610086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XN, Bahaie NS, Kang BN, Hosseinkhani RM, Ha SG, Frenzel EM, Liu FT, Rao SP, Sriramarao P. Allergen-induced airway remodeling is impaired in galectin-3 deficient mice. J Immunol. 2010;185:1205–1214. doi: 10.4049/jimmunol.1000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobe K, Inatani M, Pallerla SR, Castagnola J, Yamaguchi Y, Esko JD. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development. 2005;132:3777–3786. doi: 10.1242/dev.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S, Ge XN, Bahaie NS, Kang BN, Rao A, Rao SP, Sriramarao P. ORMDL3 promotes eosinophil trafficking and activation via regulation of integrins and CD48. Nat Commun. 2013;4:2479. doi: 10.1038/ncomms3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halwani R, Al-Muhsen S, Al-Jahdali H, Hamid Q. Role of Transforming Growth Factor-β in Airway Remodeling in Asthma. Am J Respir Cell Mol Biol. 2011;44:127–133. doi: 10.1165/rcmb.2010-0027TR. [DOI] [PubMed] [Google Scholar]
- Hamid Q, Tulic M. Immunobiology of Asthma. Annu Rev Physiol. 2009;71:489–507. doi: 10.1146/annurev.physiol.010908.163200. [DOI] [PubMed] [Google Scholar]
- Hancock A, Armstrong L, Gama R, Millar A. Production of Interleukin 13 by Alveolar Macrophages from Normal and Fibrotic Lung. Am J Respir Cell Mol Biol. 1998;18:60–65. doi: 10.1165/ajrcmb.18.1.2627. [DOI] [PubMed] [Google Scholar]
- Hardy LA, Booth TA, Lau EK, Handel TM, Ali S, Kirby JA. Examination of MCP-1 (CCL2) partitioning and presentation during transendothelial leukocyte migration. Lab Invest. 2004;84:81–90. doi: 10.1038/labinvest.3700007. [DOI] [PubMed] [Google Scholar]
- Hetzel M, Bachem M, Anders D, Trischler G, Faehling M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung. 2005;183:225–237. doi: 10.1007/s00408-004-2534-z. [DOI] [PubMed] [Google Scholar]
- Hoogewerf AJ, Kuschert GS, Proudfoot AE, Borlat F, Clark-Lewis I, Power CA, Wells TN. Glycosaminoglycans Mediate Cell Surface Oligomerization of Chemokines. Biochemistry. 1997;36:13570–13578. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- Hoshino M, Takahashi M, Aoike N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J Allergy Clin Immunol. 2001;107:295–301. doi: 10.1067/mai.2001.111928. [DOI] [PubMed] [Google Scholar]
- Hu Z, Wang C, Xiao Y, Sheng N, Chen Y, Xu Y, Zhang L, Mo W, Jing N, Hu G. NDST1-dependent heparan sulfate regulates BMP signaling and internalization in lung development. J Cell Sci. 2009;122:1145–1154. doi: 10.1242/jcs.034736. [DOI] [PubMed] [Google Scholar]
- Kang BN, Ha SG, Ge XN, Hosseinkhani MR, Bahaie NS, Greenberg Y, Blumenthal MN, Puri KD, Rao SP, Sriramarao P. The p110δ subunit of PI3 K regulates bone marrow-derived eosinophil trafficking and airway eosinophilia in allergen-challenged mice. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1179–L1191. doi: 10.1152/ajplung.00005.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasza Z, Fredlund Fuchs P, Tamm C, Eriksson AS, O'Callaghan P, Heindryckx F, Spillmann D, Larsson E, Le Jan S, Eriksson I, et al. MicroRNA-24 Suppression of N-Deacetylase/N-Sulfotransferase-1 (NDST1) Reduces Endothelial Cell Responsiveness to Vascular Endothelial Growth Factor A (VEGFA) J Biol Chem. 2013;288:25956–25963. doi: 10.1074/jbc.M113.484360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolset SO, Prydz K, Pejler G. Intracellular proteoglycans. Biochem J. 2004;379:217–227. doi: 10.1042/BJ20031230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg AR, de Boer WI, Van Krieken JH, Mooi WJ, Walters JE, Saxena PR, Sterk PJ, Sharma HS. Enhanced Expression of Fibroblast Growth Factors and Receptor FGFR-1 during Vascular Remodeling in Chronic Obstructive Pulmonary Disease. Am J Respir Cell Mol Biol. 2002;27:517–525. doi: 10.1165/rcmb.4474. [DOI] [PubMed] [Google Scholar]
- Kumar RK, Webb DC, Herbert C, Foster PS. Interferon-gamma as a possible target in chronic asthma. Inflamm Allergy Drug Targets. 2006;5:253–256. doi: 10.2174/187152806779010909. [DOI] [PubMed] [Google Scholar]
- Kurup S, Wijnhoven TJM, Jenniskens GJ, Kimata K, Habuchi H, Li JP, Lindahl U, van Kuppevelt TH, Spillmann D. Characterization of Anti-heparan Sulfate Phage Display Antibodies AO4B08 and HS4E4. J Biol Chem. 2007;282:21032–21042. doi: 10.1074/jbc.M702073200. [DOI] [PubMed] [Google Scholar]
- Kusche-Gullberg M, Kjellen L. Sulfotransferases in glycosaminoglycan biosynthesis. Curr Opin Struct Biol. 2003;13:605–611. doi: 10.1016/j.sbi.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Lee SY, Cho JY, Miller M, McElwain K, McElwain S, Sriramarao P, Raz E, Broide DH. Immunostimulatory DNA inhibits allergen-induced peribronchial angiogenesis in mice. J Allergy Clin Immunol. 2006;117:597–603. doi: 10.1016/j.jaci.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Lortat-Jacob H. Interferon and heparan sulphate. Biochem Soc Trans. 2006;34:461–464. doi: 10.1042/BST0340461. [DOI] [PubMed] [Google Scholar]
- Lyon M, Rushton G, Gallagher JT. The Interaction of the Transforming Growth Factor-betas with Heparin/Heparan Sulfate Is Isoform-specific. J Biol Chem. 1997;272:18000–18006. doi: 10.1074/jbc.272.29.18000. [DOI] [PubMed] [Google Scholar]
- Maccarana M, Casu B, Lindahl U. Minimal sequence in heparin/heparan sulfate required for binding of basic fibroblast growth factor. J Biol Chem. 1993;268:23898–23905. [PubMed] [Google Scholar]
- Massena S, Christoffersson G, Hjertstrom E, Zcharia E, Vlodavsky I, Ausmees N, Rolny C, Li JP, Phillipson M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. 2010;116:1924–1931. doi: 10.1182/blood-2010-01-266072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, Akdis C. Vascular Endothelial Growth Factor as a Key Inducer of Angiogenesis in the Asthmatic Airways. Curr Allergy Asthma Rep. 2013;13:1–9. doi: 10.1007/s11882-012-0317-9. [DOI] [PubMed] [Google Scholar]
- Minshall EM, Leung DY, Martin RY, Song YL, Cameron L, Ernst P, Hamid Q. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. Am J Respir Cell Mol Biol. 1997;17:326–333. doi: 10.1165/ajrcmb.17.3.2733. [DOI] [PubMed] [Google Scholar]
- Pallerla SR, Pan Y, Zhang X, Esko JD, Grobe K. Heparan sulfate Ndst1 gene function variably regulates multiple signaling pathways during mouse development. Dev Dyn. 2007;236:556–563. doi: 10.1002/dvdy.21038. [DOI] [PubMed] [Google Scholar]
- Pan Y, Woodbury A, Esko JD, Grobe K, Zhang X. Heparan sulfate biosynthetic gene Ndst1 is required for FGF signaling in early lens development. Development. 2006;133:4933–4944. doi: 10.1242/dev.02679. [DOI] [PubMed] [Google Scholar]
- Papakonstantinou E, Karakiulakis G. The “sweet” and “bitter” involvement of glycosaminoglycans in lung diseases:pharmacotherapeutic relevance. Br J Pharmacol. 2009;157:1111–1127. doi: 10.1111/j.1476-5381.2009.00279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006;6:633–643. doi: 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- Pease JE, Williams TJ. Chemokines and their receptors in allergic disease. J Allergy Clin Immunol. 2006;118:305–318. doi: 10.1016/j.jaci.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Redington AE, Roche WR, Madden J, Frew AJ, Djukanovic R, Holgate ST, Howarth PH. Basic fibroblast growth factor in asthma: Measurement in bronchoalveolar lavage fluid basally and following allergen challenge. J Allergy Clin Immunol. 2001;107:384–387. doi: 10.1067/mai.2001.112268. [DOI] [PubMed] [Google Scholar]
- Ringvall M, Kjellen L. Mice deficient in heparan sulfate N-Deacetylase/N-Sulfotransferase 1. Prog Mol Biol Transl Sci. 2010;93:35–58. doi: 10.1016/S1877-1173(10)93003-2. [DOI] [PubMed] [Google Scholar]
- Ringvall M, Ledin J, Holmborn K, van Kuppevelt T, Ellin F, Eriksson I, Olofsson AM, Kjellen L, Forsberg E. Defective heparan sulfate biosynthesis and neonatal lethality in mice lacking N-deacetylase/N-sulfotransferase-1. J Biol Chem. 2000;275:25926–25930. doi: 10.1074/jbc.C000359200. [DOI] [PubMed] [Google Scholar]
- Robinson CJ, Mulloy B, Gallagher JT, Stringer SE. VEGF165-binding sites within heparan sulfate encompass two highly sulfated domains and can be liberated by K5 lyase. J Biol Chem. 2006;281:1731–1740. doi: 10.1074/jbc.M510760200. [DOI] [PubMed] [Google Scholar]
- Rops AL, van den Hoven MJ, Baselmans MM, Lensen JF, Wijnhoven TJ, van den Heuvel LP, van Kuppevelt TH, Berden JH, van der Vlag J. Heparan sulfate domains on cultured activated glomerular endothelial cells mediate leukocyte trafficking. Kidney Int. 2008;73:52–62. doi: 10.1038/sj.ki.5002573. [DOI] [PubMed] [Google Scholar]
- Rostand KS, Esko JD. Microbial adherence to and invasion through proteoglycans. Infect Immun. 1997;65:1–8. doi: 10.1128/iai.65.1.1-8.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni A, Francis CW. Stimulation of endothelial cell proliferation by FGF-2 in the presence of fibrinogen requires alphavbeta3. Blood. 2004;104:3635–3641. doi: 10.1182/blood-2004-04-1358. [DOI] [PubMed] [Google Scholar]
- Schuliga M, Javeed A, Harris T, Xia Y, Qin C, Wang Z, Zhang X, Lee PVS, Camoretti-Mercado B, Stewart AG. Transforming Growth Factor-β-Induced Differentiation of Airway Smooth Muscle Cells Is Inhibited by Fibroblast Growth Factor-2. Amer J Respir Cell Mol Biol. 2013;48:346–353. doi: 10.1165/rcmb.2012-0151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Ma H, Yao C, Tao X, Gan H. Alveolar macrophage-derived vascular endothelial growth factor contributes to allergic airway inflammation in a mouse asthma model. Scand J Immunol. 2012;75:599–605. doi: 10.1111/j.1365-3083.2012.02693.x. [DOI] [PubMed] [Google Scholar]
- Spencer LA, Szela CT, Perez SAC, Kirchhoffer CL, Neves JS, Radke AL, Weller PF. Human eosinophils constitutively express multiple Th1, Th2, and immunoregulatory cytokines that are secreted rapidly and differentially. J Leukoc Biol. 2009;85:117–123. doi: 10.1189/jlb.0108058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenfeldt AL, Wennerås C. Danger signals derived from stressed and necrotic epithelial cells activate human eosinophils. Immunology. 2004;112:605–614. doi: 10.1111/j.1365-2567.2004.01906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DE, Ferris M, Nguyen H, Abboud E, Brody AR. TNF-alpha induces TGF-beta1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. Cell Mol Med. 2009;13(8B):1866–1876. doi: 10.1111/j.1582-4934.2008.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagaya E, Tamaoki J. Mechanisms of airway remodeling in asthma. Allergol Int. 2007;56:331–340. doi: 10.2332/allergolint.R-07-152. [DOI] [PubMed] [Google Scholar]
- ten Hacken NH, Oosterhoff Y, Kauffman HF, Guevarra L, Satoh T, Tollerud DJ, Postma DS. Elevated serum interferon-gamma in atopic asthma correlates with increased airways responsiveness and circadian peak expiratory flow variation. Eur Respir J. 1998;11:312–316. doi: 10.1183/09031936.98.11020312. [DOI] [PubMed] [Google Scholar]
- Teshima-Kondo S, Kondo K, Prado-Lourenco L, Gonzalez-Herrera IG, Rokutan K, Bayard F, Arnal JF, Prats AC. Hyperglycemia upregulates translation of the fibroblast growth factor 2 mRNA in mouse aorta via internal ribosome entry site. FASEB J. 2004;18(13):1583–1585. doi: 10.1096/fj.03-1118fje. [DOI] [PubMed] [Google Scholar]
- Venkatesan N, Siddiqui S, Jo T, Martin JG, Ludwig MS. Allergen-induced airway remodeling in brown norway rats. Am J Respir Cell Mol Biol. 2012;46:96–105. doi: 10.1165/rcmb.2011-0014OC. [DOI] [PubMed] [Google Scholar]
- Wang L, Fuster MM, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- Wen F-Q, Liu X, Manda W, Terasaki Y, Kobayashi T, Abe S, Fang Q, Ertl R, Manouilova L, Rennard SI. TH2 Cytokine-enhanced and TGF-[beta]-enhanced vascular endothelial growth factor production by cultured human airway smooth muscle cells is attenuated by IFN-[gamma] and corticosteroids. J Allergy Clin Immunol. 2003;111:1307–1318. doi: 10.1067/mai.2003.1455. [DOI] [PubMed] [Google Scholar]
- Westergren-Thorsson G, Chakir J, Lafrenière-Allard MJ, Boulet LP, Tremblay GM. Correlation between airway responsiveness and proteoglycan production by bronchial fibroblasts from normal and asthmatic subjects. Int J Biochem Cell Biol. 2002;34:1256–1267. doi: 10.1016/s1357-2725(02)00058-4. [DOI] [PubMed] [Google Scholar]
- Xiao L, Du Y, Shen Y, He Y, Zhao H, Li Z. TGF-beta 1 induced fibroblast proliferation is mediated by the FGF-2/ERK pathway. Front Biosci. 2012;17:2667–2674. doi: 10.2741/4077. [DOI] [PubMed] [Google Scholar]
- Xu D, Fuster MM, Lawrence R, Esko JD. Heparan sulfate regulates VEGF165- and VEGF121-mediated vascular hyperpermeability. J Biol Chem. 2011;286:737–745. doi: 10.1074/jbc.M110.177006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Eckart MR, Morgan AA, Mukai K, Butte AJ, Tsai M, Galli SJ. Identification of an IFN-γ/mast cell axis in a mouse model of chronic asthma. J Clin Invest. 2011;121:3133–3143. doi: 10.1172/JCI43598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum HY, Cho JY, Miller M, Broide DH. Allergen-Induced Coexpression of bFGF and TGF-β1 by Macrophages in a Mouse Model of Airway Remodeling: bFGF Induces Macrophage TGF- β1 Expression in vitro. Int Arch Allergy Immunol. 2011;155:12–22. doi: 10.1159/000317213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wang B, O'Callaghan P, Hjertstrom E, Jia J, Gong F, Zcharia E, Nilsson LG, Lannfelt L, Vlodavsky I, et al. Heparanase overexpression impairs inflammatory response and macrophage-mediated clearance of amyloid-β in murine brain. Acta Neuropathologica. 2012;124:465–478. doi: 10.1007/s00401-012-0997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann N, Hershey GK, Foster PS, Rothenberg ME. Chemokines in asthma: Cooperative interaction between chemokines and IL-13. J Allergy Clin Immunol. 2003;111:227–242. doi: 10.1067/mai.2003.139. [DOI] [PubMed] [Google Scholar]
- Zuberi RI, Ge X, Jiang S, Bahaie NS, Kang BN, Hosseinkhani RM, Frenzel EM, Fuster MM, Esko JD, Rao SP, et al. Deficiency of endothelial heparan sulfates attenuates allergic airway inflammation. J Immunol. 2009;183:3971–3979. doi: 10.4049/jimmunol.0901604. [DOI] [PMC free article] [PubMed] [Google Scholar]