

Abstract
Methamphetamine (METH) causes significant loss of some striatal projection and interneurons. Recently, our group reported on the proliferation of new cells 36 hours after METH and some of the new cells survive up to 12 weeks (Tulloch et al., 2011b). We hypothesized that some of these cells will differentiate and express striatal neuronal phenotypes. To test this hypothesis, mice were injected with METH (30mg/kg) followed by a single BrdU injection (100 mg/kg) 36 hours after METH. One week after METH, a population of BrdU-positive cells expressed the neuronal progenitor markers nestin (18%) and β-III-tubulin (30%). At 8 weeks, 14% of the BrdU-positive cells were also positive for the mature neuron marker, NeuN. At 12 weeks, approximately 7% of the BrdU-positive cells co-labeled with ChAT, PV or DARPP-32. We measured motor coordination on the rotarod and psychomotor activity in the open-field. At 12 weeks, METH–injected mice exhibited delayed motor coordination deficits. In contrast, open-field tests revealed that METH-injected mice compared to saline mice displayed psychomotor deficits at 2.5 days but not at 2 or more weeks after METH. Taken together, these data demonstrate that some of the new cells generated in the striatum differentiate and express the phenotypes of striatal neurons. However, the proportion of these new neurons is low compared to the proportion that died by apoptosis 24 hours after the METH injection. More studies are needed to determine if the new neurons are functional.
Keywords: methamphetamine, striatum, 5-Bromo-2′-deoxyuridine, cell proliferation, neurogenesis, behavior
INTRODUCTION
Methamphetamine (METH) is one of the most widely abused drugs worldwide and the most often synthesized illegal drug in the United States (United Nations Office on Drugs and Crime, 2008). For over 30 years it has been recognized that METH induces excessive release of dopamine (DA), which subsequently becomes depleted (Seiden et al., 1975). METH-induced DA depletion or neurotoxicity has been shown to include considerable loss of dopaminergic terminals in the striatum, as measured by loss of the terminal markers tyrosine hydroxylase (TH) and DA transporters (DAT) (Schmidt and Gibb, 1985; Ricaurte et al., 1980). More recent studies have reported striatal and cortical neuron death in animals given METH (Deng et al., 2001; Pu et al., 1996; Eisch and Marshall, 1998; Zhu et al., 2006). An important concern stems from studies in humans that suggest permanent METH-induced changes in the brain. For example, even after 3 years of abstinence Positron Emissions Tomography (PET) scans revealed DAT loss in METH users (Wilson et al., 1996; McCann et al., 1998; Volkow et al., 2001). Additionally, magnetic resonance imaging (MRI) suggests METH-induced cell death in humans (Ernst et al., 2000).
Numerous studies have also documented METH-induced effects on motor or cognitive performance in both animal models and humans who abuse METH (Friedman et al., 1998; Chang et al., 2005; Salo et al., 2009; Krasnova et al., 2009; Achat-Mendes et al., 2005; Izquierdo et al., 2010; Daberkow et al., 2007, 2008; Boger et al., 2009; Hall et al., 2008). In rodents, for example, acute METH increases open-field locomotor activity that has been shown to accompany altered DA neurotransmission (Hall et al., 2008). The long-term effect, however, includes psychomotor deficits such as decreased locomotor activity. These deficits persist even when striatal DA recovers (Boger et al., 2009; Krasnova et al., 2009). Friedman et al. (1998) demonstrated depleted DA levels in the striatum 65 days after METH, which is attenuated when measured at 265 days. However, deficits on spatial memory task recovered within 100 days suggesting that other neural events may be compensating for the attenuation of deficits. In some humans who previously abused METH, the neural changes include enlargement of the striatum and globus pallidus accompanied by cognitive and motor performance similar to controls (Chang et al., 2005). In contrast, former METH-using participants with the smallest striatal volumes had the greatest neuropsychological deficits (Chang et al., 2005). Taken together, these data suggest the possibility that enlarged basal ganglia structures like the striatum and globus pallidus may be involved in recovery or compensation for the neuropsychological impact of METH. However, until recently, few studies examined the striatal enlargement in relation to motor recovery after METH using rodent models of METH-induced toxicity.
We previously reported that one single systemic injection of METH is sufficient to induce striatal neuron loss in mice (Zhu et al., 2005, 2006). We have also recently demonstrated that METH exposure induces cell proliferation in the striatum that is accompanied by enlarged striatal volume (Tulloch et al., 2011a). Only a third of the new cells survived up to 12 weeks at which point striatal volume normalizes. Some studies have described increase in cell proliferation following injury in areas of the adult brain (Arvidsson et al., 2002; Parent; et al. 2002; Urrea et al, 2007) and one study found increased hippocampal proliferation in mice after experimenter-administered cocaine (Lloyd et al, 2010). The reported increased in hippocampal proliferation after cocaine might be due to method of drug administration. A number of studies reported different levels of progenitor cell proliferation in the adult brain (Teuchert-Noodt et al., 2000; Eisch et al., 2000; Sudai et al, 2011; Venkatesan et al., 2011; reviewed in Canales, 2013). Specifically, the hippocampus shows changes after exposure to different drugs of abuse depending on the experimental model. For example, using a self-administration model, Mandyam et al (2008) found short-term intermittent METH exposure initially increased hippocampal cell proliferation in rats. However, failure in survival of these cells resulted in no net changes in neurogenesis after short-term intermittent METH. In the same study, Mandyam et al (2008) also reported that daily METH exposure of short or long duration decreased hippocampus cell proliferation and new neuron survival. Furthermore, Venkatesan et al (2011) reported decreased progenitor cell proliferation following METH using an in vitro model. Although other injury models have suggested increased striatal cell proliferation and neurogenesis following damage (Parent et al., 2002; Bedard et al., 2002; Luzzati et al., 2011) few studies have examined proliferation following METH-induced damage in the striatum. With an overwhelming record of damaging structural and functional alterations, it is not surprising that until now, research focus has not been directed on behavioral recovery via neurogenesis after METH exposure, particularly for the striatum. To that end, we sought to characterize the type of cells generated in the striatum and measured accompanying motor performance in mice. We used immunohistochemistry for BrdU in conjunction with phenotype markers to identify progenitor and mature neurons over a 12-week time course. We also measured motor performance on the rotarod and in the open field because these tasks have long been thought to require striatal function (Jones and Roberts, 1968a, b; Whimbey and Denenberg, 1967; Denenberg, 1969).
MATERIALS AND METHODS
Animal Care and Use
All procedures regarding animal use were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Hunter College of the City University of New York. The Hunter College animal facility is accredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC). Male ICR mice (Taconic, Germantown, NY) between 11 to 12 weeks of age were housed individually on a 12-h light/dark cycle with food and water available ad libitum except where noted. Mice were habituated to the housing environment and the experimenters for two weeks prior to commencement of intraperitoneal (ip) drug administration. The work described in this article was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for animal experiments. All efforts were made to minimize animal suffering and reduce animal use. No alternatives to in vivo techniques are available for these studies and require the entire connectivity of the brain.
Drug Preparation and Treatment
(+)-Methamphetamine hydrochloride (Sigma, St. Louis, MO) was dissolved in 10 mM PBS and groups of animals were injected ip with either saline or one bolus of METH (30 mg/kg of body weight) in a volume of 200 μL. In addition to drug treatment, 100 mg/kg of the mitotic marker BrdU (Sigma, St. Louis, MO) was injected at 36 hours after METH injection. This time was chosen because it was found to be the peak of cytogenesis after METH treatment (Tulloch et al., 2011a). Subgroups of animals survived to post-METH times ranging from 2.5 days (ie, 24 hours after the BrdU injection which was given 36 hours/1.5 days post-METH) to 12 weeks at which point striatal tissue was collected and prepared for double label BrdU and phenotype immunohistochemistry. However, ChAT, PV, DARPP-32 phenotypes were assessed only at 8 and 12 weeks. These times were chosen based on the peak of mature neuron expression in the new cells (BrdU colabled with NeuN). Except for behavior animals, sacrifice and striatal tissue collection was done by anesthetizing each animal with a 1:3 mixture of ketamine/acepromazine (100mg/kg of body weight) then transcardially perfusing with 25ml of PBS followed by 25ml of 4% paraformaldehyde in PBS. Brains were dissected out and immediately post-fixed for 12 hours in 4% paraformaldehyde at 4°C, followed by cryoprotection in 30% sucrose in PBS solution at 4°C for 48 hrs. Tissue sections were then stored at −80°C until used for immunohistochemical assays. For the group of mice used in the behavior study, within 90 minutes of the last behavior task, mice were immediately sacrificed by decapitation. Striatal tissue was immediately dissected and flash frozen on dry ice. Brains were then stored in −80°C until use.
Immunohistochemistry
All staining was carried out by the free-floating method previously described in Tulloch et al. (2011b). Coronal sections from each striatal hemisphere were cut at 40μm thickness and serially collected from the striatum between bregma 0.02 and 1.4 mm. Every sixth striatal section was collected into one of six adjacent sample wells per animal. One well of samples per animal from the left hemisphere was used for each phenotype assessment. Fluorescent immunostaining was a double-label assay for BrdU and a second phenotype of interest. For all tissue sections, incubation in primary antibodies was done separately per antibody for 24 hours at 4°C. Incubation in secondary was for 1 hour at room temperature.
Labeling for BrdU used the antigen retrieval steps for BrdU immunofluorescence described previously in Tulloch et al. (2011b). In brief: After washes in PBS, tissue sections were incubated in a solution of 1:1 formamide in 0.15M 4X SSC for two hours at 65°C, followed by incubation in 2N HCL at 37°C for 30 minutes. After a 10-minute rinse in 0.1M boric acid, pH 8.5, blocking was done in 5% donkey serum in 0.2% Triton X100 in PBS at room temperature for an hour. Primary antibody solution was 1:500 sheep anti-BrdU (Novus biologicals, Littleton, CO) in 1% normal donkey serum. Secondary antibody solution was 1:500 FITC donkey anti-sheep (Novus Biologicals, Littleton, CO) for 1 hour at room temperature completed the BrdU staining.
Neuron progenitor phenotypes: β-III-tubulin, DCX, and nestin were labeled before BrdU immunostaining. For these phenotypes we used a TSA amplification protocol per TSA-CY3 kit N5000 ® (Perkin Elmer, Waltham, MA). After washing with PBS, antigen retrieval was done in 1M citric acid in dH2O at 100°C for 8 minutes. When using mouse based primary antibodies with the TSA protocol, i.e. mouse anti-β-III-tubulin and mouse anti-nestin, the one hour block step used MOM IgG from Vector Labs basic MOM kit BMK2200® (Burlingame, CA). The antibody dilutions used with MOM diluent for overnight incubation were 1:2500 mouse anti-β-III-tubulin (Millipore); 1:5000 mouse anti-nestin (Millipore). For DCX, after citric acid antigen retrieval step, blocking was done in 3% normal donkey serum in 0.3% triton X in PBS for an hour followed by incubation in primary antibody solution of 1:2000 goat anti-DCX, (Santa Cruz Biotech, Santa Cruz CA) in 1% donkey serum solution for 24 hours. For mouse primary antibodies the one-hour incubation in secondary antibody was done with a solution of 4ul/ml biotin-anti Mouse IgG from the MOM kit (Vector labs). For goat primary antibodies biotinylated-anti-goat IgG from Vector Elite ABC kit was used according to instructions from the manufacturer (Vector Labs, Burlingame, CA). Visualization of all the progenitor markers and microglia were done with incubation 1:1000 Cy3 conjugate to secondary antibody solution prepared according to TSA kit instructions (Perkin Elmer, Waltham, MA). After secondary antibody for progenitor staining, ten-minute incubation was done in a solution of 0.1% trypsin in 0.01M Tris buffer and 0.1 CaCl2 at room temperature. BrdU labeling then proceeded starting with the HCL step.
For neuronal phenotypes (NeuN, ChAT, PV and DARPP-32) the BrdU component of the assay was done first according to steps previously described for BrdU staining. On the second day after BrdU visualization, a one-hour blocking step and incubation in the phenotype primary antibody was done. For NeuN and PV phenotypes, blocking was with MOM kit IgG from (Vector labs, Burlingame CA) followed by incubation in primary antibodies: 1:1000 Mouse anti-NeuN (Novus Biologicals, Littleton CO) or 1:500 mouse anti-PV, (Millipore, Billerica, CA) over night at 4°C. NeuN and PV were visualized using secondary antibody 1:1000 CY3 goat anti-mouse (Millipore, Bellerica CA) in 1% NGS in 0.2% Triton X100 PBS. However, DARPP-32 was visualized with 1:1000 Cy3 goat anti-rabbit (Millipore) in 1% NGS in 0.2% triton X PBS. ChAT and DARPP-32, blocking was done in 3% normal donkey serum in 0.2% triton X100 in PBS. After blocking step, primary incubation was done in 1:500 goat anti-ChAT (Millipore, Billerica, MA). ChAT was visualized using secondary antibody dilution 1:500 Cy3 donkey anti-goat (Millipore) in 1% donkey serum in 0.2% Triton X100-PBS. For DARPP-32, blocking was done in 5% goat serum in 0.2% Triton X100-PBS and then incubated in 1:500 rabbit anti-DARPP-32 (Millipore, Bellerica MA) in a solution of 1% goat serum in 0.2% Triton X-PBS. DARPP-32 was visualized with 1:500 Cy3 goat anti-rabbit (Millipore, Bellerica, MA). All secondary antibody incubations were for an hour. All tissue sections were mounted on coded superfrost glass slides and cover slipped with vectashield hardset mounting medium for fluorescence (Vecta Labs, Burlingame, CA).
The number of BrdU nuclei co-labeled with a given phenotype marker was determined using a Leica TCS ™ confocal microscope and corresponding Leica software system (Leica microsystems, Heidelberg, Germany). FITC and CY-3 signals corresponded to single wavelength laser line 488 (green) and 568 (red), respectively. The striatum was divided into four regions corresponding to dorsal medial (DM), dorsal lateral (DL), ventromedial (VM) and ventrolateral (VL). Z-stack images from each striatal region were taken in four animals per group, six tissue sections per animal. To avoid cross detection between the signals, the pinhole setting was less than 2μm and z-stacks between fifteen and thirty μm thick were recorded sequentially between frames at 100X in a raster pattern per series. Confirmation of co-label was done by reconstruction and orthogonal rotation of the images using the Leica confocal software in view and analysis modes. Four striatal sections per animal were analyzed.
To determine the proportion of cells with BrdU positive nuclei that were also positive for the second marker of interest, a modification of the stereology technique for the confocal microscope as described by Peterson (1999) was employed as follows: Using the quantification mode in the Leica software a sampling frame measuring an area of 50 mm2 per z-stack with inclusion and exclusion lines was drawn on the computer monitor and counts were taken at 100X magnification after outlining the striatum at 5X. Only cells that fell within the inclusion area of each sampling frame and clearly visible within the z-axis were counted. Partially visible nuclei or those that touched the exclusion lines of the sampling frame were not counted. Separate counts of BrdU-positive and BrdU-colabeled nuclei from the same tissue sections were taken from 4 mice per time point. After stereological quantification was complete, the total BrdU-positive nuclei per animal and the total phenotype immunostain co-labeled with BrdU were used to calculate the percentage of BrdU nuclei co-labeled. Once phenotype percentages were done, coding was revealed for statistical analysis. With the exception of PV and ChAT, statistical analysis was done using a one-way ANOVA followed by Bonferroni post-hoc to determine the differences in percent of a given phenotype at various times after METH-treatment (2.5 days to 12 weeks). For PV, ChAT, and DARPP-32 one-way ANOVAs compared each phenotype to the alternative hypothesis value of zero replacement of striatal neurons that die by apoptosis 24 hours after METH injections. Significant one-way ANOVA was followed by Dunnett’s post-hoc test.
Rotarod
Motor coordination tests were done on the rotarod because performance is known to correlate with the level of striatal cell death (Haelewyn et al., 2007). All animals were tested before treatment to get baseline performance when naive. Tests were done on a five station ENV-577 mouse ROTA-ROD treadmill ™ by Med Associates (St Albans, VT). Rotarod tasks lasted for 3 minutes in the home cage room. Each animal was individually placed on one of five treadmill stations on the rotarod, which began rotating after 30 seconds of placement. The length of time that animals remained on the rotating treadmill at fixed speed (20 rpm) followed two hours later by the variable speed (2-.20 rpm) test was measured. The mean pre-treatment performance on the rotarod was calculated from the average of the fixed versus variable speed latency to fall of the rotating rod. After rotarod tasks animals were randomly placed in either METH or saline condition and treated by intraperitoneal injections. Subgroups of these animals were tested again at one of several survival post-treatment times (2.5 days, 1, 2, 4, 8 and 12 weeks). The average of fixed and variable speed was again used as the post-treatment score and compared with pre-treatment latency to fall off the rotarod. It was important to analyze the pre-injection performance separately to ensure that the latencies to fall were relatively the same regardless of what group the mice were later randomly assigned for the post-treatment tests. This was done with a two-way ANOVA for pre-treatment performance per assigned treatment group and post-test time. Non-significant pre-treatment result was followed by post-treatment analysis. Post-treatment comparisons for the mean latency to fall off the rotating rod for the post-injection condition per time point (2.5 days to 12 weeks) were done with a two-way ANOVA. For significant interaction and simple effects, Bonferroni post-hoc was done to determine differences between treatment conditions per post-treatment time.
Open-field
The open field has been reliably used to test psychomotor activity (Brooks and Dunnett, 1999). Additionally, activity in the center provides a rudimentary measure of anxiety (Denenberg, 1969), thus behaviors that corresponded to motor activity (distance traveled) and anxiety (entries into the center zone) were assessed. Psychomotor assessments were done in the same animals used for the rotarod task. However, each subgroup of mice was tested once because we were interested in activity in a completely novel situation. Post-injection times tested in subgroups of mice in the open-field corresponded with phenotype assessment time course (2.5 days, 1, 2, 4, 8 and 12 weeks). Dimmed room lights signaled the start of the five-minute open field test. Mice were individually placed in the center of a 32″ × 16″ × 32″ rectangular space surrounded by a Kinder Scientific Smart Frame ® open field system. The system was powered with 32 Infared PhotoBeams connected to Motor Monitor 5.05 software to measure the behavior parameters (Kinder Scientific, Poway CA). Photobeam breaks corresponding to the total distance travelled in the open-field and the number of entries into the center zone was used in statistical analysis. These two measures were analyzed using two-way ANOVA with Bonferonni post-hoc comparisons after significant effects.
Statistical analysis
Type of test used and group comparisons were indicated under each section above. One-way and Two-way ANOVA was used with Dennett and Bonferroni post hoc tests.
RESULTS
Phenotypes of Newly Generated Cells
To determine the cellular phenotype of the newly generated cells, mice received a single injection of METH (30 mg/kg, ip) previously demonstrated to induce apoptosis in striatal neurons 24 hours after treatment (Zhu et al., 2005). Mice also received a single injection of BrdU (100 mg/kg, ip) 36 hours after METH in order to label newly generated cells previously reported to proliferate at this time post-METH (Tulloch et al., 2011b). Neuron progenitors were observed throughout the striatum (Figure 1). BrdU-positive nuclei expressing the neural progenitor markers nestin (Figure 1) and β-III-tubulin (Figure 2) but not DCX (Figure 3) were present in the striatum after METH treatment. Significantly more nestin-positive cells were observed at 2.5 days and 1 week with a mean of 18.1% and 17.7%, respectively (Figure 1). BrdU with nestin immunostain declined and by 12 weeks only 4.8% remained. One-way ANOVA demonstrated that these differences were significant [F (5, 18)=6.63, p = 0.0012]. Bonferroni post-hoc tests showed significant differences between early post-treatment times (2.5 days, and 1 week) when compared to later post-treatment times (8 and 12 but not 4 weeks) with p< 0.05 (Figure 1). The β-III-tubulin phenotype appeared early at 2.5 days and peaked at 30% one week after METH (Figure 2). BrdU-positive nuclei co-labeled with β-III-tubulin declined over the rest of the time course to 6.6% at 12 weeks (Figure 2). One-way ANOVA revealed these differences were significant over time [F (5, 18) = 11.5, p< 0.0001]. Post-hoc revealed significant differences between the peak of new β-III-tubulin cells at one week and every other time point measured (p< 0.01; Figure 2). BrdU-positive nuclei expressing the DCX progenitor marker were only observed in the SVZ (Figure 3). Although it appears that some DCX staining may have been in the striatum, orthogonal analysis demonstrated no BrdU co-labeled with DCX in this structure over the entire time course (data not shown).
Figure 1.
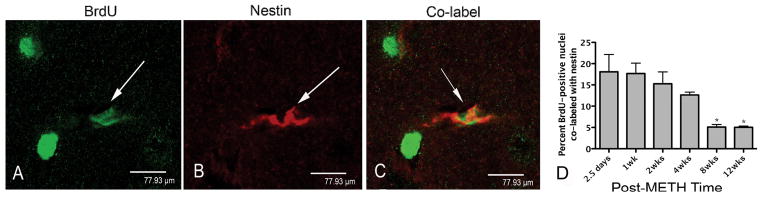
BrdU-positive nucleus co-labeled with nestin in Z-stack images from representative striatal tissue sections taken 2 weeks after METH. In panel A BrdU-positive nuclei are labeled in green with FITC. The white arrow points to one particular BrdU nucleus that is then examined for nestin and co-label in panels B and C, respectively. Panel B has a white arrow pointing to the same cell with nestin-positive stain labeled red with Cy3. Panel C shows the same cell co-labeled and indicated by the white arrow. Panels (A–C) scale bar 77.9 μm at 100x magnification. Significant one-way ANOVA revealed that the percentage of BrdU-positive nuclei co-labeled with nestin peaked between 2.5 days and 1 week. Post-hoc analysis demonstrated that they declined significantly at 8 and 12 weeks compared to 2.5 days and 1 week (panel D). *p<0.05 (n=4).
Figure 2.
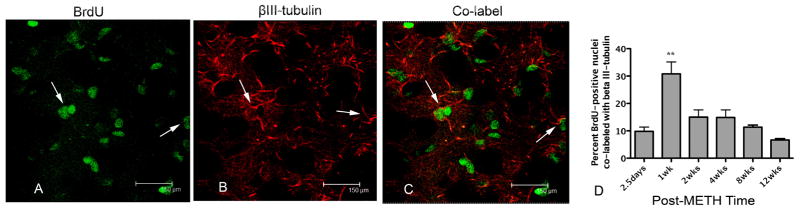
BrdU-positive nuclei co-localized with β-III-tubulin. Confocal z-stack image taken from representative striatal tissue section 1 week after METH. BrdU-positive nuclei are labeled in green with FITC (A) and β-III-tubulin-positive progenitors are labeled with Cy3 in red (B). Co-labeled cells can be seen in the same striatal tissue section (C) identified by arrows. Significant one-way ANOVA revealed the percentage of BrdU-positive nuclei co-labeled with β-III-tubulin peaked at 1 week but was significantly less at all other times after METH (D), **p<0.01 (n=4).
Figure 3.

BrdU-positive nuclei co-labeled with DCX. Confocal z-stack images were taken from representative striatal tissue sections at 2.5 days after METH. BrdU-positive nuclei are labeled in green with FITC (A, D) and DCX-positive progenitors are labeled with Cy3 in red (B, E). No co-labeled cells can be seen in the same striatal tissue section but are present in the SVZ (C, F). Top panel scale bar 300 μm taken at 5x magnification. Magnified SVZ image (D–F) scale bar 47.3 μm taken at 63x magnification (n=4). CC, corpus callosum; SVZ, subventricular zone.
Differentiation of Newly Generated Cells
New cells differentiate to express mature neuron markers in the striatum after METH. BrdU-positive nuclei co-labeled with NeuN were present in the striatum at later compared to earlier post-METH times (Figure 4). Few BrdU- positive nuclei co-labeling with NeuN were observed 2.5 days after METH. However, there was a slow and steady increase to a peak of 13.8% at 8 weeks, which then decreased to 8.7% at 12 weeks (Figure 4). One-way ANOVA demonstrated that these differences were significant [F (5, 18) = 10.06, p = 0.0001]. Bonferonni post-hoc revealed significantly more cells adopting neuronal phenotype at the 4, and 8 weeks post-METH time points, p< 0.01 and p< 0.001, and p<0.05, respectively.
Figure 4.
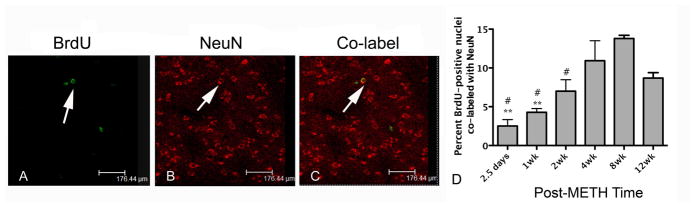
BrdU-positive nuclei co-localized with NeuN. Confocal z-stack images from representative tissue section 12 weeks after METH. BrdU-positive nuclei are labeled in green with FITC (A) and NeuN-positive neurons are labeled with Cy3 in red (B). Co-labeled cells can be seen in the same tissue section (panel C). Few co-labeled cells were present at 2.5 days and peak co-label was at 8 weeks. BrdU-positive NeuN cells declined slightly at 12 weeks (D). **p<0.01, asterisk symbol compared to 4 weeks. #p<0.05, number symbol compared to 8 weeks.
A very small percentage of the BrdU-positive cells were seen to express markers for interneuron and projection neuron phenotypes at 8 and 12 weeks. For interneurons, a one-way ANOVA revealed that BrdU-positive nuclei co-labeled with ChAT was significantly different from zero [F (2,9)= 73.04, p<0.0001; Figure 5]. New ChAT cells were 1.3% of BrdU-positive nuclei at both 8 and 12 weeks after METH (Figure 5). BrdU-positive nuclei expressing the interneuron marker PV were also significantly different from zero as revealed by one-way ANOVA [F (2, 9) = 14.7, p=0.001]. At 8 and 12 weeks, respectively, 0.89% and 1.2% of the BrdU- positive nuclei were co-labeled with PV and post-hoc test revealed they were both significantly different from zero (p<0.01) at each post-METH time point (Figure 6). At 8 and 12 weeks after METH, 9.9 and 6 % of the BrdU-positive cells expressed the striatal projection neuron marker DARPP-32. One-way ANOVA revealed these proportions to be significantly different from hypothesized value of zero DARPP-32 labeled BrdU positive cells at 12 weeks, [F(2,11)=36.7, p<0.001] (Figure 7).
Figure 5.
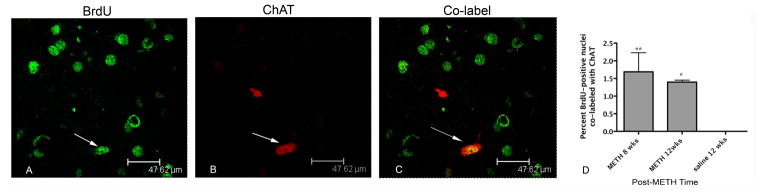
BrdU-positive nuclei co-localized with ChAT. Confocal z-stack image from representative tissue section taken 12 weeks after METH. BrdU-positive nuclei are labeled in green with FITC (A). ChAT-positive interneurons are labeled with Cy3 in red (B). Co-labeled cells can be seen in the same tissue section in panel C. Arrows indicate co-labeled interneurons. Few BrdU-positive cells co-labeled with ChAT can be seen at 8 and 12 weeks post-METH (D). *p<0.001, **p<0.01 (n=4).
Figure 6.
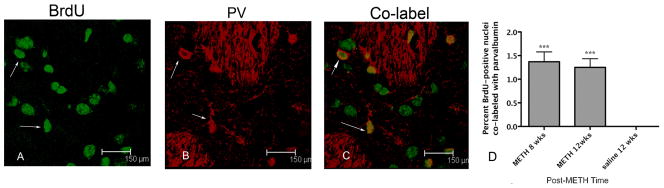
BrdU-positive nuclei co-localized with PV. BrdU-positive nuclei were labeled with FITC (A) and PV with Cy3 (B). Overlay of images A and B is shown in C. Arrow denotes a co-labeled cell. Graph represents the mean percent ± SEM (n=4) BrdU-positive nuclei co-labeled with PV were observed at 8 and 12 weeks after METH (D). One-way ANOVA ***p<0.0005 (one-way ANOVA, N=4).
Figure 7.
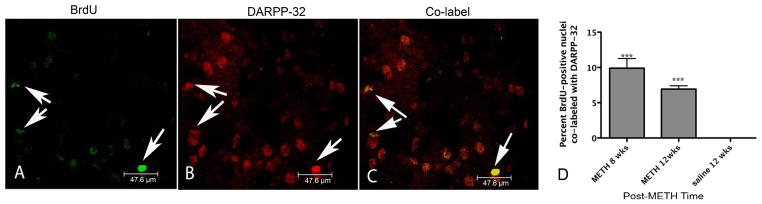
BrdU-positive nuclei co-localized with DARPP-32. Confocal z-stack image taken from representative striatal tissue section 8 weeks after METH (A–C). BrdU-positive nuclei are labeled in green with FITC (A) and DARPP-32 cells are labeled with Cy3 in red (B). Co-labeled cells can be seen in the same striatal tissue section (C) identified by short arrows. Scale bar 47.6 μm taken at 100x. Graph represents the mean percent ± SEM (n=4) BrdU-positive nuclei co-labeled with DARPP-32 at 8 and 12 weeks after METH (D). One-way ANOVA ***p<0.005 (one-way ANOVA).
Rotarod and Open Field Behavioral Studies
To assess functional recovery, motor coordination on the rotarod (latency to fall) and psychomotor activity (distance traveled and center one entries) in the open field were also measured over the same 12-week time course observed in neuronal phenotype assessments. All mice were tested on the rotarod before random assignment to post-METH or saline rotarod test groups then tested again at various post-treatment times (Figure 8). The mice were matched on pre-injection performance when they were assigned to the various post-treatment test groups. Analysis with a two-way ANOVA revealed no interaction effects between the assigned drug treatment and post-test time conditions, when tested for pre-treatment latencies to fall [F (5, 50) = 0.21, p= 0.96]. No effect of the post-treatment test time later assigned [F (5. 50) = 1.3, p= 0.28] was observed, or later drug-injection assigned [F (1, 50) = 0.28, p = 0.60]. In contrast, significant differences in performance were observed between METH and saline-treated mice (Figure 8). A two-way ANOVA revealed that METH-injected animals performed worse on the task by scoring shorter latencies to fall off the rotarod. Although the comparisons showed no significant interactions between post-treatment time and the type of treatment [F (5, 50) = 1.04, p= 0.40], or the post-treatment time effect [F (5, 50) = 1.06, p = 0.39], a significant effect of drug treatment [F (1, 50) =33.4, p< 0.001] was observed. The Bonferroni corrected post-hoc test showed that METH-treated animals fell off the rotarod significantly earlier than saline animals at 12 weeks post-injection [MD = −79.8 seconds, p<0.05] (Figure 8).
Figure 8.

Assessment of latency to fall off the rotarod after METH treatment. Mice received an injection of METH (30 mg/kg. ip) and were assessed behaviorally at times indicated under the x-axis. Two-way ANOVA revealed METH-injected mice performed worse and post-hoc revealed the significant deficit at 12 weeks compared to saline injected mice. *p<0.05 for post-hoc test (n=6).
For psychomotor performance, subgroups of METH and saline-treated mice were tested once in the open-field at randomly assigned post-treatment times. METH-treated mice differed from saline-treated mice at specific post-treatment times (Figure 9). These differences were revealed by two-way ANOVA which demonstrated a significant effect of post-treatment test time [F(5, 60)=2.9, p=0.02] and the type of drug treatment [F(1,60)=5.4, p=0.02] on total distance traveled (Figure 9A). Post-hoc test revealed METH-treated mice traveled for significantly less distance than saline-treated mice at 2.5 days (p<0.01) but did not differ at any other post-treatment times. However, there was no significant interaction effect of post-treatment time tested and type of drug treatment time together [F(5,60)=1.7, p=0.14]. METH treated mice entered the center zone of the open field significantly less than saline treated mice when assessed by two-way ANOVA for interaction effects of post-treatment time tested and type of drug treatment [F(5,60)=3.6, p=0.006] (Figure 9B). The type of drug treatment alone had a significant effect [F(1,60)= 8.5, p=0.005] and the post-treatment time alone had a significant effect on number of center zone entries [F(5,60)= 21.2, p<0.001]. Post-hoc test revealed that METH-treated mice entered the center zone significantly less than saline-treated mice at 2.5 days after treatment (p<0.001) but by 2 weeks returned to similar levels as saline-treated mice (Figure 9B).
Figure 9.
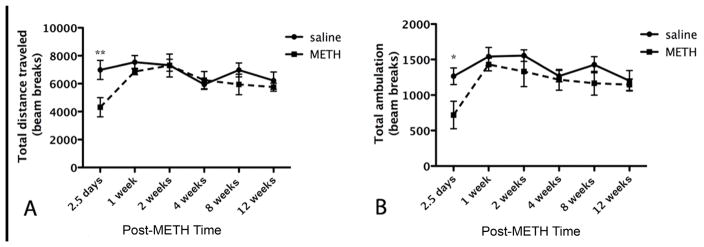
(A) Assessment of distance traveled in the open-field. Mice injected with METH traveled for a significantly less distance at 2.5 days compared to mice that received saline injections. **p<0.05 (n=6). (B) Assessment of ambulation showed a deficit in the METH treated group that normalized after one week. *p<0.01 (n=6).
DISCUSSION
BrdU-positive nuclei co-labeled with various phenotype markers were observed over a twelve-week time course of new cell differentiation following the previously reported peak of METH-induced cell death (Zhu et al., 2006). Previous work published by our group and others demonstrated that the incorporation of BrdU occurred in newly generated and not in apoptotic neurons. For example, histological evidence utilizing TUNEL or Fluoro-Jade C staining showed that at 24 hours post-METH the apoptotic cells displayed pyknotic nuclei that completely disappeared by 48 hours post-METH (Zhu et al., 2006; Bowyer et al., 2008). The incorporation of BrdU begins at 24 hours after acute exposure to METH (Tulloch et al., 2011b). Moreover, biochemical studies demonstrate that damage to mitochondria and endoplasmic reticulum occurred just a few hours after exposure to METH (for review see Krasnova and Cadet, 2009). METH-induced oxidative stress and nitrosylation have also been shown to increase apoptosis and decrease neural progenitor cell proliferation in the dentate gyrus of the hippocampus (Venkatesan et al., 2011). Although progenitor cell proliferation decreased, particularly after 48 hours of METH exposure, four days after treating the cells with METH Venkatesan et al. found that METH did not alter the amount of cells adopting neural or glial phenotypes. Given the results observed in Tulloch et al. 2011a and 2011b and the current study, a time course of in vivo events following METH suggests that compensatory progenitor cell proliferation in the mammalian brain begins approximately 24 hours after the peak of METH-induced apoptosis as demonstrated by our previous published studies (Zhu et al., 2005, 2006).
The present study shows that most of the new cells first appear as neural progenitors expressing nestin and β-III-tubulin, reaching peak levels within one week after METH. By the second week the number of BrdU cells expressing these markers declined. The corresponding increases in BrdU-positive nuclei co-labeled with NeuN suggest that a significant proportion of the progenitors differentiated into mature neurons. However, the number of mature neurons does not sufficiently account for the loss of cells expressing progenitor markers, suggesting a proportion of the nestin and β-III-tubulin-positive progenitors might have been lost via apoptosis. Interestingly, a small subpopulation of the new cells displayed histochemical markers for interneurons. This surprising result provides a basis for future studies of recovery from toxic damage.
We did not observe BrdU co-localization with DCX in the striatum or a gradient of BrdU cells from the SVZ extending into the adjacent striatum. This lack of BrdU/DCX-positive nuclei was not due to assay failure because BrdU/DCX-positive cells were seen in the SVZ leading us to conclude that either no migratory progenitors were present or we failed to capture morphological evidence or migratory cells at the time points measured. Given the significant increase in Brdu-positive cells within the striatum that co-expressed other progenitor markers, we suggest new cells generated in the aftermath of METH did not derive from the SVZ. We suggest the new cells might derive from dormant striatal progenitors some of which differentiate into neurons at later time points post-METH. Furthermore, many of the remaining new cells are unidentified and may include glial cells.
Striatal neurogenesis following injury has been well studied (Reynolds and Weiss, 1992; Parent et al., 2002; Bédard et al., 2002; Luzatti et al., 2011; DeCaudo et al., 2012; Sun et al, 2012; Supeno et al., 2013; Ignarro et al., 2013). Although suggestions of a dormant population of striatal progenitors is controversial, Bédard et al. (2002) reported a low level of neurogenesis in the normal monkey striatum thought to derive from striatal progenitors. The same group later found that the number of new neurons derived from progenitors was enhanced with brain-derived neurotropic factor after the damage (Bedard et al., 2006). Furthermore, the new cells observed by this group expressed morphological and chemical markers for striatal projection neurons. In contrast to our results, however, the Bédard group did not observe new striatal interneurons. More recent evidence suggestive of dormant striatal progenitors proliferating after striatal degeneration was reported by Luzatti et al. (2011) and DiCaudo et al. (2012). Luzatti et al. used a slow striatal degeneration model of mice lacking Creb1 gene. They found SVZ-derived DCX-positive progenitors that entered the striatum individually, DCX-positive striatal progenitors, and a percentage of progenitors in the striatum that lacked DCX labeling. These findings might explain our failure to observe a gradient of DCX-positive cells from the SVZ indicative of migrating cells if they entered the striatum singly. Furthermore, the Luzatti group suggested that neuroblasts with an SVZ origin could also later divide within the striatum producing clusters of new cells but that a percentage of the progenitors appear to be endogenous to the striatum. In contrast, DiCaudo et al. used a MPTP-primate model of Parkinson’s disease to induce striatal damage and found increased numbers of cells expressing phenotypes of damaged striatal cells. Although they suggested the new cells likely derived from the SVZ, they could not rule out a striatal origin. Because we previously observed clusters of dividing cells in the striatum following METH treatment and no spontaneous proliferation in response to saline (Tulloch et al., 2011b), we suggest that the low levels of striatal neurogenesis and lack of DCX-positive progenitors within the striatum might represent a specific response to METH treatment involving dormant striatal progenitors. The generation of PV, ChAT and DARPP-32 neurons suggests compensatory neurogenesis to replace cell types lost early after METH. With the exception of a few studies such as those by Van Kampen et al. (2004, 2005, 2006) and Bedard et al. (2006), most research to date has shown that after striatal damage, the SVZ-derived progenitors migrate to the site of injury and mature into neuronal and glial phenotypes (Kojima et al., 2010; Yamashita et al., 2006; Kuhn et al., 2001 for review). Furthermore, similar to studies involving MPTP lesions to the nigrostrial DA pathway (Kay and Blum, 2000), many of the neuronal progenitors we observed failed to survive as determined from significant decrease in BrdU co-labeled with nestin or β-III-tubulin at later time points. Therefore, the novelty of our results is the generation of striatal neurons of the same phenotype reported to die early after METH treatment (Zhu et al., 2006) and the observation of likely dormant progenitors in the striatum capable of activation under special conditions. Dormant progenitors represent a potential source of new cells to replace the loss of some striatal neurons in neurological disorders involving neurodegeneration or chronic METH use.
A chronic two-week regimen of D3 receptor agonist promotes adult neurogenesis in the nigrostriatal pathway (Van Kampen et al., 2005, 2006). Maturation of the new neurons and recovery of striatal DA innervation accompanied recovery of motor performance measured by amphetamine-induced rotation and skilled paw reaching on the “staircase test” (Van Kampen et al., 2005, 2006). In contrast, Parent (2003) suggested that increased proliferation and neurogenesis following neural damage may be compensatory but not indicative of behavioral recovery. Analysis of behaviors that accompany striatal neurogenesis in the current work revealed that METH-treated mice had significant motor coordination deficits compared to saline-injected mice on the rotarod at the time new cells expressed markers for previously lost striatal neurons. Surprisingly, the psychomotor deficits in the open field while disrupted at 2.5 days, recovered beginning at 2 weeks post-METH. It is possible that the behavioral deficits we observed might be due to altered DA function in the striatum. For example, one group reported that METH reduces levels of dopamine transporter binding sites in mice for over two weeks after METH and is accompanied by deficits on conditioned place preference task (Achat-Mendes et al., 2005). Thus measures of dopamine transporter binding would be an intriguing area of study in the context of METH-induced neurogenesis and motor performance. Interestingly, reduction of PV interneurons in the striatum is associated with persistent motor deficits (Marrone et al., 2006; Kataoka et al., 2010). In a previous study we demonstrated that the striatal neuronal population most severely affected by a bolus high dose of METH was the PV interneurons (Zhu et al., 2006). The low number of newly generated PV neurons is insufficient for recovery of fine motor coordination and thus the rotarod deficits persist. Another interesting area for further analysis is whether treatment with agents to increase the survival of PV and other new neurons would attenuate the motor deficits on the rotarod task.
In conclusion, the results of this study demonstrate the phenomenon of cell proliferation in the striatum of mice in the aftermath of a high dose of METH. The newly generated cells are not derived from the SVZ but arise from the activation of dormant cells throughout the striatum. A small fraction of the newly generated cells differentiate and express phenotypic markers of projection and interneurons, the latter population has been shown to sustain considerable cell loss in the aftermath of METH. It needs to be determined if the newly generated neurons are functional. It is important to further investigate the mechanism that triggers the proliferation of dormant striatal precursors because this can be of value in the treatment of neurological disorders involving neuronal degeneration and traumatic brain injury.
Acknowledgments
This work was supported by R01 DA020142 from the National Institute on Drug Abuse to JAA. Support for infrastructure came from a grant from the National Center for Research Resources (G12 RR003037) and the National Institute on Minority Health Disparities (8 G12 MD007599) awarded to Hunter College by the NIH. We would like to thank Drs. Cheryl Harding and Peter Serrano for use of their behavior testing equipment.
Abbreviations used
- BrdU
5-Bromo-2߰-deoxyuridine
- ICR
Institute for Cancer Research
- ip
intraperitoneal
- METH
(+)-methamphetamine hydrochloride
- PBS
phosphate-buffered saline, pH 7.4
- IB4
isolectin B4
- NeuN
neuron nuclear protein
- ChAT
choline acetyltransferase
- DA
dopamine
- DARPP-32
dopamine and cAMP-regulated phosphoprotein of 32 kDA
- PV
Parvalbumin
- SVZ
sub-ventricular zone
References
- Achat-Mendes C, Ali SF, Itzhak Y. Differential effects of amphetamines-induced neurotoxicity on appetitive and aversive Pavlovian conditioning in mice. Neuropsychopharmacol. 2005;30:1128–1137. doi: 10.1038/sj.npp.1300675. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–967. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Bédard A, Cossette M, Lévesque M, Parent A. Proliferating cells can differentiate into neurons in the striatum of normal adult monkey. Neurosci Lett. 2002;328:213–216. doi: 10.1016/s0304-3940(02)00530-x. [DOI] [PubMed] [Google Scholar]
- Bédard A, Gavel C, Parent A. Chemical characterization of newly generated neurons in the striatum of adult primates. Exptl Brain Res. 2006;170:501–512. doi: 10.1007/s00221-005-0233-5. [DOI] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Granholm AC, McGinty JF. Minocycline restores striatal tyrosine hydroxylase in GDNF heterozygous mice but not in methamphetamine-treated mice. Neurobiol Disease. 2009;33:459–466. doi: 10.1016/j.nbd.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer JF, Robinson B, Ali S, Schmued LC. Neurotoxic-related changes in tyrosine hydroxylase, microglia, myelin, and the blood-brain barrier in the caudate-putamen from acute methamphetamine exposure. Synapse. 2008;62:193–204. doi: 10.1002/syn.20478. [DOI] [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB. Tests to assess motor phenotype in mice: a user’s guide. Nature Rev Neurosci. 2009;10:519–529. doi: 10.1038/nrn2652. [DOI] [PubMed] [Google Scholar]
- Canales JJ. Deficient plasticity in the hippocampus and the spiral of addiction: focus on adult neurogenesis. Cur Topics Beh Neurosci. 2013;15:293–312. doi: 10.1007/7854_2012_230. [DOI] [PubMed] [Google Scholar]
- Chang L, Cloak C, Patterson K, Grob C, Miller E, Ernst T. Enlarged striatum in abstinent methamphetamine abusers: A possible compensatory response. Biol Psychiatry. 2005;57:967–974. doi: 10.1016/j.biopsych.2005.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daberkow DP, Riedy MD, Kesner RP, Keefe KA. Effect of methamphetamine neurotoxicity on learning-induced arc mRNA expression in identified striatal efferent neurons. Neurotox Res. 2008;14:307–315. doi: 10.1007/BF03033855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daberkow DP, Riedy MD, Kesner RP, Keefe KA. Arc mRNA induction in striatal efferent neurons associated with response learning. Eur J Neurosci. 2007;26:228–241. doi: 10.1111/j.1460-9568.2007.05630.x. [DOI] [PubMed] [Google Scholar]
- DiCaudo C, Riverol M, Mundiñano IC, Ordoñez C, Hernández M, Marcilla I, Luquin MR. Chronic levodopa administration followed by a washout period increased number and induced phenotypic changes in striatal dopaminergic cells in MPTP-monkeys. PlosOne. 2012;7(11):e50842. doi: 10.1371/journal.pone.0050842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Wang Y, Chou J, Cadet JL. Methamphetamine causes widespread apoptosis in the mouse brain: evidence from using an improved TUNEL histochemical method. Brain Res Mol Brain Res. 2001;93:64–69. doi: 10.1016/s0169-328x(01)00184-x. [DOI] [PubMed] [Google Scholar]
- Denenberg VH. Open-field behavior in the rat: what does it mean? Ann N Y Acad Sci. 1969;159:852–859. doi: 10.1111/j.1749-6632.1969.tb12983.x. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Marshall JF. Methamphetamine neurotoxicity: dissociation of striatal dopamine terminal damage from parietal cortical cell body injury. Synapse. 1998;30:433–445. doi: 10.1002/(SICI)1098-2396(199812)30:4<433::AID-SYN10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Ernst T, Chang L, Leonido-Yee M, Speck O. Evidence for long-term neurotoxicity associated with methamphetamine abuse: a 1H MRS study. Neurol. 2000;54:1344–1349. doi: 10.1212/wnl.54.6.1344. [DOI] [PubMed] [Google Scholar]
- Friedman SD, Castañeda E, Hodge GK. Long-term monoamine depletion, differential recovery, and subtle behavioral impairment following methamphetamine-induced neurotoxicity. Pharmacol Biochem Beh. 1998;61:35–44. doi: 10.1016/s0091-3057(98)00066-5. [DOI] [PubMed] [Google Scholar]
- Haelewyn B, Freret T, Pacary E, Schumann-Bard P, Boulouard M, Bernaudin M, Bouët V. Long-term evaluation of sensorimotor and amnesic behaviour following striatal NMDA-induced unilateral excitotoxic lesion in the mouse. Beh Brain Res. 2007;178:235–243. doi: 10.1016/j.bbr.2006.12.023. [DOI] [PubMed] [Google Scholar]
- Hall DA, Stanis JJ, Marquez AH, Gulley JM. A comparison of amphetamine- and methamphetamine-induced locomotor activity in rats: evidence for qualitative differences in behavior. Psychopharmacol. 2008;195:469–478. doi: 10.1007/s00213-007-0923-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro RC, Vieira AS, Sartori CR, Langone F, Rogerio F, Parada CA. JAK2 inhibition is neuroprotective and reduces astrogliosis after quinolinic acid striatal lesion in adult mice. J Chem Neuroanat. 2013;48–49:14–22. doi: 10.1016/j.jchemneu.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Izquierdo A, Belcher AM, Scott L, Cazares VA, Chen J, O’Dell SJ, Malvaez M, Wu T, Marshall JF. Reversal-specific learning impairments after a binge regimen of methamphetamine in rats: possible involvement of striatal dopamine. Neuropsychopharmacol. 2010;35:505–514. doi: 10.1038/npp.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. A rotarod suitable for quantitative measurements of motor incoordination in naive mice. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1968a;259:211. doi: 10.1007/BF00537801. [DOI] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. The quantitative measurement of motor inco-ordination in naive mice using an accelerating rotarod. J Pharm Pharmacol. 1968b;20:302–304. doi: 10.1111/j.2042-7158.1968.tb09743.x. [DOI] [PubMed] [Google Scholar]
- Kataoka Y, Kalanithi PS, Grantz H, Schwartz ML, Saper C, Leckman JF, Vaccarino FM. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J Comp Neurol. 2010;518:277–291. doi: 10.1002/cne.22206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay JN, Blum M. Differential response of ventral midbrain and striatal progenitor cells to lesions of the nigrostriatal dopaminergic projection. Dev Neurosci. 2000;22:56–67. doi: 10.1159/000017427. [DOI] [PubMed] [Google Scholar]
- Krasnova IN, Hodges AB, Ladenheim B, Rhoades R, Phillip CG, Cesena A, Ivanova E, Hohmann CF, Cadet JL. Methamphetamine treatment causes delayed decrease in novelty-induced locomotor activity in mice. Neurosci Res. 2009;65:160–165. doi: 10.1016/j.neures.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnova IN, Cadet JL. Methamphetamine toxicity and messengers of death. Brain Res Rev. 2009;60:379–407. doi: 10.1016/j.brainresrev.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn HG, Palmer TD, Fuchs E. Adult neurogenesis: a compensatory mechanism for neuronal damage. Eur Arch Psychiatry Clinical Neurosci. 2001;251:152–158. doi: 10.1007/s004060170035. [DOI] [PubMed] [Google Scholar]
- Kojima T, Hirota Y, Ema M, Takahashi S, Miyoshi I, Okano H, Sawamoto K. Subventricualr zone-derived neural progenitor cells migrate along a blood vessel scaffold toward the post-stroke striatum. Stem Cells. 2010;28:545–554. doi: 10.1002/stem.306. [DOI] [PubMed] [Google Scholar]
- Luzzati F, De Marchis S, Parlato R, Gribaudo S, Schutz G, et al. New striatal neurons in a mouse model of progressive striatal degeneration are generated in both the subventricular zone and the striatal parenchyma. PloS One. 2011;6:e25088. doi: 10.1371/journal.pone.0025088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandyam CD, Wee S, Crawford EF, Eisch AJ, Richardson HN, Koob GF. Varied access to intravenous methamphetamine self-administration differentially alters adult hippocampal neurogenesis. Biol Psychiatry. 2008;64:958–965. doi: 10.1016/j.biopsych.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone MC, Marinelli S, Biamonte F, Keller F, Sgobio CA, Ammassari-Teule M, Bernardi G, Mercuri NB. Altered cortico-striatal synaptic plasticity and related behavioural impairments in reeler mice. Eur J Neurosci. 2006;24:2061–2070. doi: 10.1111/j.1460-9568.2006.05083.x. [DOI] [PubMed] [Google Scholar]
- McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci. 1998;18:8417–8422. doi: 10.1523/JNEUROSCI.18-20-08417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol. 2002;52:802–813. doi: 10.1002/ana.10393. [DOI] [PubMed] [Google Scholar]
- Parent JM. Injury-induced neurogenesis in the adult mammalian brain. Neuroscientist. 2003;9(4):261–272. doi: 10.1177/1073858403252680. [DOI] [PubMed] [Google Scholar]
- Peterson DA. Quanititative histology using confocal microscopy: implementation of unbiased stereology procedures. Methods. 1999;18:493–507. doi: 10.1006/meth.1999.0818. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Pu C, Broening HW, Vorhees CV. Effect of methamphetamine on glutamate-positive neurons in the adult and developing rat somatosensory cortex. Synapse. 1996;23:328–334. doi: 10.1002/(SICI)1098-2396(199608)23:4<328::AID-SYN11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: A regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- Salo R, Irsu S, Buonocore MH, Leamon MH, Carter C. Impaired prefrontal cortical function and disrupted adaptive cognitive control in methamphetamine abusers: a functional magnetic resonance imaging study. Biol Psychiatry. 2009;65:706–709. doi: 10.1016/j.biopsych.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CJ, Gibb JW. Role of the dopamine uptake carrier in the neurochemical response to methamphetamine: Effects of amfonelic acid. Eur J Pharmacol. 1985;109:73–80. doi: 10.1016/0014-2999(85)90541-2. [DOI] [PubMed] [Google Scholar]
- Seiden LS, MacPhail RC, Oglesby MW. Catecholamines and drug-behavior interactions. Fed Proc. 1975;34:1823–1831. [PubMed] [Google Scholar]
- Sudai E, Croitoru O, Shaldubina A, Abraham L, Gispan I, Flaumenhaft Y, Roth-Deri I, Kinor N, Aharoni S, Ben-Tzion M, Yadid G. High cocaine dosage decreases neurogenesis in the hippocampus and impairs working memory. Addict Biol. 2011;16(2):251–260. doi: 10.1111/j.1369-1600.2010.00241.x. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhang QW, Xu M, Guo JJ, Shen SW, Wang YQ, Sun FY. New striatal neurons form projections to substantia nigra in adult rat brain after stroke. Neurobiol Dis. 2012;45:601–609. doi: 10.1016/j.nbd.2011.09.018. [DOI] [PubMed] [Google Scholar]
- Supeno NE, Pati S, Hadi RA, Ghani AR, Mustafa Z, Abdullah JM, Idris FM, Han X, Jaafar H. IGF-1 acts as controlling switch for long-term proliferation and maintenance of EGF/FGF-responsive striatal neural stem cells. Int J Med Sci. 2013;10:522–531. doi: 10.7150/ijms.5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuchert-Noodt G, Dawirs RR, Hildebrandt K. Adult treatment with methamphetamine transiently decreases dentate granule cell proliferation in the gerbil hippocampus. J Neural Trans. 2000;107:133–143. doi: 10.1007/s007020050012. [DOI] [PubMed] [Google Scholar]
- Tulloch IK, Afanador L, Zhu J, Angulo JA. Methamphetamine induces striatal cell death followed by the generation of new cells and a second round of cell death in mice. Cur Neuropharmacol. 2011a;9:79–83. doi: 10.2174/157015911795017281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulloch I, Afanador L, Mexhitaj I, Ghazaryan N, Garzagongora AG, Angulo JA. A single high dose of methamphetamine induces apoptotic and necrotic striatal cell loss lasting up to 3 months in mice. Neurosci. 2011b;193:162–169. doi: 10.1016/j.neuroscience.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United Nations Office on Drugs and Crime. 2008 World Drug Report Annual overview 2008: UNDC Research and Analysis 2008 http://www.unodc.org/documents/wdr/WDR_2008/WDR2008_Overview.
- Widespread Urrea C, Castellanos DA, Sagen J, Tsoufas P, Bramlett HM, Dietrich WD. Widespread cellular proliferation and focal neurogenesis after traumatic brain injury in the rat. Restor Neurol Neurosci. 2007;25(1):65–76. [PubMed] [Google Scholar]
- Van Kampen JM, Hagg T, Robertson HA. Induction of neurogenesis in the adult rat subventricular zone and neostriatum following dopamine D3 receptor stimulation. Eur J Neurosci. 2004;19:2377–2387. doi: 10.1111/j.0953-816X.2004.03342.x. [DOI] [PubMed] [Google Scholar]
- Van Kampen JM, Robertson HA. Possible role for dopamine D3 receptor stimulation in the induction of neurogenesis in the adult rat substantia nigra. Neurosci. 2005;136:381–386. doi: 10.1016/j.neuroscience.2005.07.054. [DOI] [PubMed] [Google Scholar]
- Van Kampen JM, Eckman CB. Dopamine D3 receptor agonist delivery to a model of Parkinson’s disease restores the nigrostriatal pathway and improves locomotor behavior. J Neurosci. 2006;26:7272–7280. doi: 10.1523/JNEUROSCI.0837-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan A, Uzasci L, Chen Z, Rajbhandari L, Anderson C, Lee MH, Bianchet MA, Cotter R, Song H, Nath A. Impairment of adult hippocampal neural progenitor proliferation by methamphetamine: role for nitrotyrosination. Mol Brain. 2011;27(4):28. doi: 10.1186/1756-6606-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, Sedler MJ, Gatley SJ, Hitzemann R, Ding YS, Logan J, Wong C, Miller EN. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry. 2001;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Whimbey AE, Denenberg VH. Two independent behavioral dimensions in open-field performance. J Comp Physiol Psychol. 1967;63:500–504. doi: 10.1037/h0024620. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nature Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Ninomiya M, Hernández Acosta P, García-Verdugo JM, Sunabori T, Sakaguchi M, Adachi K, Kojima T, Hirota Y, Kawase T, Araki N, Abe K, Okano H, Sawamoto K. Subventricular zone neuroblasts migrate and differentiate into mature neurons in the post-stroke striatum. J Neurosci. 2006;26:6627–6636. doi: 10.1523/JNEUROSCI.0149-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JPQ, Xu W, Angulo JA. Disparity in the temporal appearance of methamphetamine-induced apoptosis and depletion of dopamine terminal markers in the striatum of mice. Brain Res. 2005;1049:171–181. doi: 10.1016/j.brainres.2005.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JPQ, Xu W, Angulo JA. Methamphetamine-induced cell death:selective vulnerability in neuronal subpopulations of the striatum in mice. Neurosci. 2006;140:607–622. doi: 10.1016/j.neuroscience.2006.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]