

Abstract
Summary: Driven by dramatic technological improvements, large-scale characterization of lymphocyte receptor repertoires via high-throughput sequencing is now feasible. Although promising, the high germline and somatic diversity, especially of B-cell immunoglobulin repertoires, presents challenges for analysis requiring the development of specialized computational pipelines. We developed the REpertoire Sequencing TOolkit (pRESTO) for processing reads from high-throughput lymphocyte receptor studies. pRESTO processes raw sequences to produce error-corrected, sorted and annotated sequence sets, along with a wealth of metrics at each step. The toolkit supports multiplexed primer pools, single- or paired-end reads and emerging technologies that use single-molecule identifiers. pRESTO has been tested on data generated from Roche and Illumina platforms. It has a built-in capacity to parallelize the work between available processors and is able to efficiently process millions of sequences generated by typical high-throughput projects.
Availability and implementation: pRESTO is freely available for academic use. The software package and detailed tutorials may be downloaded from http://clip.med.yale.edu/presto.
Contact: steven.kleinstein@yale.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
High-throughput sequencing technologies now enable large-scale characterization of lymphocyte receptor repertoires (Rep-Seq) (Benichou et al., 2012). Rep-Seq studies have used a variety of next-generation sequencing platforms, including Roche’s 454 and Illumina’s Genome Analyzer. Researchers are now beginning to take advantage of newer platforms, such as the MiSeq offered by Illumina, which can generate >10 million paired-end 300 base-pair reads in a single run. Many experimental protocols take advantage of these high read volumes by tagging reads with sample-specific barcode sequences and multiplexing sample groups. More recently, protocols to barcode single mRNA molecules with unique identifiers (UID) before PCR amplification have emerged (Shiroguchi et al., 2012; Vollmers et al., 2013), allowing PCR amplification effects to be resolved and offering the potential to dramatically reduce sequencing-dependent error rates using single-molecule consensus reads.
We have developed a suite of utilities, the REpertoire Sequencing TOolkit (pRESTO), that provides an integrated framework to handle all stages of sequence processing prior to germline segment assignment, which may then be handled by other available software such as IMGT/HighV-QUEST (Alamyar et al., 2012). pRESTO is designed to handle either single- or paired-end reads, has been tested on data from both the Roche 454 and Illumina MiSeq platforms and includes a wide range of features designed to meet the needs of various Rep-Seq protocols; see Supplementary Material Section 1.1 and Supplementary Tables S1 and S2.
2 FEATURES
2.1 Overview and implementation
The pRESTO software package is provided as a set of command-line utilities, all of which are implemented as platform-independent Python modules. pRESTO is designed to allow for maximum flexibility in workflow organization to meet the unique needs of different sequencing projects. Particular emphasis is placed on providing support for multiplexed primers, multiplexed sample pools and emerging technologies that use UID barcoding. However, pRESTO is equally suitable for experimental protocols that do not use complex mixtures of molecular tags. Each tool accepts sequences in the form of FASTA or FASTQ (with Phred scoring scheme) files. The more computationally expensive tools in the pRESTO suite are natively parallelized, allowing users to take advantage of multicore systems by specifying the number of subprocesses to execute. Additionally, pRESTO allows users to easily integrate automatic file segregation into their workflow for distributed processing on cluster computing resources. A detailed description of each pRESTO tool is provided in the Supplementary Material (summarized in Supplementary Table S1). Example datasets with step-by-step tutorials covering the workflows illustrated in Figure 1 can be found on the pRESTO website.
Fig. 1.
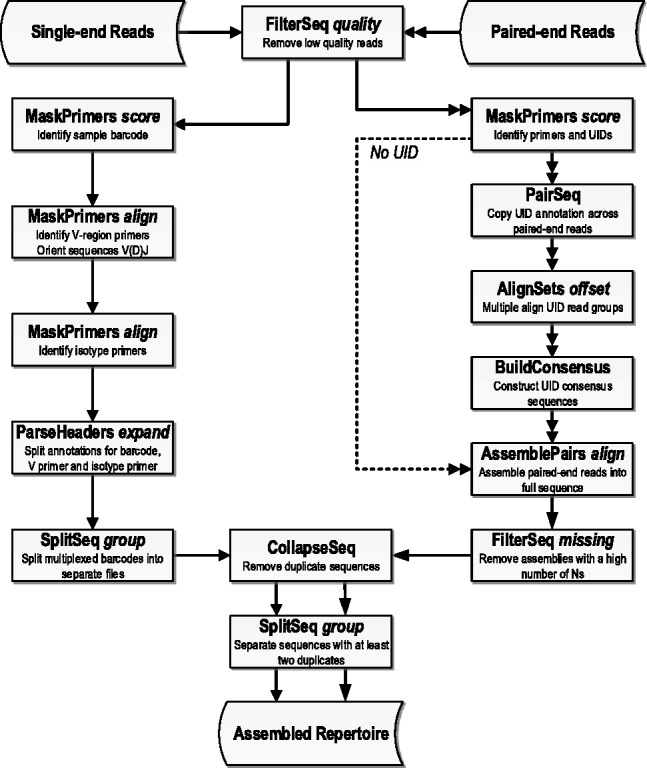
Example workflow diagram. Example workflows for single-end read sequencing protocols with sample barcoding (left) and paired-end read protocols with/without UID barcoding (right). Single sequence file inputs are shown with single arrowheads, and parallel processing of two paired-end read files are shown with a double arrowhead
2.2 Annotation
To meet the particular needs of Rep-Seq projects, pRESTO uses an annotation scheme that labels individual reads by extending the sequence descriptions (Supplementary Material Section 2.1 and Supplementary Fig. S1). pRESTO’s annotation features allow users to sort and subset the sequences in multiplexed runs, simplifying the workflow and reducing the chance of human error. For example, within a single multiplexed run, the receptor isotype is often determined by the particular constant region primer sequence. pRESTO’s annotation system associates this information with each read, rather than requiring a complex system of separate files for each set of annotations, thereby simplifying comparative analysis.
pRESTO provides several methods for manipulating these annotations, allowing pipelines to be customized by integrating textual or arithmetic sequence filters into the workflow (Supplementary Material Sections 2.10 and 2.11). Beyond the default annotations, more detailed sequence-specific information can be captured through pRESTO’s logging features. pRESTO can convert both sequence-embedded and logged annotations into data tables suitable for automated analysis and plotting (Supplementary Material Sections 2.11 and 2.12).
2.3 Quality control and error profiling
pRESTO provides comprehensive quality control tools to filter reads based on sequence properties such as Phred quality scores (Supplementary Material Section 2.2), valid barcode labeling (Supplementary Material Section 2.3), primer identity (Supplementary Material Section 2.3) and abundance of duplicate reads (Supplementary Material Section 2.9). pRESTO also provides tools to measure the diversity and error profiles of sets of annotated reads (Supplementary Material Sections 2.5 and 2.8); such information may be used to estimate sequencing error rates and remove highly variable UID read groups from the analysis.
2.4 UID barcoding and paired-end reads
Recent advances in Rep-Seq protocols are allowing researchers to improve both the sequencing and quantification accuracy of repertoire data by labeling each starting nucleic acid sequence with a unique single-molecule identifier (UID) before amplification (Shiroguchi et al., 2012; Vollmers et al., 2013). The pRESTO suite includes special operations tailored for UID barcoding technologies, including tools to multiple align UID read groups (Supplementary Material Section 2.5) and generate consensus sequences from UID read groups (Supplementary Material Section 2.6).
pRESTO also provides support for de novo assembly of overlapping paired-end reads (Supplementary Material Section 2.7). pRESTO does not require maintenance of file ordering across pair-end read files, facilitating independent filtering of separate paired-end read files. However, the sequence sampling and sorting tools may operate in a paired-end mode, allowing users to create uniformly ordered paired-end files (Supplementary Material Section 2.10). pRESTO allows users to propagate annotations between paired-end records (Supplementary Material Section 2.4), which is required for protocols where the sample barcode or UID are found on only one read of the mate pair.
3 CONCLUSIONS
We have developed a flexible toolkit, pRESTO, for rapid processing of high-throughput lymphocyte receptor sequencing data originating from either mRNA or DNA templates. pRESTO is compatible with the latest molecular barcoding and sequencing technologies and provides a means to generate high-fidelity repertoire datasets. This output can be used to drive subsequent analysis, such as (i) identifying V(D)J gene segments (Alamyar et al., 2012; Gaëta et al., 2007; Thomas et al., 2013; Volpe et al., 2006; Ye et al., 2013), (ii) clustering clonally related sequences (Chen et al., 2010), (iii) inferring individual genotypes (Kidd et al., 2012), (iv) creating lineage trees (Barak et al., 2008), (v) quantifying affinity-dependent selection (Yaari et al., 2012) and (vi) analysis of somatic hypermutation patterns (Yaari et al., 2013). The software package, detailed example workflows and sample datasets are available online at http://clip.med.yale.edu/presto.
Supplementary Material
ACKNOWLEDGEMENT
The authors thank the Yale High Performance Computing Center (funded by NIH grant: RR19895) for use of their computing resources.
Funding: National Library of Medicine grant (T15 LM07056); United States-Israel Binational Science Foundation grant (2009046); National Institutes of Health grant (U19AI089992); National Institutes of Health grant (U19AI050864); EMD/Merck/Serono sponsored Grant for Multiple Sclerosis Research Innovation; Race to Erase MS.
Conflict of Interest: none declared.
REFERENCES
- Alamyar E, et al. IMGT/HighV-QUEST: the IMGT web portal for immunoglobulin (IG) or antibody and T cell receptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Res. 2012;8:26. [Google Scholar]
- Barak M, et al. IgTree: creating immunoglobulin variable region gene lineage trees. J. Immunol. Methods. 2008;338:67–74. doi: 10.1016/j.jim.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Benichou J, et al. Rep-seq: uncovering the immunological repertoire through next-generation sequencing. Immunology. 2012;135:183191. doi: 10.1111/j.1365-2567.2011.03527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, et al. Clustering-based identification of clonally-related immunoglobulin gene sequence sets. Immunome Res. 2010;6(Suppl. 1):S4. doi: 10.1186/1745-7580-6-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaëta BA, et al. iHMMune-align: hidden Markov model-based alignment and identification of germline genes in rearranged immunoglobulin gene sequences. Bioinformatics. 2007;23:1580–1587. doi: 10.1093/bioinformatics/btm147. [DOI] [PubMed] [Google Scholar]
- Kidd MJ, et al. The inference of phased haplotypes for the immunoglobulin h chain v region gene loci by analysis of VDJ gene rearrangements. J. Immunol. 2012;188:1333–1340. doi: 10.4049/jimmunol.1102097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiroguchi K, et al. Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc. Natl Acad. Sci. USA. 2012;109:1347–1352. doi: 10.1073/pnas.1118018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas N, et al. Decombinator: a tool for fast, efficient gene assignment in T-cell receptor sequences using a finite state machine. Bioinformatics. 2013;29:542–550. doi: 10.1093/bioinformatics/btt004. [DOI] [PubMed] [Google Scholar]
- Vollmers C, et al. Genetic measurement of memory B-cell recall using antibody repertoire sequencing. Proc. Natl Acad. Sci. USA. 2013;110:13463–13468. doi: 10.1073/pnas.1312146110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JM, et al. SoDA: implementation of a 3D alignment algorithm for inference of antigen receptor recombinations. Bioinformatics. 2006;22:438–444. doi: 10.1093/bioinformatics/btk004. [DOI] [PubMed] [Google Scholar]
- Yaari G, et al. Quantifying selection in high-throughput immunoglobulin sequencing datasets. Nucleic Acids Res. 2012;40:e134. doi: 10.1093/nar/gks457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaari G, et al. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front. Immunol. 2013;4:358. doi: 10.3389/fimmu.2013.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, et al. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41:W34–W40. doi: 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.