

Synopsis
GISTs are unique tumors that arise largely due to oncogenic mutations in KIT or PDGFRA tyrosine kinases. Although surgery remains the most effective treatment, the remarkable clinical success achieved with kinase inhibition in both the adjuvant and metastatic settings has made GIST one of the most successful examples of targeted therapy for the treatment of cancer. The insight gained from this approach has allowed a deeper understanding of the molecular biology driving kinase dependent cancers, and the adaptations to kinase inhibition, linking genotype to phenotype. Mutation tailored kinase inhibition with second generation TKI’s, and combination immunotherapy to harness the effects of TKIs and achieve longer durable responses, remain exciting areas of investigation.
Keywords: gastrointestinal stromal tumor, sarcoma, KIT, imatinib, tyrosine kinase
Introduction
Gastrointestinal stromal tumor (GIST) is the most common sarcoma, accounting for approximately 18% of all sarcomas and 1% of all intestinal neoplasms.1 The annual incidence of GIST as determined by population-based studies is approximately 10 cases per million.2–4 GISTs have historically portended a poor prognosis. Up to 50% of patients have recurrent disease 5 years after complete resection. Median survival in metastatic GIST used to be approximately 9 months as it is inherently resistant to chemotherapy and radiation.5–7 The discovery of oncogenic tyrosine kinase mutations in GIST, and the successful application of kinase inhibitor therapies have made GIST a model of targeting aberrant signal transduction to treat cancer. Lessons learned from this approach have allowed new insight into the molecular biology and mechanisms of resistance of kinase driven cancers. It has spurred development of novel targeted inhibitors and uncovered exciting possibilities for combination therapy with other systemic agents.
Oncogenic kinase mutations and GIST pathogenesis
KIT
In 1998, two important discoveries were made that furthered our understanding of GIST biology. Hirota and colleagues described their landmark discovery of gain-of-function mutations in KIT in 5 GIST patients.8 They hypothesized that these were oncogenic driver mutations, as Ba/F3 lymphoid cells transfected with mutant KIT cDNA underwent malignant transformation. Shortly thereafter, two groups reported that 95% of GISTs are immunohistochemically positive for the receptor tyrosine kinase KIT, also known as CD117.9, 10 Since then, a causal relationship between KIT mutations and GIST pathogenesis has been further supported by many lines of evidence. Mutant KIT induces constitutive kinase activation without ligand binding.8, 11, 12 KIT mutations have been discovered in very small GISTs, suggesting it occurs as a very early event.13, 14 GIST tumor extracts almost universally demonstrate phosphorylated KIT.15 Transgenic Kit knock-in mouse models develop spindle cell tumors that are morphologically similar to human GIST.16, 17 Finally, KIT blockade in vitro and in vivo inhibits tumor growth.12, 18–21
KIT, a receptor tyrosine kinase, binds KIT ligand (stem cell factor), which results in receptor dimerization, phosphorylation and activation of downstream signaling pathways that promote cell proliferation and survival. It is now known that 70–80% of GISTs harbor a KIT mutation that induces constitutive kinase activation. Mutations most commonly occur in the juxtramembrane domain in exon 11 (Table 1, Figure 1), which normally inhibits the kinase activation loop in the absence of ligand binding. Exon 11 mutations include in-frame deletions, insertions, and substitutions, but deletions are the most common. Mutations also occur in the extracellular domains (exons 8 (rarely) and 9), and infrequently in the kinase domains (exons 13 and 17) (Table 1, Figure 1).22 The downstream signaling pathways activated include the MAPK, PI3K-AKT, and STAT3 pathways, which lead to inhibition of apoptosis and cell proliferation.22 Recently, ETV1, a lineage survival factor in interstitial cells of Cajal (ICC), the hypothesized cell of origin for GIST, was shown to cooperate with activated KIT to induce GIST tumorigenesis.23
Table 1.
Molecular classification of GIST.
| Gene | Incidence | Anatomic location | Imatinib sensitivity |
|---|---|---|---|
| Mutations in KIT (80%) | |||
| Exon 9 | 7% | Small intestine, colon | Yes, consider 800mg/day |
| Exon 11 | 65% | All locations | Yes |
| Exon 13 | 1% | All locations | Variable |
| Exon 17 | 1% | All locations | Variable |
| Mutations in PDGFRA (5–8%) | |||
| Exon 12 | 2% | All locations | Yes |
| Exon 14 | <1% | Stomach | Yes |
| Exon 18 | 7% | Stomach, mesentary, omentum | D842V insensitive, most other sensitive |
| WT (12–15%) | |||
| BRAF V600E | 7–15%* | Stomach, small intestine | Possibly |
| SDHA, SDHB, SDHC, SDHD | 12%* | Stomach, small intestine | Usually not |
| Familiar GIST | |||
| KIT, rarely PDGFRA | Very rare | Small intestine | Usually not |
| Syndromic GIST | |||
| Unknown gene (Carney Triad) | Very rare | Stomach | Usually not |
| SDHB, SDHC, SDHD (Carney-Stratakis) | Rare | Stomach | Usually not |
| NF1 (Neurofibromatosis-1) | Rare | Small intestine | Usually not |
indicates % of WT GISTs.
Data from Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247–258.
Figure 1.
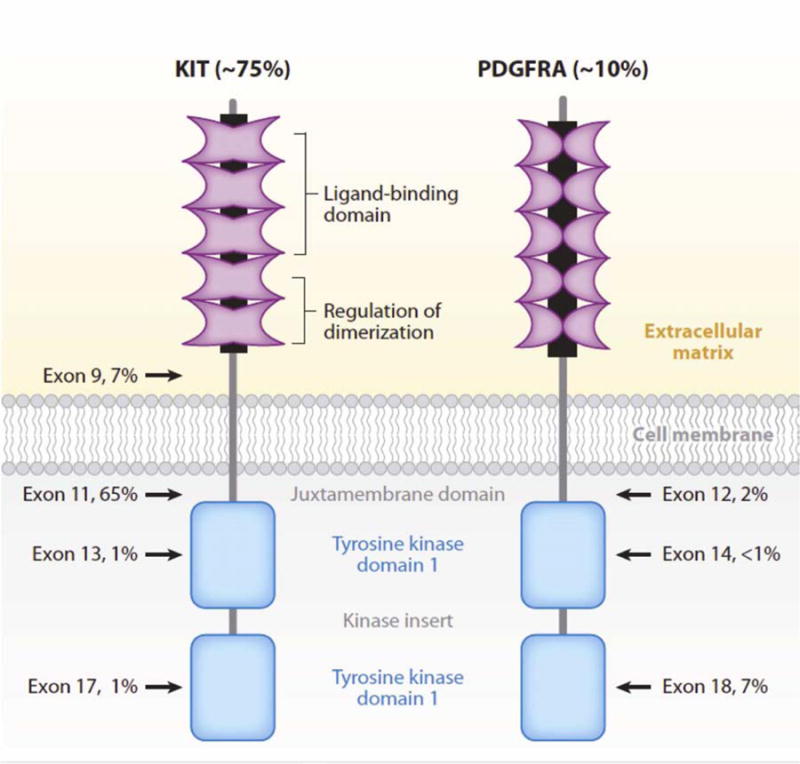
Schematic structures of KIT and PDGFR. The percentages indicate the frequency of mutations detected in each exon of the gene that encodes for the protein.
From Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247–258; with permission.
PDGFRA
Approximately one third of GISTs that do not have a mutation in KIT (8% of all GISTs) harbor a mutation in a closely related tyrosine kinase, platelet-derived growth factor receptor alpha (PDGFRA).24, 25 PDGFRA and KIT mutations are mutually exclusive in GIST. Like mutations in KIT, PDGFRA mutations are found in its juxtramembrane domain (Table 1, Figure 1), ATP binding domain, or activation loop, and cause ligand independent receptor activation. An oncogenic role for these mutations in GIST has followed evidence similar to that for KIT - mutant PDGFRA induces ligand independent receptor activation, and PDGFRA inhibition induces cellular arrest.24–26 PDGFRA mutant GISTs do however have unique clinical profiles, including gastric location, epithelioid morphology, variable KIT expression, and a more indolent clinical course.27
Wild type GIST
10–15% of tumors do not have mutations in KIT and PDGFRA (WT GIST). Other mutations that may contribute to tumorigenesis have been recently uncovered (Table 1). Similar to BRAF mutations in melanoma, papillary thyroid cancer, and colorectal cancer, GIST BRAF mutations have also been identified in 7–15% of WT GISTS within the exon 15 V600E hot-spot.28, 29 BRAF proteins and constituents of the MAPK signaling pathway can stimulate cell growth independent of KIT and are a possible cause of resistance to KIT and PDGFRA kinase inhibitors. Mutations in the succinate dehydrogenase (SDH) respiratory chain complex have also been discovered in WT GIST. SDH mutations were initially identified in the germline in subunits SDHB, SDHC, and SDHD, predisposing affected individuals to GIST and paraganglionomas (Carney-Stratakis syndrome). They have since been identified in 12% of WT GIST (Table 1).30 Mutations in SDHA have also since been reported.31 The precise oncogenic role of SDH mutations in GIST remains to be elucidated. Expression of insulin-like growth factor 1 receptor (IGF1R), that signals through MAPK and PI3K-AKT pathways, has also been detected and may contribute to GIST pathogenesis.32 WT GISTs are also found in 7% of patients with neurofibromatosis type I (NF1), who harbor germline mutations in the neurofibromin 1 gene (Table 1).33
Targeting kinase pathways in GIST
Until 2000, outcomes in patients with metastatic GIST were extremely poor. Median survival was approximately 9 months, and responses to conventional chemotherapy was < 5%.5–7 The discovery of oncogenic KIT mutations in GIST coincided with the successful clinical development and application of the tyrosine kinase inhibitor imatinib (Gleevec) for the treatment of chronic myelogenous leukemia. It was noted that the kinases KIT and ABL shared structural similarity, prompting the first clinical application of imatinib in a 50-year-old female with advanced GIST, which was met with a dramatic clinical response.34 This led to phase I, II, and two international phase III trials to investigate the benefit of imatinib in the metastatic setting. Overall, imatinib achieved disease control in 70–85% of patients with KIT-positive GIST, with a median progression-free-survival of 20–24 months, and an estimated overall survival over 36 months (Figure 2).6, 7, 35, 36 The advent of imatinib therapy for metastatic GIST has dramatically altered prognosis - currently, median survival is 5 years with 34% of patients surviving more than 9 years.33 Imatinib is first line treatment in patients with metastatic GIST, and treatment is recommended to continue indefinitely as long as there is clinical benefit, as interruption is associated with high rate of relapse.37
Figure 2.
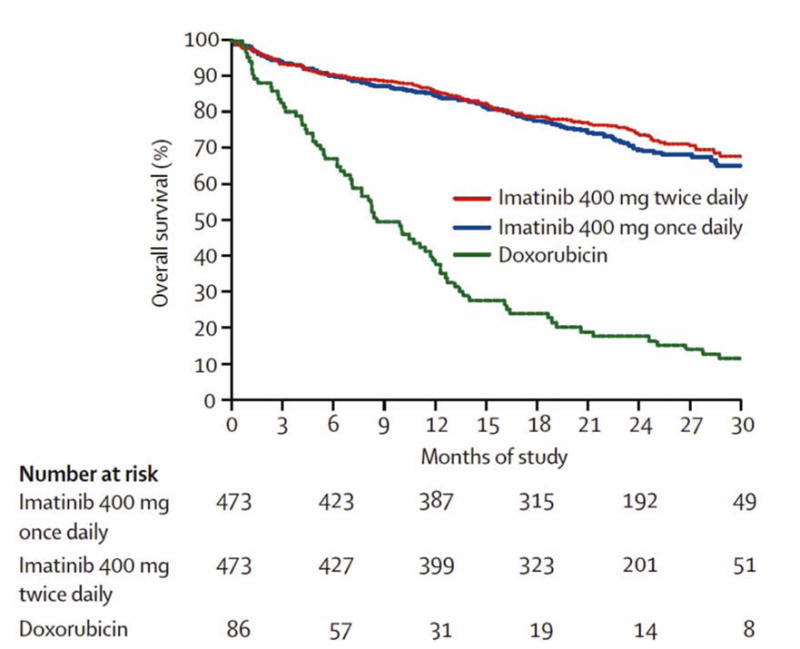
Overall survival for study population of EORTC 62005 compared with historical controls from EORTC database.
From Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial*. The Lancet. 2004; 364(9440):1127–1134; with permission.
Paralleling the success in GIST, a molecular approach to systemic therapy has been adopted in many other solid tumors. Genomic analyses have uncovered biologically relevant and druggable kinase mutations in other solid malignancies. Although the success achieved in these cancers has not replicated the GIST success, it has validated a molecular approach to systemic treatment and has heralded kinase based therapies as an integral component of cancer care (Table 2).
Table 2.
Tyrosine kinase mutations and targeted agents in solid tumors.
| Gene | Tumor | Agent |
|---|---|---|
| KIT | Melanoma, seminoma, small cell lung cancer, synovial sarcoma, thymic carcinoma | Imatinib81–89 |
| PDGFRA | Dermatofibrosarcoma protuberans | Imatinib90,91 |
| EGFR | Non-small cell lung cancer | Gefitinib, Erlotinib92–96 |
| Squamous cell, ovarian, renal cell, and colorectal cancer, glioblastoma multiforme | Erlotinib97, gefitinib98, Lapatanib99, Cetuximab100 Panitumumab101 | |
| BRAF | Melanoma, papillary thyroid cancer, colon cancer | Vemurafenib102,103 |
| HER-2 | Breast cancer, lung cancer | Trastuzumab104,105 |
| VEGFR | Non-small cell lung, breast, prostate, renal, colorectal | Bevacizumab, VEGF inhibitors106 |
| RET | Multiple endocrine neoplasia 2A, 2B, Familial Medullary Thyroid Cancer, Radiation-associated papillary thyroid cancer | Cabozantinib107, Vandetanib108, Sorafenib109 |
Assessing response to kinase therapy
Responses to systemic therapy in solid tumors have been traditionally assessed using the response evaluation criteria in solid tumors (RECIST), which incorporates unidirectional tumor size. However assessing responses using RECIST has been shown to be insensitive in GIST.38 PET scans had been traditionally used to assess continuing responses to TKI treatment, as significant decreases in FDG signal is seen within 24 hours in patients responding to imatinib.39 However, Choi and colleagues proposed using CT determined tumor size and density in assessing treatment response - responding tumors demonstrate homogeneous and hypodense features, losing solid elements and neovascularity.40, 41 The Choi criteria correlate with PET, are superior to RECIST, and are a significant improvement in our understanding of assessing clinical responses to systemic agents in solid tumors.
Combining targeted therapy with surgery
Adjuvant imatinib
While TKI therapy induces tumor regression in the majority of patients, it rarely induces complete responses. Even long-term TKI therapy fails to eradicate GIST cells, with viable tumor cells detected even in tumors with good histologic responses.42 In contrast, surgery for patients with primary GIST without metastases cures over 50% of patients.43 In a double-blind, placebo-controlled, multicenter, randomized trial, the American College of Surgeons Oncology Group (ACOSOG) demonstrated that one year of adjuvant imatinib following resection of GISTs at least 3 cm in size significantly improved 1-year RFS (83% in placebo arm versus 98% in imatinib arm, Figure 3).44 Based on these results, the Food and Drug Administration (FDA) approved imatinib for use in the adjuvant setting. Recently, it was shown that patients at high risk of recurrence treated with 3 years of adjuvant imatinib following surgical resection have 5-year RFS and overall survival (OS) rates of 65.6% and 92% respectively, compared to 47.9% and 81.7% in patients treated with 1 year of adjuvant imatinib.45 However, there was no difference in disease-specific survival between 1 and 3 years of therapy. An additional phase III trial is currently examining the outcomes after 2 years of adjuvant imatinib following surgery. A phase II, non-randomized, multicenter trial is also evaluating the efficacy of 5 years of adjuvant imatinib following complete resection of primary GIST. The success of adjuvant imatinib in GIST ranks with trastuzumab as one the most successful applications of kinase inhibitor therapy for the adjuvant treatment of solid tumors.46
Figure 3.
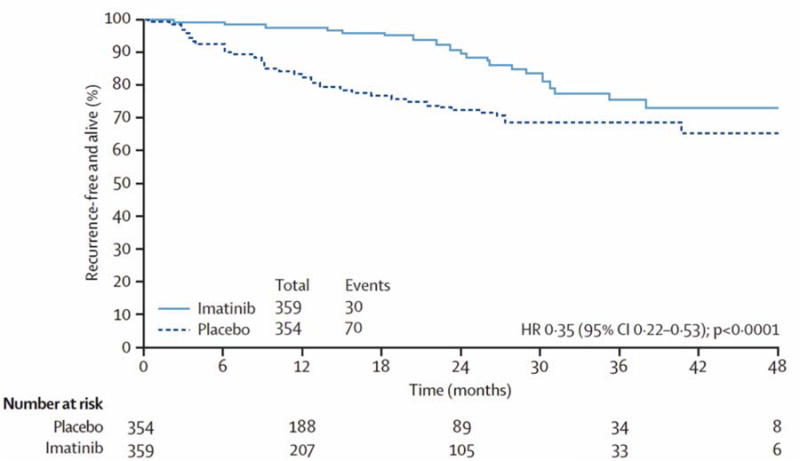
Recurrence-free survival in the American College of Surgeons Group (ACOSOG) trial Z9001 evaluating the efficacy of one year of adjuvant imatinib compared to placebo.
From DeMatteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localized, primary gastrointestinal stromal tumour; a randomized, double-blind, placebo-controlled trial. Lancet. Mar 28 2009; 373(9669):1097–1104; with permission.
Neoadjuvant imatinib
When primary GIST appears borderline resectable or unresectable, neoadjuvant imatinib treatment may allow for tumor shrinkage and a subsequent R0 resection. Preliminary phase II trials have demonstrated the safety and efficacy of preoperative imatinib.47–49 However, there are no published phase III data on neoadjuvant imatinib for unresectable GIST. This is an area of ongoing investigation.
Molecular biology and risk stratification
Similar to other sarcomas, tumor size, mitotic index, and location have been shown to determine biological aggressivenesss in GIST.50 However, the discovery of oncogenic kinase mutations has allowed new insight into links between molecular biology and clinical behavior. It is now clear that recurrence patterns after primary resection are also governed by mutation type - deletion and insertion mutations in KIT exon 11, and exon 9 confer higher recurrence rates compared to other mutations.50, 51 Within exon 11 mutations, deletions (specifically in amino acids 557 and/or 558) have worse outcome.50, 52, 53 Currently, our understanding of risk stratification to predict the natural history of resected disease is achieved through prognostic nomograms. We developed a nomogram predicting 2-year and 5-year RFS factoring tumor size, mitotic index, and location (Figure 4).54 Dei Tos and colleagues have reported a nomogram predicting 10-year overall survival.55 Currently, the relationship between mutation type, adjuvant imatinib, and other factors in the nomogram remain unclear.
Figure 4.
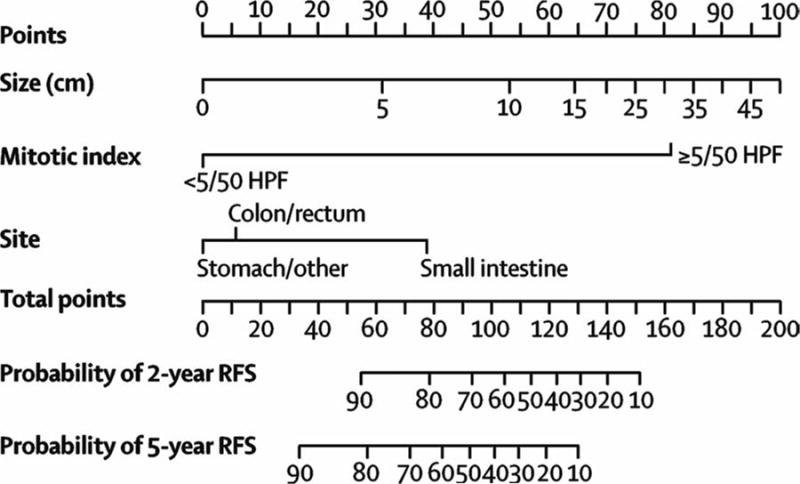
Nomogram predicting 2 and 5-year recurrence-free survival in patients with resected primary GIST. Points are assigned based on tumor size, mitotic index, and site by drawing an upward vertical line to the “Points” bar. Based on the sum of the points generated, a downward vertical line is drawn from the “Total Points” line to calculate 2 and 5-year RFS.
From Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumor: a retrospective analysis. Lancet Oncol 2009; 10:1045–1052; with permission.
TKI resistance
Although most patients initially respond to TKI therapy, the majority develops resistance. Over 50% of patients develop disease progression by 2 years.56 Primary resistance, defined as progression within the first 6 months of treatment, occurs in 10% of patients. Resistance is linked to kinase genotype and TKI sensitivity - patients with KIT exon 11 or 9 mutation or WT GISTs have a 5%, 16%, and 23% probability of demonstrating primary imatinib resistance.57 PDGFRA D842V mutations are strongly resistant to imatinib in vitro and in vivo. The mechanism of primary resistance remains unclear.
Patients with secondary resistance develop disease progression after an initial benefit from imatinib, predominantly due to secondary mutations in the identical gene and allele as the primary oncogenic driver mutation.33, 56, 58–64 More than 80% of drug-resistant GIST tumors harbor secondary mutations.33, 65–67 Secondary mutations may disrupt imatinib binding, or stabilize the active conformation of the KIT kinase.56, 60 The mechanism of development of second site mutations remains unclear. Long-term imatinib therapy can also lead to “polyclonal acquired resistance”, whereby different tumor nodules acquire different secondary mutations, and progress independently.56, 63, 68, 69 Additionally, up to one-third of secondary resistant GIST lack secondary mutations, where possible mechanisms of resistance include KIT genomic amplification and alternate tyrosine kinase activation.42 These findings have provided invaluable insight into a common endpoint of kinase inhibitor therapy in solid tumors and have guided the development of second line TKIs. However, the genetic complexity of acquired resistance argues against second line TKI monotherapy providing durable clinical benefit.
Strategies to combat TKI resistance
Second line TKIs
Imatinib dose escalation is the initial recommendation for patients progressing on imatinib as 20–30% of patients may have 1 year or more of disease control.36 Multiple salvage TKIs are in development to combat imatinib resistance (Table 3). Currently, sunitinib, a TKI that inhibits KIT, PDGFRA, PDGFRB, Fms-like tyrosine kinase-3 receptor, RET, and vascular endothelial growth factor receptors (VEGFR) 1, 2, and 3, is the second line TKI of choice in patients with generalized disease progression who have failed imatinib dose escalation or who are imatinib intolerant. Demetri et al. demonstrated that patients with imatinib-resistant GIST treated with sunitinib had a median time to progression of 27.3 weeks compared to 6.4 weeks for placebo.70 Despite the remarkable success with imatinib, results with second line TKIs in GIST have been poor, underscoring the need for new treatment strategies.71–74 Regorafenib was recently FDA approved as a third line agent.74
Table 3.
New targeted agents under investigation for GIST.
| Tyrosine kinase inhibitors | Molecular Target | Development phase |
|---|---|---|
| Nilotinib | KIT, PDGFR, BCR-ABL110–112 | Phase III |
| Dasatinib | KIT, ABL, SRC113 | Phase III |
| Sorafenib | KIT, PDGFR, VEGFR, BRAF114–116 | Phase II |
| Regorafenib | KIT, PDGFRA, VEGFR, BRAF, FLT-3, Raf-174,117 | Phase III |
| Masitinib | KIT, PDGFR, LYN71,118 | Phase III |
| Pazopanib | KIT, PDGFRA, VEGFR | Phase II |
| Vatalanib | KIT, PDGFRA, VEGFR73,119 | Phase II |
| Crenolanib | PDGFRA D842V120 | Phase II |
Surgery and TKI therapy for metastatic disease
TKI therapy has been combined with surgery in the metastatic setting. We found that metastatic GIST patients with focal resistance (1 tumor growing) on imatinib who were treated with surgery had a 2-year OS of 36% compared to 100% in patients with imatinib-responsive or stable tumors. Patients with multifocal resistance (more than 1 tumor growing) had a 1-year OS of 36%.75 Other groups have also reported a lack of clinical benefit for patients progressing on imatinib treated with surgery.76, 77 Identifying the patient cohort and quantifying the precise benefit from surgery after imatinib in the metastatic setting remains an area needing further examination.
Combination targeted therapy and immunotherapy
In addition to inhibition of oncogenic signaling pathways, targeted agents are potent immunomodulators. They promote dendritic cell maturation and T cell priming, increase death receptor expression on tumor cells sensitizing them to immune-mediated tumor clearance, and diminish tumor-induced immunosuppression.78 The immune system has also been shown to be important in GIST. In imatinib treated GIST patients, progression-free survival correlated with IFN-γ secretion by natural killer (NK) cells in the blood.79 We demonstrated that the antitumor effects of imatinib, previously thought to act exclusively via oncogenic kinase inhibition in tumor cells, relies partially on indirect effects of the immune system. Using a mouse model of spontaneous GIST, we found that imatinib therapy activated CD8+ T cells and induced inhibitory regulatory T cell (Treg) apoptosis, thereby increasing the intratumoral CD8+ T cell/ Treg ratio, a hallmark of immunologic outcome.80 The mechanism relied on imatinib inhibiting tumor-cell expression of the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO), by reducing expression of the transcription factor ETV4, and disrupting its ability to bind the IDO promoter. Extending these findings in vivo, we correlated the intratumoral CD8+ T cell/ Treg ratio to imatinib response and intratumoral IDO expression in freshly analyzed human GIST tumors. Our results link acquired resistance to imatinib to restoration of intratumoral immunosuppression. Hence molecular and immune resistance in GIST appear to be intertwined. To investigate whether imatinib synergizes with immune modulating agents, we combined imatinib therapy with blockade of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), a known T cell and IDO modulator. Tumor size was significantly decreased in mouse GIST compared to either treatment alone. These data demonstrate the rationale and potential of combining targeted therapy with immunotherapy to improve outcomes in not only GIST, but also other solid tumors treated with TKIs. Currently, we are conducting an NCI sponsored phase I trial examining the effects of CTLA-4 inhibition with dasatanib in GIST and other sarcomas. Multiple other groups are investigating combining targeted agents and immune agents, including a phase II trial examining vemurafenib and CTLA-4 blockade in patients with melanoma who have V600E BRAF mutations.78
Key Points.
GISTs are unique solid tumors as they are driven predominantly by oncogenic mutations in KIT or PDGFRA tyrosine kinases.
Surgery is the most effective treatment for localized, primary GIST. Adjuvant tyrosine kinase inhibition (TKI) with imatinib substantially decreases recurrence rates but does not appear to affect overall survival.
Imatinib is initial therapy for metastatic GIST however acquired mutations frequently lead to resistance after initial responses. The role of surgery and TKI in metastatic GIST remains unclear.
Imatinib dose escalation, sunitinib, and regorafenib are the initial therapeutic options for imatinib resistant GIST, with many novel TKIs currently under investigation.
Preclinical data suggest antitumor effects of imatinib in GIST are partially dependent on host immune responses. Combination imatinib and immunotherapy may be effective in GIST and other solid tumors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have nothing to disclose.
References
- 1.Ducimetiere F, Lurkin A, Ranchere-Vince D, et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS One. 2011;6(8):e20294. doi: 10.1371/journal.pone.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nilsson B, Bumming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era–a population-based study in western Sweden. Cancer. 2005 Feb 15;103(4):821–829. doi: 10.1002/cncr.20862. [DOI] [PubMed] [Google Scholar]
- 3.Steigen SE, Eide TJ. Trends in incidence and survival of mesenchymal neoplasm of the digestive tract within a defined population of northern Norway. APMIS. 2006 Mar;114(3):192–200. doi: 10.1111/j.1600-0463.2006.apm_261.x. [DOI] [PubMed] [Google Scholar]
- 4.Mucciarini C, Rossi G, Bertolini F, et al. Incidence and clinicopathologic features of gastrointestinal stromal tumors. A population-based study. BMC Cancer. 2007;7:230. doi: 10.1186/1471-2407-7-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg. 2000 Jan 1;231(1):51–58. doi: 10.1097/00000658-200001000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002 Aug 15;347(7):472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 7.Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial*. The Lancet. 2004;364(9440):1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 8.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998 Jan 23;279(5350):577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 9.Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998 May;152(5):1259–1269. [PMC free article] [PubMed] [Google Scholar]
- 10.Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, Miettinen M. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol. 1998 Aug;11(8):728–734. [PubMed] [Google Scholar]
- 11.Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000 Aug 1;96(3):925–932. [PubMed] [Google Scholar]
- 12.Tuveson DA, Willis NA, Jacks T, et al. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001 Aug 16;20(36):5054–5058. doi: 10.1038/sj.onc.1204704. [DOI] [PubMed] [Google Scholar]
- 13.Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol. 2002 May;160(5):1567–1572. doi: 10.1016/S0002-9440(10)61103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007 Jan;31(1):113–120. doi: 10.1097/01.pas.0000213307.05811.f0. [DOI] [PubMed] [Google Scholar]
- 15.Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001 Nov 15;61(22):8118–8121. [PubMed] [Google Scholar]
- 16.Sommer G, Agosti V, Ehlers I, et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci USA. 2003 May 27;100(11):6706–6711. doi: 10.1073/pnas.1037763100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubin BP, Antonescu CR, Scott-Browne JP, et al. A knock-in mouse model of gastrointestinal stromal tumor harboring kit K641E. Cancer Res. 2005 Aug 1;65(15):6631–6639. doi: 10.1158/0008-5472.CAN-05-0891. [DOI] [PubMed] [Google Scholar]
- 18.Nakatani H, Kobayashi M, Jin T, et al. STI571 (Glivec) inhibits the interaction between c-KIT and heat shock protein 90 of the gastrointestinal stromal tumor cell line, GIST-T1. Cancer Sci. 2005 Feb;96(2):116–119. doi: 10.1111/j.1349-7006.2005.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarn C, Skorobogatko YV, Taguchi T, Eisenberg B, von Mehren M, Godwin AK. Therapeutic effect of imatinib in gastrointestinal stromal tumors: AKT signaling dependent and independent mechanisms. Cancer Res. 2006 May 15;66(10):5477–5486. doi: 10.1158/0008-5472.CAN-05-3906. [DOI] [PubMed] [Google Scholar]
- 20.Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006 Oct 10;24(29):4764–4774. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 21.Rossi F, Ehlers I, Agosti V, et al. Oncogenic Kit signaling and therapeutic intervention in a mouse model of gastrointestinal stromal tumor. Proc Natl Acad Sci USA. 2006 Aug 22;103(34):12843–12848. doi: 10.1073/pnas.0511076103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet. 2007 May 19;369(9574):1731–1741. doi: 10.1016/S0140-6736(07)60780-6. [DOI] [PubMed] [Google Scholar]
- 23.Chi P, Chen Y, Zhang L, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010 Oct 14;467(7317):849–853. doi: 10.1038/nature09409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003 Jan 31;299(5607):708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 25.Hirota S, Ohashi A, Nishida T, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003 Sep;125(3):660–667. doi: 10.1016/s0016-5085(03)01046-1. [DOI] [PubMed] [Google Scholar]
- 26.Kang HJ, Nam SW, Kim H, et al. Correlation of KIT and platelet-derived growth factor receptor alpha mutations with gene activation and expression profiles in gastrointestinal stromal tumors. Oncogene. 2005 Feb 3;24(6):1066–1074. doi: 10.1038/sj.onc.1208358. [DOI] [PubMed] [Google Scholar]
- 27.Lasota J, Dansonka-Mieszkowska A, Sobin LH, Miettinen M. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest. 2004 Jul;84(7):874–883. doi: 10.1038/labinvest.3700122. [DOI] [PubMed] [Google Scholar]
- 28.Agaram NP, Wong GC, Guo T, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008 Oct;47(10):853–859. doi: 10.1002/gcc.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hostein I, Faur N, Primois C, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol. 2010 Jan;133(1):141–148. doi: 10.1309/AJCPPCKGA2QGBJ1R. [DOI] [PubMed] [Google Scholar]
- 30.Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011 Jan 4;108(1):314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pantaleo MA, Astolfi A, Indio V, et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst. 2011 Jun 22;103(12):983–987. doi: 10.1093/jnci/djr130. [DOI] [PubMed] [Google Scholar]
- 32.Lasota J, Wang Z, Kim SY, Helman L, Miettinen M. Expression of the receptor for type i insulin-like growth factor (IGF1R) in gastrointestinal stromal tumors: an immunohistochemical study of 1078 cases with diagnostic and therapeutic implications. Am J Surg Pathol. 2013 Jan;37(1):114–119. doi: 10.1097/PAS.0b013e3182613c86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011 Dec;11(12):865–878. doi: 10.1038/nrc3143. [DOI] [PubMed] [Google Scholar]
- 34.Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001 Apr 5;344(14):1052–1056. doi: 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- 35.van Oosterom AT, Judson I, Verweij J, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001 Oct 27;358(9291):1421–1423. doi: 10.1016/s0140-6736(01)06535-7. [DOI] [PubMed] [Google Scholar]
- 36.Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. Journal of Clinical Oncology. 2008;26(4):626. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 37.Le Cesne A, Ray-Coquard I, Bui BN, et al. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. Lancet Oncol. 2010 Oct;11(10):942–949. doi: 10.1016/S1470-2045(10)70222-9. [DOI] [PubMed] [Google Scholar]
- 38.Benjamin RS, Choi H, Macapinlac HA, et al. We should desist using RECIST, at least in GIST. J Clin Oncol. 2007 May 1;25(13):1760–1764. doi: 10.1200/JCO.2006.07.3411. [DOI] [PubMed] [Google Scholar]
- 39.Van den Abbeele AD, Badawi RD. Use of positron emission tomography in oncology and its potential role to assess response to imatinib mesylate therapy in gastrointestinal stromal tumors (GISTs) Eur J Cancer. 2002 Sep;38(Suppl 5):S60–65. doi: 10.1016/s0959-8049(02)80604-9. [DOI] [PubMed] [Google Scholar]
- 40.Choi H. Critical issues in response evaluation on computed tomography: lessons from the gastrointestinal stromal tumor model. Curr Oncol Rep. 2005 Jul;7(4):307–311. doi: 10.1007/s11912-005-0055-4. [DOI] [PubMed] [Google Scholar]
- 41.Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007 May 1;25(13):1753–1759. doi: 10.1200/JCO.2006.07.3049. [DOI] [PubMed] [Google Scholar]
- 42.Antonescu CR. The GIST paradigm: lessons for other kinase-driven cancers. J Pathol. 2011 Jan;223(2):251–261. doi: 10.1002/path.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247–258. doi: 10.1146/annurev-med-043010-091813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeMatteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009 Mar 28;373(9669):1097–1104. doi: 10.1016/S0140-6736(09)60500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joensuu H, Eriksson M, Sundby Hall K, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012 Mar 28;307(12):1265–1272. doi: 10.1001/jama.2012.347. [DOI] [PubMed] [Google Scholar]
- 46.Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005 Oct 20;353(16):1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- 47.Mcauliffe JC, Hunt KK, Lazar AJF, et al. A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann Surg Oncol. 2009 Apr 1;16(4):910–919. doi: 10.1245/s10434-008-0177-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doyon C, Sideris L, Leblanc G, Leclerc YE, Boudreau D, Dube P. Prolonged Therapy with Imatinib Mesylate before Surgery for Advanced Gastrointestinal Stromal Tumor Results of a Phase II Trial. Int J Surg Oncol. 2012;2012:761576. doi: 10.1155/2012/761576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D, Zhang Q, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumors: long-term follow-up results of Radiation Therapy Oncology Group 0132. Ann Surg Oncol. 2012 Apr;19(4):1074–1080. doi: 10.1245/s10434-011-2190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeMatteo RP, Gold JS, Saran L, et al. Tumor mitotic rate, size, and location independently predict recurrence after resection of primary gastrointestinal stromal tumor (GIST) Cancer. 2008 Feb 1;112(3):608–615. doi: 10.1002/cncr.23199. [DOI] [PubMed] [Google Scholar]
- 51.Singer S, Rubin BP, Lux ML, et al. Prognostic value of KIT mutation type, mitotic activity, and histologic subtype in gastrointestinal stromal tumors. J Clin Oncol. 2002 Sep 15;20(18):3898–3905. doi: 10.1200/JCO.2002.03.095. [DOI] [PubMed] [Google Scholar]
- 52.Wardelmann E, Losen I, Hans V, et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer. 2003 Oct 10;106(6):887–895. doi: 10.1002/ijc.11323. [DOI] [PubMed] [Google Scholar]
- 53.Martin J, Poveda A, Llombart-Bosch A, et al. Deletions affecting codons 557–558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS) J Clin Oncol. 2005 Sep 1;23(25):6190–6198. doi: 10.1200/JCO.2005.19.554. [DOI] [PubMed] [Google Scholar]
- 54.Gold J, Gönen M, Gutiérrez A, Broto J. Development and validation of a prognostic nomogram for recurrence-free survival after. Lancet Oncology. 2009 Jan 1; doi: 10.1016/S1470-2045(09)70242-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rossi S, Miceli R, Messerini L, et al. Natural History of Imatinib-naive GISTs: A Retrospective Analysis of 929 Cases With Long-term Follow-up and Development of a Survival Nomogram Based on Mitotic Index and Size as Continuous Variables. The American journal of surgical pathology. 2011 Nov 1;35(11):1646–1656. doi: 10.1097/PAS.0b013e31822d63a7. [DOI] [PubMed] [Google Scholar]
- 56.Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005 Jun 1;11(11):4182–4190. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 57.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003 Dec 1;21(23):4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 58.Chen LL, Trent JC, Wu EF, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. 2004 Sep 1;64(17):5913–5919. doi: 10.1158/0008-5472.CAN-04-0085. [DOI] [PubMed] [Google Scholar]
- 59.Wakai T, Kanda T, Hirota S, Ohashi A, Shirai Y, Hatakeyama K. Late resistance to imatinib therapy in a metastatic gastrointestinal stromal tumour is associated with a second KIT mutation. Br J Cancer. 2004 Jun 1;90(11):2059–2061. doi: 10.1038/sj.bjc.6601819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005 Feb;128(2):270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 61.Grimpen F, Yip D, McArthur G, et al. Resistance to imatinib, low-grade FDG-avidity on PET, and acquired KIT exon 17 mutation in gastrointestinal stromal tumour. Lancet Oncol. 2005 Sep;6(9):724–727. doi: 10.1016/S1470-2045(05)70321-1. [DOI] [PubMed] [Google Scholar]
- 62.Wardelmann E, Thomas N, Merkelbach-Bruse S, et al. Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol. 2005 Apr;6(4):249–251. doi: 10.1016/S1470-2045(05)70097-8. [DOI] [PubMed] [Google Scholar]
- 63.Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006 Oct 10;24(29):4764–4774. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 64.Koyama T, Nimura H, Kobayashi K, et al. Recurrent gastrointestinal stromal tumor (GIST) of the stomach associated with a novel c-kit mutation after imatinib treatment. Gastric Cancer. 2006;9(3):235–239. doi: 10.1007/s10120-006-0368-5. [DOI] [PubMed] [Google Scholar]
- 65.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008 Sep;216(1):64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lim KH, Huang MJ, Chen LT, et al. Molecular analysis of secondary kinase mutations in imatinib-resistant gastrointestinal stromal tumors. Med Oncol. 2008;25(2):207–213. doi: 10.1007/s12032-007-9014-2. [DOI] [PubMed] [Google Scholar]
- 67.Nishida T, Kanda T, Nishitani A, et al. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci. 2008 Apr;99(4):799–804. doi: 10.1111/j.1349-7006.2008.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006 Mar 15;12(6):1743–1749. doi: 10.1158/1078-0432.CCR-05-1211. [DOI] [PubMed] [Google Scholar]
- 69.Loughrey MB, Waring PM, Dobrovic A, Demetri G, Kovalenko S, McArthur G. Polyclonal resistance in gastrointestinal stromal tumor treated with sequential kinase inhibitors. Clin Cancer Res. 2006 Oct 15;12(20 Pt 1):6205–6206. doi: 10.1158/1078-0432.CCR-06-1079. author reply 6206–6207. [DOI] [PubMed] [Google Scholar]
- 70.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006 Oct 14;368(9544):1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 71.Le Cesne A, Blay JY, Bui BN, et al. Phase II study of oral masitinib mesilate in imatinib-naive patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST) Eur J Cancer. 2010 May;46(8):1344–1351. doi: 10.1016/j.ejca.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 72.Benjamin RS, Schoffski P, Hartmann JT, et al. Efficacy and safety of motesanib, an oral inhibitor of VEGF, PDGF, and Kit receptors, in patients with imatinib-resistant gastrointestinal stromal tumors. Cancer Chemother Pharmacol. 2011 Jul;68(1):69–77. doi: 10.1007/s00280-010-1431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Joensuu H, De Braud F, Grignagni G, et al. Vatalanib for metastatic gastrointestinal stromal tumour (GIST) resistant to imatinib: final results of a phase II study. Br J Cancer. 2011 May 24;104(11):1686–1690. doi: 10.1038/bjc.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013 Jan 26;381(9863):295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DeMatteo RP, Maki RG, Singer S, Gonen M, Brennan MF, Antonescu CR. Results of tyrosine kinase inhibitor therapy followed by surgical resection for metastatic gastrointestinal stromal tumor. Ann Surg. 2007 Mar 1;245(3):347–352. doi: 10.1097/01.sla.0000236630.93587.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raut CP, Posner M, Desai J, et al. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol. 2006 May 20;24(15):2325–2331. doi: 10.1200/JCO.2005.05.3439. [DOI] [PubMed] [Google Scholar]
- 77.Gronchi A, Fiore M, Miselli F, et al. Surgery of residual disease following molecular-targeted therapy with imatinib mesylate in advanced/metastatic GIST. Ann Surg. 2007 Mar 1;245(3):341–346. doi: 10.1097/01.sla.0000242710.36384.1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012 Apr;12(4):237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ménard C, Blay J-Y, Borg C, et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res. 2009 Apr 15;69(8):3563–3569. doi: 10.1158/0008-5472.CAN-08-3807. [DOI] [PubMed] [Google Scholar]
- 80.Balachandran VP, Cavnar MJ, Zeng S, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011 Jan 1;17(9):1094–1100. doi: 10.1038/nm.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006 Sep 10;24(26):4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 82.Beadling C, Jacobson-Dunlop E, Hodi FS, et al. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008 Nov 1;14(21):6821–6828. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- 83.Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011 Jun 8;305(22):2327–2334. doi: 10.1001/jama.2011.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kemmer K, Corless CL, Fletcher JA, et al. KIT mutations are common in testicular seminomas. Am J Pathol. 2004 Jan;164(1):305–313. doi: 10.1016/S0002-9440(10)63120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pedersini R, Vattemi E, Mazzoleni G, Graiff C. Complete response after treatment with imatinib in pretreated disseminated testicular seminoma with overexpression of c-KIT. Lancet Oncol. 2007 Nov;8(11):1039–1040. doi: 10.1016/S1470-2045(07)70344-3. [DOI] [PubMed] [Google Scholar]
- 86.Johnson BE, Fischer T, Fischer B, et al. Phase II study of imatinib in patients with small cell lung cancer. Clin Cancer Res. 2003 Dec 1;9(16 Pt 1):5880–5887. [PubMed] [Google Scholar]
- 87.Boldrini L, Ursino S, Gisfredi S, et al. Expression and mutational status of c-kit in small-cell lung cancer: prognostic relevance. Clin Cancer Res. 2004 Jun 15;10(12 Pt 1):4101–4108. doi: 10.1158/1078-0432.CCR-03-0664. [DOI] [PubMed] [Google Scholar]
- 88.Tamborini E, Bonadiman L, Greco A, et al. Expression of ligand-activated KIT and platelet-derived growth factor receptor beta tyrosine kinase receptors in synovial sarcoma. Clin Cancer Res. 2004 Feb 1;10(3):938–943. doi: 10.1158/1078-0432.ccr-03-0059. [DOI] [PubMed] [Google Scholar]
- 89.Strobel P, Hartmann M, Jakob A, et al. Thymic carcinoma with overexpression of mutated KIT and the response to imatinib. N Engl J Med. 2004 Jun 17;350(25):2625–2626. doi: 10.1056/NEJM200406173502523. [DOI] [PubMed] [Google Scholar]
- 90.Simon MP, Pedeutour F, Sirvent N, et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet. 1997 Jan;15(1):95–98. doi: 10.1038/ng0197-95. [DOI] [PubMed] [Google Scholar]
- 91.Rutkowski P, Van Glabbeke M, Rankin CJ, et al. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol. 2010 Apr 1;28(10):1772–1779. doi: 10.1200/JCO.2009.25.7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003 Oct 22;290(16):2149–2158. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- 93.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004 May 20;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 94.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004 Sep 7;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004 Jun 4;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 96.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005 Jul 14;353(2):123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 97.Cohen EE, Davis DW, Karrison TG, et al. Erlotinib and bevacizumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck: a phase I/II study. Lancet Oncol. 2009 Mar;10(3):247–257. doi: 10.1016/S1470-2045(09)70002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pautier P, Joly F, Kerbrat P, et al. Phase II study of gefitinib in combination with paclitaxel (P) and carboplatin (C) as second-line therapy for ovarian, tubal or peritoneal adenocarcinoma (1839IL/0074) Gynecol Oncol. 2010 Feb;116(2):157–162. doi: 10.1016/j.ygyno.2009.10.076. [DOI] [PubMed] [Google Scholar]
- 99.Rini BI. Lapatinib therapy for patients with advanced renal cell carcinoma. Nat Clin Pract Oncol. 2008 Nov;5(11):626–627. doi: 10.1038/ncponc1220. [DOI] [PubMed] [Google Scholar]
- 100.Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004 Jul 22;351(4):337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 101.Peeters M, Price TJ, Cervantes A, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010 Nov 1;28(31):4706–4713. doi: 10.1200/JCO.2009.27.6055. [DOI] [PubMed] [Google Scholar]
- 102.Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010 Sep 30;467(7315):596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011 Jun 30;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001 Mar 15;344(11):783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 105.Oxnard GR, Binder A, Janne PA. New Targetable Oncogenes in Non-Small-Cell Lung Cancer. J Clin Oncol. 2013 Feb 11; doi: 10.1200/JCO.2012.42.9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005 Jul 14;353(2):172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 107.Kurzrock R, Sherman SI, Ball DW, et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol. 2011 Jul 1;29(19):2660–2666. doi: 10.1200/JCO.2010.32.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wells SA, Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012 Jan 10;30(2):134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010 May 10;28(14):2323–2330. doi: 10.1200/JCO.2009.25.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sawaki A, Nishida T, Doi T, et al. Phase 2 study of nilotinib as third-line therapy for patients with gastrointestinal stromal tumor. Cancer. 2011 Mar 31; doi: 10.1002/cncr.26120. [DOI] [PubMed] [Google Scholar]
- 111.Cauchi C, Somaiah N, Engstrom PF, et al. Evaluation of nilotinib in advanced GIST previously treated with imatinib and sunitinib. Cancer Chemother Pharmacol. 2012 Apr;69(4):977–982. doi: 10.1007/s00280-011-1785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reichardt P, Blay JY, Gelderblom H, et al. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann Oncol. 2012 Jul;23(7):1680–1687. doi: 10.1093/annonc/mdr598. [DOI] [PubMed] [Google Scholar]
- 113.Demetri GD, Lo Russo P, MacPherson IR, et al. Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res. 2009 Oct 1;15(19):6232–6240. doi: 10.1158/1078-0432.CCR-09-0224. [DOI] [PubMed] [Google Scholar]
- 114.Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004 Oct 1;64(19):7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 115.Italiano A, Cioffi A, Coco P, et al. Patterns of care, prognosis, and survival in patients with metastatic gastrointestinal stromal tumors (GIST) refractory to first-line imatinib and second-line sunitinib. Ann Surg Oncol. 2012 May;19(5):1551–1559. doi: 10.1245/s10434-011-2120-6. [DOI] [PubMed] [Google Scholar]
- 116.Park SH, Ryu MH, Ryoo BY, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs. 2012 Dec;30(6):2377–2383. doi: 10.1007/s10637-012-9795-9. [DOI] [PubMed] [Google Scholar]
- 117.George S, Wang Q, Heinrich MC, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012 Jul 1;30(19):2401–2407. doi: 10.1200/JCO.2011.39.9394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Soria JC, Massard C, Magne N, et al. Phase 1 dose-escalation study of oral tyrosine kinase inhibitor masitinib in advanced and/or metastatic solid cancers. Eur J Cancer. 2009 Sep;45(13):2333–2341. doi: 10.1016/j.ejca.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 119.Joensuu H, De Braud F, Coco P, et al. Phase II, open-label study of PTK787/ZK222584 for the treatment of metastatic gastrointestinal stromal tumors resistant to imatinib mesylate. Ann Oncol. 2008 Jan;19(1):173–177. doi: 10.1093/annonc/mdm419. [DOI] [PubMed] [Google Scholar]
- 120.Heinrich MC, Griffith D, McKinley A, et al. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res. 2012 Aug 15;18(16):4375–4384. doi: 10.1158/1078-0432.CCR-12-0625. [DOI] [PubMed] [Google Scholar]