

Abstract
Subpopulations of dopaminergic (DA) neurons within the substantia nigra pars compacta (SNpc) display a differential vulnerability to loss in Parkinson’s disease (PD); however, it is not clear why these subsets are preferentially selected in PD-associated neurodegeneration. In rodent SNpc, DA neurons can be divided into two subpopulations based on the expression of aldehyde dehydrogenase 1 (ALDH1A1). Here, we have shown that, in α-synuclein transgenic mice, a murine model of PD-related disease, DA neurodegeneration occurs mainly in a dorsomedial ALDH1A1-negative subpopulation that is also prone to cytotoxic aggregation of α-synuclein. Notably, the topographic ALDH1A1 pattern observed in α-synuclein transgenic mice was conserved in human SNpc. Postmortem evaluation of brains of patients with PD revealed a severe reduction of ALDH1A1 expression and neurodegeneration in the ventral ALDH1A1-positive DA subpopulations. ALDH1A1 expression was also suppressed in α-synuclein transgenic mice. Deletion of Aldh1a1 exacerbated α-synuclein–mediated DA neurodegeneration and α-synuclein aggregation, whereas Aldh1a1-null and control DA neurons were comparably susceptible to 1-methyl-4-phenylpyridinium–, glutamate-, or camptothecin-induced cell death. ALDH1A1 overexpression appeared to preferentially protect against α-synuclein–mediated DA neurodegeneration but did not rescue α-synuclein–induced loss of cortical neurons. Together, our findings suggest that ALDH1A1 protects subpopulations of SNpc DA neurons by preventing the accumulation of dopamine aldehyde intermediates and formation of cytotoxic α-synuclein oligomers.
Introduction
Parkinson’s disease (PD), the most common degenerative movement disorder, is pathologically characterized by a preferential loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and the presence of intracytoplasmic inclusions, named Lewy bodies and Lewy neurites, throughout the central and peripheral nervous systems (1). Interestingly, SNpc DA neurons exhibit regionally selective susceptibility in PD (2–4). This differential vulnerability of SNpc DA neurons in PD indicates the existence of different subpopulations of DA neurons in the SNpc, which may therefore possess the molecular clues critical for understanding the cause of selective SNpc DA neurodegeneration and designing new therapeutics.
Previous genetic and pathological studies establish a prominent role of α-synuclein in the pathogenesis of PD (5–9). When the PD-related α-synuclein A53T (A53T) missense mutation is introduced into the midbrain DA neurons, the resulting mutant mice develop profound motor disabilities and robust SNpc DA neurodegeneration (10). However, it is unclear whether the loss of DA neurons occurs randomly in the SNpc of mutant mice or shows any regional preference, as observed in the PD cases (2–4). Notably, the rodent SNpc consists of two subpopulations of DA neurons based on the expression of aldehyde dehydrogenase 1 (ALDH1A1) (11). ALDH1A1 mediates the oxidation of reactive dopamine-3,4-dihydroxyphenylacetaldehyde (DOPAL) into 3,4-dihydroxyphenylacetic acid (DOPAC), which is then degraded to form homovanillic acid in DA neurons (12). A lack of ALDH1A1 dehydrogenase activity may lead to accumulation of reactive DOPAL that promotes cytotoxic polymerization of α-synuclein (13). Higher exposure to fungicide benomyl, an inhibitor of aldehyde dehydrogenase (ALDH) via its bioactivated thiocarbamate sulfoxide metabolite, increases the risk of PD (14). However, Aldh1a1 knockout (Aldh1a1–/–) mice show no overt motor behavioral phenotypes and develop no apparent SNpc DA neurogeneration (15, 16). By contrast, Aldh1a1 and Aldh2 double-knockout mice develop age-related SNpc DA neuron loss, suggesting complementary functions of ALDH family proteins in maintaining the survival of SNpc DA neurons (16). In the meantime, whether ALDH1A1-positive and -negative SNpc DA neurons show differential vulnerability to PD-related α-synuclein–mediated cytotoxicity is unknown.
In this study, we found that ALDH1A1-positive SNpc DA neurons were more resistant to α-synuclein–mediated neurodegeneration compared with the ALDH1A1-negative SNpc DA neurons in α-synuclein transgenic mice (10). ALDH1A1 may exert its protective function via suppressing the accumulation of cytotoxic α-synuclein aggregates, likely resulting from the DOPAL-mediated covalent modifications. We further revealed a conserved topographic distribution of ALDH1A1-positive SNpc DA neurons in the human SNpc and observed substantial reduction of ALDH1A1 expression and severe loss of ALDH1A1-positive SNpc DA neurons in the postmortem PD brains. Supporting a protective role of ALDH1A1 against DA neuron loss, genetic inhibition of Aldh1a1 enhanced the formation of α-synuclein aggregates and exacerbated the overall degeneration of SNpc DA neurons, whereas overexpression of ALDH1A1 was neuron protective. Finally, we tested the specificity of ALDH1A1 in α-synuclein–induced midbrain DA neuron loss. We found that Aldh1a1+/+ and Aldh1a1–/– midbrain DA neurons showed no difference in 1-methyl-4-phenylpyridinium+– (MPP+–), glutamate-, and camptothecin-mediated cell death, whereas overexpression of ALDH1A1 did not rescue the α-synuclein–induced loss of cortical neurons. Our findings demonstrate that ALDH1A1 is an important molecular determinant responsible for the differential susceptibility of SNpc DA neurons in PD and further support the notion that the reactive aldehyde byproducts derived from abnormal cytosolic dopamine oxidation may contribute to the DA neuron loss by facilitating the formation of cytotoxic α-synuclein oligomers (17).
Results
ALDH1A1-negative DA neurons in the dorsal SNpc are preferentially degenerated in A53T transgenic mice.
As shown previously (10), a progressive loss of tyrosine hydroxylase–positive (TH-positive) DA neurons was observed in the SNpc of 18-month-old A53T transgenic mice as compared with that in age-matched nontransgenic (nTg) littermate controls (Figure 1, A and B). To determine whether the loss of DA neurons occurs randomly throughout SNpc or shows any regional preference, we calculated the total space occupied by the SNpc DA neurons and measured the shortest distance between neighboring neurons. If neurons died randomly in the SNpc, we expected to observe a significant increase of distance between remaining neurons, while the volume of SNpc might not change substantially. In contrast, we observed more than 40% reduction of SNpc volume but only a mild increase of neuron spacing in the mutant mice (Figure 1, C and D). These data suggest that DA neurons may not degenerate randomly in the SNpc of A53T transgenic mice, implying that regional selectivity of SNpc DA neuron loss in the mutant mice may exist.
Figure 1. ALDH1A1-negative nigrostriatal DA neurons in the dorsal SNpc are preferentially degenerated in A53T transgenic mice.

(A) TH staining (brown) of midbrain coronal sections (top, bregma –2.90, bottom, bregma –3.20) of 18-month-old A53T and littermate nTg female mice. (B) Number of TH-positive neurons remaining in SNpc of 1-month-old (n = 3 per genotype) and 18-month-old (n = 5 per genotype) A53T and littermate nTg female mice. (C) Volume of SNpc of 18-month-old A53T and littermate nTg female mice (n = 5 per genotype). (D) Minimal distance between neighboring DA neurons in SNpc of 18-month-old A53T and littermate nTg female mice. 1,336 and 1,060 nigrostriatal DA neurons from 3 pairs of A53T and nTg mice were analyzed, respectively. (E) 3D reconstruction of TH-positive neurons distributed in SNpc, VTA, and retrorubral field (RRF) of 18-month-old A53T and control nTg female mice. Arrows point to DM tier of DA neurons in SNpc of nTg and A53T mice. D, dorsal; L, lateral; Ro, rostral. (F) Representative images show TH and ALDH1A1 staining in midbrain coronal sections (bregma –2.92) of 1- and 18-month-old A53T and 18-month-old nTg female mice. Arrowheads indicate DM ALDH1A1-negative DA neurons. Arrowheads point to the DM tier of SNpc. (G) Representative images show ALDH1A1 and TH staining in DA neurons of 1-month-old A53T nTg mice. (H) Numbers of TH-positive/ALDH1A1-positive DA neurons and (I) TH-positive/ALDH1A1-negative DA neurons remaining in SNpc of 1-month-old (n = 3 per genotype) and 18-month-old (n = 5 per genotype) A53T and nTg female mice. (J–L) Numbers of total (J) TH-positive, (K) TH-positive/ALDH1A1-positive, and (L) TH-positive/ALDH1A1-negative DA neurons remaining in VTA of 1-month-old (n = 3 per genotype) and 18-month-old (n = 5 per genotype) A53T and nTg female mice. Scale bar: 100 μm (A and F); 20 μm (G). (A and F) The dashed line marks the boundary between SNpc and VTA. *P < 0.05, **P < 0.01, ***P < 0.001.
To identify the regions that show more substantial loss of DA neurons in the SNpc, we generated 3D reconstructions of DA neurons in the midbrains of 18-month-old A53T and nTg mice. We found the loss of DA neurons mainly occurred in the dorsomedial (DM) tier of SNpc in A53T mice (arrows, Figure 1E and Supplemental Videos 1 and 2; supplemental material available online with this article; doi:10.1172/JCI72176DS1). This observation suggests that DA neurons from the DM and ventrolateral (VL) tiers of SNpc may exhibit differential susceptibility to α-synuclein–induced cytotoxicity.
We then tried to identify the molecular determinants in the VL SNpc DA neurons that are responsible for the increased resistance to α-synuclein–mediated cytotoxicity. A previous study shows that ALDH1A1 is selectively expressed by a subset of DA neurons located predominantly in the VL tier of rodent SNpc (11). We therefore examined the distribution of both TH- and ALDH1A1-positive (TH+/ALDH1A1+) DA neurons in the SNpc of A53T and control mice. Consistent with the previous finding (11), these double-positive neurons were located primarily in the VL tier of SNpc (Figure 1F). At the subcellular level, ALDH1A1 showed a similar cytosolic distribution pattern as TH in DA neurons (Figure 1G). There was a modest but marked loss of TH+/ALDH1A1+ DA neurons in the SNpc of 1-month-old A53T mice; however, no additional loss of these neurons was found in 18-month-old A53T mice (Figure 1, F and H). By contrast, the TH-positive but ALDH1A1-negative (TH+/ALDH1A1–) DA neurons, located mainly in the DM tier of SNpc, displayed remarkably progressive degeneration in the mutant mice from 1 to 18 months of age (arrowheads, Figure 1, F and I). The ALDH1A1-positive and -negative DA neurons also maintained a conserved topographic distribution in the ventral tegmental area (VTA) of midbrain (Supplemental Figure 1). In addition, a similarly selective loss of ALDH1A1-negative DA neurons was found in the DM tier of VTA in 18-month-old A53T mice (Figure 1, J–L). These observations demonstrate the differential susceptibility of ALDH1A1-positive and -negative SNpc DA neurons in response to α-synuclein–mediated genetic insults during the aging process, in which the ALDH1A1-positive DA neurons in the VL tier of SNpc are more resistant to α-synuclein–induced neurodegeneration.
ALDH1A1-negative SNpc DA neurons contain more aggregated α-synuclein.
ALDH1A1 regulates the metabolism of dopamine through oxidation of its biogenic aldehyde intermediate, DOPAL (12). DOPAL is a potent cross-linker of α-synuclein, which promotes the polymerization of α-synuclein (12, 13, 18). Consistently, DOPAL increased the oligomerization of recombinant α-synuclein protein in a cell-free assay (Figure 2A). We speculate that ALDH1A1-negative DA neurons may contain higher levels of DOPAL than the ALDH1A1-positive ones, which may lead to a greater accumulation of cytotoxic α-synuclein aggregates in the ALDH1A1-negative neurons. In support of this notion, we found substantially more proteinase K–resistant (PK-resistant) α-synuclein aggregates in the soma of ALDH1A1-negative SNpc neurons, although the levels of total transgenic α-synuclein were comparable between ALDH1A1-positive and -negative neurons (Figure 2, B–D). The levels of transgenic α-synuclein and ALDH1A1 expression were also plotted for each neuron under nontreated and PK-treated conditions (Figure 2E). There is no correlation between the levels of transgenic α-synuclein and ALDH1A1 expression in nontreated samples (Spearman correlation test, P = 0.6083). By contrast, a reverse correlation between the levels of aggregated α-synuclein and ALDH1A1 was found in PK-treated samples (P < 0.0001).
Figure 2. ALDH1A1-negative nigrostriatal DA neurons contain more PK-resistant α-synuclein aggregates in soma and axon fibers.

(A) Western blot shows α-synuclein aggregation induced by application of DOPAL (1 mM) for various time periods as well as the level of monomeric α-synuclein with shorter exposure. HMW, high molecular weight. (B) Human α-synuclein (hα-syn) (green) and ALDH1A1 (red) costaining in the nontreated or PK-treated SNpc coronal sections of 12-month-old female A53T mice. Topro3 staining (blue) was used to mark the nucleus. The asterisks indicate an ALDH1A1-negative nigrostriatal DA neuron with substantial deposition of α-synuclein aggregates. Arrows mark the ALDH1A1-positive nigrostriatal DA neurons with less accumulation of α-synuclein aggregates. (C and D) The average intensity of α-synuclein signals in the soma of (C) nontreated and (D) PK-treated ALDH1A1-positive (+) and negative (–) nigrostriatal DA neurons of 12-month-old A53T female mice (n ≥ 3 animals per genotype and ≥40 neurons per animal). ***P < 0.001. (E) Dot plot correlates the level of α-synuclein (y axis) and ALDH1A1 (x axis) expression in 115 nontreated and 89 PK-treated neurons in SNpc sections of 12-month-old A53T female mice. (F) Human α-synuclein (green) and ALDH1A1 (red) costaining in the nontreated or PK-treated DL striatum sections of 12-month-old A53T female mice. The numbers mark the cross points of white lines with nigrostriatal DA axon fibers that correlate with the peaks of histograms of α-synuclein (green) and ALDH1A1 (red) signals at individual fibers. Scale bar: 10 μm (B and F).
We next examined the level of PK-resistant α-synuclein aggregates in the axons of SNpc DA neurons that project to the dorsal striatum. ALDH1A1 and transgenic α-synuclein were extensively colocalized in the midbrain DA axons and axon terminals at the striatum (Figure 2F), confirming a highly selective expression of Aldh1a1 among Aldh family genes in the midbrain DA neurons (Supplemental Figure 2). The total transgenic human α-synuclein was comparable in both ALDH1A1-positive and -negative axon fibers in the nontreated sections, whereas PK-resistant α-synuclein aggregates appeared only in the ALDH1A1-negative fibers (Figure 2F). PK-resistant α-synuclein aggregates have been shown to generate higher levels of oxidative stress and are more toxic to cells (19–21). Therefore, these results suggest that ALDH1A1 may improve the survival of DA neurons via attenuating the formation of PK-resistant α-synuclein aggregates.
ALDH1A1-positive DA neurons exhibit a conserved topographic distribution in human SNpc.
To investigate whether ALDH1A1 plays a similar role in defining two anatomically distinct subpopulations of DA neurons in the human SNpc, we stained normal human SNpc sections with antibodies against ALDH1A1. The SNpc DA neurons were identified by the presence of brown-colored neuromelanin (NM) in the soma, while the staining of ALDH1A1 appeared as a purple color present in both soma and neurites (Figure 3A). A clear clustering of ALDH1A1-positive and -negative DA neurons was found in the control human SNpc (Figure 3A). The existence of distinguished subpopulations of ALDH1A1-positive and -negative DA neurons in the control human SNpc was further confirmed by immunofluorescence staining with antibodies against ALDH1A1 and TH (Figure 3B). To reveal the topographic distribution of ALDH1A1-positive and -negative DA neurons in the human SNpc, we stained a series of transverse sections evenly sampled from the entire SNpc of 10 sporadic PD and 9 age- and gender-matched control cases (Supplemental Table 1) and generated 3D maps of these two populations of DA neurons along the dorsal-ventral and medial-lateral axis of each SNpc (Figure 3C and Supplemental Figure 3). In all control cases, ALDH1A1-positive DA neurons were concentrated in the ventral tier of SNpc, whereas ALDH1A1-negative DA neurons were primarily located in the dorsal tier (Figure 3C, Table 1, and Supplemental Figure 3A). Therefore, ALDH1A1-positive DA neurons display a topographically conserved distribution pattern in both rodent and human SNpc.
Figure 3. ALDH1A1-positive DA neurons exhibit a conserved topographic distribution in human SNpc and show reduction of ALDH1A1 expression in PD.

(A) Representative bright-field image shows ALDH1A1 staining (purple) in SNpc transverse sections of control human brains. Nigrostriatal DA neurons (dark brown) were marked by the presence of NM in the soma. Arrow points to a cluster of ALDH1A1-negative DA neurons in the dorsal tier of SNpc. Arrowhead indicates a group of ALDH1A1-positive DA neurons in the ventral tier of SNpc. (B) Representative fluorescent images show TH and ALDH1A1 costaining in the transverse section of SNpc in control human brains. (C) 3D reconstruction of DA neurons in human SNpc shows the distribution of ALDH1A1 and NM double-positive (purple) as well as ALDH1A1-negative/NM-positive (brown) neurons remaining in the SNpc of control (Ctrl) and PD human brains. (D) Sketch of transverse section of human SNpc. RN, red nucleus. (E–I) Remaining DA neurons in the (E) SNpc, (F) dorsal, (G) PL, (H) VM, and (I) VL parts of control (n = 9) and PD (n = 10) brains. Numbers over the PD bars show the percentage of DA neuron loss compared with the controls. (J and K) Percentages of ALDH1A1+/NM+ and ALDH1A1–/NM+ DA neurons in SNpc, dorsal, PL, VM, and VL parts of (J) control human (n = 9) and (K) PD cases (n = 10). (L) Percentages of ALDH1A1+/NM+ and ALDH1A1–/NM+ DA neurons in SNpc, dorsal, PL, VM, and VL parts of control (n = 9) and PD cases (n = 10). (M–P) Total number and percentage of ALDH1A1+ and ALDH1A1–/NM+ neurons in the (M and N) SNpc and (O and P) VL subregion of control, mild, moderate, and severe PD cases. Scale bar: 70 μm (A); 100 μm (B). *P < 0.05, **P < 0.01, ***P < 0.001.
Table 1.
List of the number and percentage of remaining DA neurons in the SNpc and its subdivisions in control and PD cases
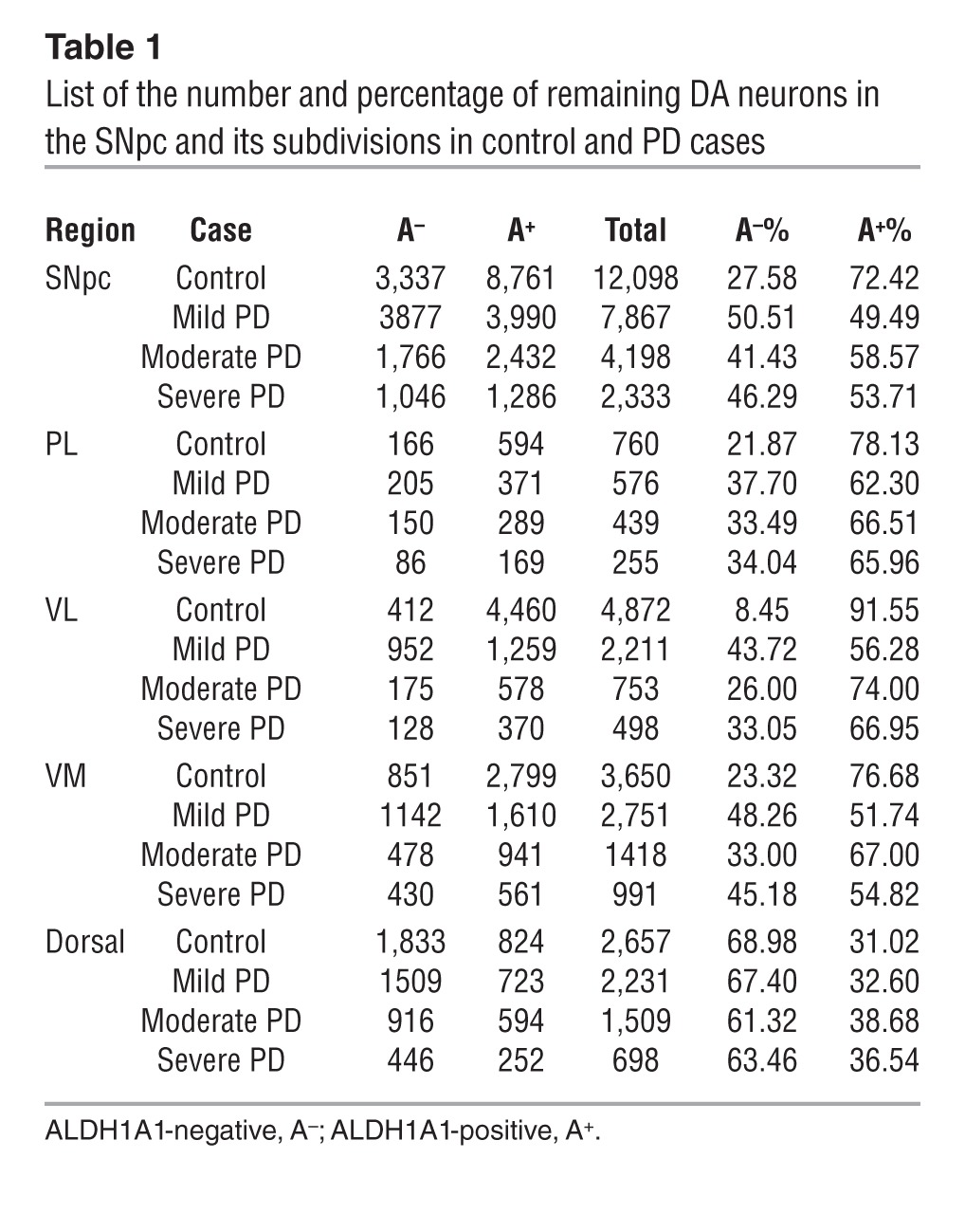
Human SNpc can be divided into 6 morphometric regions: in the dorsal tier, the medial (DM) part, lateral (DL) part, and pars lateralis (PL); in the ventral tier, the medial (VM) part, intermediate (VI) part, and lateral (VL) part (3). Since PD-related neurodegeneration is comparable between DM and DL in the dorsal tier and between VI and VL in the ventral tier (3), we merged DM and DL as dorsal part and VI and VL as VL part in this study (Figure 3D). We calculated the percentage of DA neurons that remained in each SNpc region of control and PD cases (Figure 3, E–I, and Table 1). Our results indicate that, in normal human SNpc, the dorsal part had more ALDH1A1-negative DA neurons (69%), whereas the VL part contained the majority of ALDH1A1-positive neurons (92%, Table 1 and Figure 3, J and L).
Reduction of ALDH1A1 expression in the ventral ALDH1A1-positive subpopulations of nigrostriatal DA neurons in PD.
There was an average of 66% loss of DA neurons in the SNpc of PD brains compared with that in age-matched control cases (Figure 3E). We further estimated the severity of neurodegeneration in each SNpc subdivision. Consistent with the early findings (3), the dorsal, PL, and VM parts of SNpc had 50%–60% loss of DA neurons in PD brains, whereas the VL region showed greater than 80% of loss compared with that in the control brains (Figure 3, F–I). When examining the 3D distribution of ALDH1A1-positive and -negative DA neurons in the SNpc of PD brains, the segregation of these two subpopulations of DA neurons became less obvious; ALDH1A1-negative DA neurons seemed to replace the ALDH1A1-positive ones in the ventral tier of SNpc, especially in the mild PD cases (Figure 3C, Table 1, and Supplemental Figure 3B). Moreover, substantial reduction of ALDH1A1 staining was observed in the soma and neurites of DA neurons located in the PL, VM, and VL regions (Supplemental Figure 4). An overall increase of the percentage of ALDH1A1-negative neurons and a decrease of the percentage of ALDH1A1-positive of DA neurons were observed in the SNpc of PD cases as compared with that in the controls (P < 0.01, Figure 3, J–L). We then calculated the percentage of ALDH1A1-positive and -negative DA neurons in each SNpc region of PD cases. The ratio of ALDH1A1-positive and ALDH1A1-negative DA neurons in the dorsal region was comparable between controls and PD cases (Figure 3, J–L). In contrast, a significant increase of the percentage of ALDH1A1-negative DA neurons and decrease of the percentage of ALDH1A1-positive DA neurons was found in the VL part of PD SNpc as compared with that in the controls (P < 0.05, Figure 3, J–L). We further examined the number of remaining DA neurons in the SNpc and VL part of control and PD cases with mild, moderate, and severe depigmentation (Figure 3, M–P). Interestingly, in the mild PD cases, the total numbers of ALDH1A1-negative neurons in the SNpc and VL part were higher than those in the control cases (Table 1 and Figure 3, M and O). These extra ALDH1A1-negative neurons may come from ALDH1A1-positive neurons that lost a substantial amount of ALDH1A1 expression (Supplemental Figure 4). Together, these results suggest that, in PD cases, a reduction of ALDH1A1 expression may occur prior to the loss of DA neurons in the PL, VM, and VL regions of SNpc, which further supports the notion that inhibition of ALDH1A1 may render DA neurons more susceptible to degeneration.
Overexpression of α-synuclein suppresses ALDH1A1 expression in the midbrain DA neurons of A53T transgenic mice.
Reduction of ALDH1A1 mRNA expression has also been reported previously in postmortem PD brains (22). We then examined whether the expression of Aldh1a1 mRNA and protein was affected in the SNpc DA neurons of A53T transgenic mice. We used striatal tissues for protein expression analysis instead of midbrain tissues, because the dissection of striatal tissues is more consistent between different animals and the striatal ALDH1A1 and transgenic α-synuclein proteins were derived mainly from the axons and axon terminals of midbrain DA neurons projected to the striatum (ref. 10 and Figure 2E). We collected the striatum from 2-month-old nTg and A53T mice and checked the expression of ALDH1A1, α-synuclein, and dopamine- and cAMP-regulated neuronal phosphoprotein 32 (DARPP32). DARPP32 serves as a maker for the striatal tissues (23). We found that the expression of ALDH1A1 protein was significantly decreased in the striatum of A53T mice compared with that in the control mice (Figure 4, A and B). Therefore, overexpression of α-synuclein in DA neurons may suppress the expression of ALDH1A1 protein in these neurons. We further tested this hypothesis directly by quantifying the expression of Aldh1a1 mRNA in the SNpc DA neurons isolated by laser capture microdissection (LCM) from 2-week-old control and A53T mice. We observed a substantial reduction of Aldh1a1 mRNA expression in the SNpc DA neurons of A53T mice compared with that in the controls (Figure 4C). A similar decrease of Aldh1a1 mRNA expression was also observed in the midbrain tissues of 2-month-old A53T mice (P = 0.0289). We next examined the expression of ALDH1A1 in the striatum of 1-, 6-, and 18-month-old controls and α-synuclein transgenic mice (Figure 4, D and E). Both genotypes (2-way ANOVA, interaction: F1,18 = 130.10, P < 0.0001) and age (interaction: F2,18 = 25.17, P < 0.0001) affect the expression of ALDH1A1; the expression level of ALDH1A1 was substantially decreased in 18-month-old mice compared with that in the 1-month-old mice in both genotypes (Figure 4E). Together, these data suggest that both aging and pathogenic α-synuclein overexpression suppress the expression of ALDH1A1 in midbrain DA neurons.
Figure 4. ALDH1A1 expression is reduced in A53T transgenic mice.

(A) Western blot shows expression of ALDH1A1 protein in the striatum of 3 independent pairs of 2-month-old A53T transgenic and littermate nTg female mice. (B) The relative expression levels of ALDH1A1 in the striatal homogenate of 2-month-old nTg (n = 3) and A53T (n = 3) female mice. (C) Aldh1a1 mRNA expression in nigrostriatal DA neurons isolated from 2-week-old control and littermate A53T transgenic mice (n > 1,000 cells per genotype). β-Actin was used to normalize the gene expression from different samples. (D) Western blot analysis shows ALDH1A1 expression in the striatum of 1-, 6-, and 18-month-old nTg control and A53T female mice. (E) The level of ALDH1A1 expression. Four animals were used per genotype per age group. One-way ANOVA plus Tukey post test and 2-way ANOVA plus Bonferroni post test were used for data analyses. *P < 0.05, **P < 0.01, ***P < 0.001.
Genetic deletion of Aldh1a1 exacerbates SNpc DA neurodegeneration and motor behavioral impairments in A53T transgenic mice.
To further establish the function of ALDH1A1 in protecting against SNpc DA neuron loss, we crossbred A53T transgenic mice with Aldh1a1–/– mice to generate A53T transgenic mice in both the Aldh1a1-null (A53T/Aldh1a1–/–) and wild-type (A53T/Aldh1a1+/+) background. Aldh1a1–/– mice did not exhibit any loss of SNpc DA neurons at 18 months of age (refs. 16, 24, and Figure 5, A and B). By contrast, A53T/Aldh1a1–/– mice developed more severe loss of SNpc DA neurons compared with control A53T/Aldh1a1+/+ mice (Figure 5, A and B). Similarly, Aldh1a1–/– mice showed no obvious motor behavioral phenotypes in both open-field and rotarod tests (Figure 5, C–E). However, A53T/Aldh1a1–/– mice developed further motor impairments in both of these two tests compared with A53T/Aldh1a1+/+ mice (Figure 5, C–E). Therefore, these in vivo studies provide additional evidence for and support a protective function of ALDH1A1 against α-synuclein–induced SNpc DA neurodegeneration.
Figure 5. Genetic ablation of ALDH1A1 exacerbates the SNpc DA neurodegeneration and motor behavioral impairments in A53T transgenic mice.

(A) Representative images show ALDH1A1 and TH staining in the midbrain coronal sections of 18-month-old Aldh1a1+/+, Aldh1a1–/–, A53T/Aldh1a1+/+, and A53T/Aldh1a1–/– mice. Scale bar: 100 μm. (B) Numbers of all TH-positive neurons remaining in the SNpc of 18-month-old Aldh1a1+/+, Aldh1a1–/–, A53T/Aldh1a1+/+, and A53T/Aldh1a1–/– mice (n = 5 per genotype, 2 males and 3 females per genotype). (C and D) The 6-month-old Aldh1a1+/+ (n = 28, 20 males and 8 females), Aldh1a1–/– (n = 16, 11 males and 5 females), A53T/Aldh1a1+/+ (n = 31, 22 males and 9 females), and A53T/Aldh1a1–/– (n = 13, 8 males and 5 females) mice were subjected to open-field tests. The (C) ambulatory and (D) rearing activities were quantified. (E) The latency to fall was quantified by the rotarod test in 6-month-old Aldh1a1+/+ (n = 23, 18 males and 5 females), Aldh1a1–/– (n = 13, 8 males and 5 females), A53T/Aldh1a1+/+ (n = 22, 15 males and 7 females), and A53T/Aldh1a1–/– (n = 12, 8 males and 4 females) mice. *P < 0.05, **P < 0.01.
Genetic inhibition of Aldh1a1 promotes α-synuclein aggregation in A53T transgenic mice.
We next examined the impact of Aldh1a1 deficiency on the aggregation of α-synuclein in the axonal fibers of SNpc DA neurons from A53T/Aldh1a1–/– mice. The overall expression level of transgenic human α-synuclein seemed modestly decreased in A53T/Aldh1a1–/– mice, whereas the accumulation of PK-resistant α-synuclein aggregates was substantially increased in the A53T/Aldh1a1–/– mice as compared with that in A53T/Aldh1a1+/+ mice (Figure 6, A–C). Western blot analysis further revealed an increase of high molecular weight α-synuclein–positive species in the striatum of A53T/Aldh1a1–/– mice compared with that in A53T/Aldh1a1+/+ mice (Figure 6, D and E). Moreover, protein pull-down experiments with aminophenylboronic acid (APBA) resin, which isolates proteins covalently modified by DOPAL (25), showed a marked increase of α-synuclein monomers and polymers modified by DOPAL (Figure 6, F and G). Taken collectively, these data further suggest that ALDH1A1 plays a protective function against the formation of PK-resistant α-synuclein aggregates through removal of DOPAL from DA neurons.
Figure 6. Genetic inhibition of Aldh1a1 promotes α-synuclein aggregation in A53T transgenic mice.

(A) Human α-synuclein and ALDH1A1 costaining in the nontreated or PK-treated DL striatum sections of 18-month-old A53T/Aldh1a1+/+ and A53T/Aldh1a1–/– mice. Scale bar: 10 μm. (B and C) The average intensity of α-synuclein signals in the axon fibers of (B) nontreated and (C) PK-treated DL striatum sections of 18-month-old A53T/Aldh1a1+/+ and A53T/Aldh1a1–/– female mice (n = 3 animals per genotype). (D) Western blot shows the formation of α-synuclein–positive HMW bands in the striatum homogenate of 6-month-old A53T/Aldh1a1+/+ and A53T/Aldh1a1–/– mice. (E) The relative intensity of HMW α-synuclein bands (n = 3 female mice per genotype). (F) A Western blot of α-synuclein from the APBA pull-down experiment shows the enrichment of DOPAL-modified α-synuclein in 6-month-old A53T/Aldh1a1+/+ and A53T/Aldh1a1–/– mice. (G) The graph indicates an increase of accumulation of HMW DOPAL-modified α-synuclein in brains of A53T/Aldh1a1–/– mice compared with littermate A53T/Aldh1a1+/+ mice (n = 3 pairs females). *P < 0.05, ***P < 0.001.
Overexpression of ALDH1A1 protects midbrain DA neurons against α-synuclein–mediated cytotoxicity.
To directly test the protective function of ALDH1A1 in DA neurodegeneration, we transduced cultured midbrain DA neurons from newborn A53T and control pups with adeno-associated viruses (AAV) carrying human ALDH1A1 or control GFP. Overexpression of transgenic α-synuclein triggered the accumulation of cleaved caspase-3, an active form of caspase-3, in TH-positive neurons (Figure 7A). Around 30% to 40% loss of TH-positive neurons was observed from the A53T cultures as compared with that from the littermate controls, whereas administration of doxycycline (DOX) that suppresses the expression of α-synuclein transgene prevented the DA neuron loss (ref. 10 and Figure 7, B and C). We found that overexpression of ALDH1A1 significantly increased the survival of DA neurons derived from both A53T and control pups (Figure 7, B and C). Moreover, the presence of extra ALDH1A1 effectively rescued the loss of DA neurons derived from A53T transgenic mice (Figure 7C). In addition, an ALDH1A1 activator, 6-methyl-2-phenylazo-3-pyridinol (SIB1757) (26), also significantly improved the survival of TH-positive neurons in A53T cultures (Figure 7D). Together, these observations provide direct support for a protective function of ALDH1A1 in maintenance of the survival of midbrain DA neurons against α-synuclein–induced cytotoxicity.
Figure 7. Overexpression of ALDH1A1 protects midbrain DA neurons against α-synuclein–mediated cytotoxicity.

(A) Representative images show cleaved caspase-3, TH, and human α-synuclein in control and A53T cultures on DIV8. Arrowheads indicate cleaved caspase-3–positive TH+ neurons. Arrows point to cleaved caspase-3–positive astrocytes. The death of astrocytes was caused by cytosine β-D-arabinofuranoside in the medium. (B) Expression of ALDH1A1 and TH in the A53T midbrain cultures infected with either AAV-ALDH1A1 or AAV-GFP virus. The noninfected or AAV-GFP virus–infected cultures serve as the control. Scale bar: 20 μm (A and B). (C) Survival rate (percentage) of TH-positive neurons treated with AAV-ALDH1A1 or AAV-GFP viruses. Six independent nTg and A53T cultures per condition were analyzed. (D) The number of surviving TH-positive neurons treated with vehicle DMSO, SIB1757, and DOX in control and A53T mouse midbrain neuron cultures. Seven independent nTg and A53T cultures per condition were analyzed. *P < 0.05, **P < 0.01, ***P < 0.001.
ALDH1A1 preferentially protects against α-synuclein–mediated midbrain DA neuron loss.
To further test a specific role of ALDH1A1 in α-synuclein–induced midbrain DA neuron loss, we treated cultured midbrain DA neurons derived from Aldh1a1+/+ and Aldh1a1–/– mice with 3 different types of cell death inducers: MPP+, a known mitochondrial complex I inhibitor that causes DA neuron death (27); glutamate, which induces excitotoxicity (28); and camptothecin, a DNA enzyme topoisomerase I inhibitor that causes apoptosis (29). We found that Aldh1a1+/+ and Aldh1a1–/– midbrain DA neurons showed comparable susceptibility to MPP+-, glutamate-, and camptothecin-induced cell death (Figure 8, A–C). These data provide evidence that ALDH1A1 may preferentially protect against α-synuclein–induced neuron loss.
Figure 8. ALDH1A1 selectively protects midbrain DA neurons against α-synuclein–mediated cytotoxicity.

(A–C) Line graphs show survival rate (percentage) of cultured Aldh1a1+/+ and Aldh1a1–/– TH-positive neurons treated with (A) MPP+, (B) glutamate, and (C) camptothecin for (B and C) 24 hours and (A) 48 hours. Six independent Aldh1a1+/+ and Aldh1a1–/– cultures per condition were analyzed. (D) Scatter blot compares the survival rate of transgenic human α-synuclein–expressing cortical neurons after treatment with DOX, AAV-GFP, and AAV-ALDH1A1 for 7 days. Twelve independent A53T cultures per treatment were analyzed. ***P < 0.001.
Next, we examined whether ALDH1A1 protects against α-synuclein–induced neuron loss preferentially in DA neurons. We cultured cortical neurons from CaMKII-tTA/tetO-A53T transgenic mice, which overexpress PD-related A53T α-synuclein in the forebrain neurons and develop extensive loss of cortical neurons (30). We then tested whether ALDH1A1 prevents the α-synuclein–induced loss of cortical neurons by infecting these neurons with ALDH1A1 and control GFP–expressing AAV vectors. We found that overexpression of ALDH1A1 did not improve the survival of cortical neurons compared with the GFP controls (Figure 8D). In contrast, DOX treatment that suppressed the expression of transgenic α-synuclein significantly improved the survival rate of these cortical neurons (Figure 8D and Supplemental Figure 5). These results suggest a preferential role of ALDH1A1 in protecting α-synuclein–induced DA neuron loss.
Discussion
Previous studies reveal some very special characteristics of the SNpc DA neurons, such as having a long, unmyelinated and highly ramified axonal structure; using dopamine as the transmitter; and being pace-making neurons that keep firing through a l-type calcium channel (31). As the result of these characteristics, the high neural activity, the accumulation of biogenic dopamine metabolites and other reactive oxygen species, and the alteration of calcium homeostasis may make the SNpc DA neurons more vulnerable to PD-related degeneration (31–33). In addition, DA neurons residing in different subdivisions of SNpc also display differential vulnerability in PD (2–4). However, the underlying molecular mechanism is unclear. Here, we report that a preferential degeneration of DA neurons also occurred in the DM tier of SNpc in α-synuclein transgenic mice. Furthermore, we have shown that these neurons were primarily ALDH1A1-negative DA neurons, by which ALDH1A1 provides an important molecular marker that divides SNpc DA neurons into two subpopulations corresponding to their differential susceptibility in α-synuclein–induced DA neurodegeneration. Interestingly, ALDH1A1-positive and -negative DA neurons exhibited a very similar topographic distribution in the SNpc of human brains, i.e., ALDH1A1-positive neurons were mainly located in the ventral tier of the SNpc, whereas ALDH1A1-negative neurons were concentrated in the dorsal tier. An early study shows that those dorsal SNpc DA neurons are more prone to aging-related neuronal loss, while the ventral DA neurons are largely spared in human SNpc (3). In our mouse studies, we found that the expression of ALDH1A1 was decreased in control mice during aging; however, we did not observe any significant loss of SNpc DA neurons in the aged control mice. Furthermore, genetic deletion of Aldh1a1 in mice also did not cause degeneration of SNpc DA neurons in aged Aldh1a1–/– mice. Together, no age-related loss of SNpc DA neurons was found in both control and Aldh1a1–/– mice. By contrast, in our α-synuclein transgenic mice, we found that ALDH1A1-negative but not -positive SNpc DA neurons were degenerated during aging. Since the expression of transgenic α-synuclein was comparable between these two populations of SNpc DA neurons, we conclude that a combination of Aldh1a1 deficiency, α-synuclein overexpression, and aging may contribute to the loss of ALDH1A1-negative neurons in these mice through a “multiple-hit” mechanism. Take collectively, these mouse studies suggest that a lack of Aldh1a1 is not sufficient to cause SNpc DA neuron loss in normal control and Aldh1a1–/– mice but that it contributes to the α-synuclein–induced SNpc DA neuron loss during aging. In line with this notion, genetic deletion of Aldh1a1 exacerbates SNpc DA neuron loss in α-synuclein transgenic mice, whereas overexpression of ALDH1A1 is protective. Enhancement of ALDH1A1 activities may thereby serve as a potential therapeutic strategy for the treatment of SNpc DA neuron loss. It is worth pointing out that all mice used in this study were in C57/BL6 congenic background. Whether the same phenotypes would appear in different strains or species remain to be determined.
An early study demonstrates a potential pathogenic interaction between cytosolic dopamine and α-synuclein in causing cell death (17, 34). The autoxidation of cytosolic dopamine may produce cytotoxic quinones and free radicals. Particularly, quinone may covalently modify α-synuclein and favor the formation of more cytotoxic α-synuclein protofibrils (17). However, alterations of other enzymatic processes can also lead to the accumulation of cytotoxic aldehydes. In DA neurons, a battery of enzymes, including monoamine oxidases, ALDHs, and catechol-O-methyl transferases, are used to convert dopamine into the inactive metabolite, DOPAC. ALDH1A1, one of main ALDHs in the SNpc DA neurons, likely exerts its protective function through removal of the biogenic dopamine derivative DOPAL (12, 35). DOPAL is highly reactive and may modify and impair many cellular proteins critical for the normal function and survival of SNpc DA neurons (13). The presence of excessive α-synuclein in DA neurons may impair the synthesis, uptake, and degradation of dopamine (10, 36, 37); the increase of cytosolic dopamine may increase the likelihood of the formation of cytotoxic dopamine quinones and dopamine aldehydes, like DOPAL. DOPAL promotes α-synuclein polymerization in forming more cytotoxic α-synuclein aggregates (18, 21). Indeed, more α-synuclein aggregates were present in ALDH1A1-negative nigrostriatal DA neurons of α-synuclein transgenic mice. Additionally, genetic deletion of Aldh1a1 caused further accumulation of α-synuclein aggregates. Notably, the protective function of ALDH1A1 seems specific to α-synuclein–induced midbrain DA neuron loss. Aldh1a1+/+ and Aldh1a1–/– midbrain DA neurons showed similar vulnerability to MPP+-, glutamate-, and camptothecin-mediated cell death. Meanwhile, overexpression of ALDH1A1 did not rescue the α-synuclein–induced loss of cortical neurons. In line with the previous findings (17), our studies further support the notion that the reactive byproducts derived from the abnormal cytosolic dopamine oxidation, such as quinones and DOPAL, may cause the SNpc DA neuron loss by facilitating the formation of cytotoxic α-synuclein oligomers. It remains intriguing why ALDH1A1 is selectively expressed by a subpopulation of SNpc DA neurons. ALDH1A1 may not only oxidize reactive DOPAL, but also affect the steady level of dopamine in DA neurons (15, 16); a lack of ALDH1A1 in these neurons may slow down the catabolism of dopamine and enhance dopamine transmission. Future studies may be interesting to determine whether ALDH1A1-positive and -negative SNpc DA neurons have different connectivity and functionalities in regulating the physiological activities of neurons in the striatum for various motor controls. In addition, it is also intriguing why fewer ALDH1A1-positive DA neurons were found in the SNpc of 1-month-old A53T transgenic mice. An early study suggests that ALDH1A1 may be involved with the differentiation of midbrain DA neurons during development via producing retinoic acid (38). We speculate that the early onset of transgenic α-synuclein overexpression (10) may interfere with the expression and function of ALDH1A1, resulting in less production of ALDH1A1-positive DA neurons during development.
ALDH1A1-positive SNpc DA neurons were spared from degeneration in α-synuclein transgenic mice. On the contrary, in addition to degeneration of dorsal ALDH1A1-negative neurons, a more severe loss of VL ALDH1A1-positive SNpc DA neurons seems to be a prominent pathological feature of the postmortem PD brains (3). Notably, ALDH1A1-negative DA neurons seem to replace the positive ones in the ventral tier of SNpc in the mild PD cases, reflecting a decrease of ALDH1A1 expression in the ALDH1A1-positive DA neurons prior to the neuronal loss. A reduction of ALDH1A1 mRNA expression has also been reported previously in the SNpc of postmortem PD brains (22). However, the mechanism of reduced expression of ALDH1A1 mRNA in PD brains is unclear. It may reflect the loss of SNpc DA neurons, a reduction of ALDH1A1 mRNA expression in individual neurons, or a combination of both neuronal loss and gene expression decrease. In α-synuclein transgenic mice, both Aldh1a1 mRNA and protein levels were substantially decreased in DA neurons. However, ALDH1A1 remained detectable in the SNpc DA neurons in aged α-synuclein transgenic mice, which may explain why no significant loss of ALDH1A1-positive population of SNpc DA neurons was observed in these mice. Our findings suggest that ALDH1A1 itself is also a target in α-synuclein–mediated pathogenic processes in PD. The reduction of ALDH1A1 expression in PD may weaken the protective function of ALDH1A1 against DA neurodegeneration in the ventral tier of SNpc. In support of this notion, genetic ablation of Aldh1a1 exacerbates DA neuron loss in α-synuclein transgenic mice, whereas overexpression of ALDH1A1 is protective.
While ALDH1A1 plays an important role in protecting α-synuclein–induced loss of DA neurons, we cannot exclude the involvement of other genetic factors important for the survival of ventral ALDH1A1-positive DA neurons. A recent study suggests that calbindin-positive and -negative SNpc DA neurons show differential vulnerability to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treatment in mice (39). We carried out ALDH1A1, calbindin, and TH costaining in the midbrain sections of 2-month-old wild-type C57BL/6 mice (Supplemental Figure 1). There were a few calbindin-positive cells in the SNpc, which were distributed mainly in the medial area adjacent to the VTA. However, these calbindin-positive cells were negative to both TH and ALDH1A1 staining. The identity of these cells remains elusive. In the VTA, there were more calbindin-positive cells. However, the majority of these cells were not positive to either TH or ALDH1A1 staining. Our data do not support the notion that calbindin is a valid molecular marker for different subpopulations of SNpc DA neurons in mouse brains. It remains interesting to identify other genes selectively expressed by subpopulations of SNpc DA neurons, which may provide additional molecular insights into this rather unique pathophysiological property of ALDH1A1-positive DA neurons in PD-related neurodegeneration.
In summary, the present study identified ALDH1A1 as an important molecular marker for the subdivision of DA neurons in the SNpc that show differential susceptibility in PD-related DA neurodegeneration. Our findings also support a protective function of ALDH1A1 in maintaining the normal function and survival of SNpc DA neurons, proposing ALDH1A1 as a potential therapeutic target in preventing PD-related SNpc DA neuron loss.
Methods
Animals
Pitx3+/IRES2–tTA (Pitx3-tTA) knockin mice and tetO-A53T transgenic mice were created as described previously (10, 30). Aldh1a–/– (24) and CaMKII-tTA (40) mice were obtained from The Jackson Laboratory. The Pitx3-tTA mice were crossbred with tetO-A53T transgenic mice to get Pitx3-tTA/tetO-A53T (A53T) double-transgenic mice. Pitx3-tTA and tetO-A53T transgenic mice were also crossbred with Aldh1a–/– mice to generate Pitx3-tTA/Aldh1a+/– and tetO-A53T/Aldh1a+/– mice. These two lines of mice were intercrossed to get A53T/Aldh1a–/– and A53T/Aldh1a1+/+ animals. All of the mice were housed in a 12-hour-light/dark cycle and fed regular diet ad libitum. All mouse work follows the guidelines approved by the Institutional Animal Care and Use Committees of the National Institute of Child Health and Human Development, NIH.
Genotyping
Genomic DNA was prepared from tail biopsy using DirectPCR Lysis Reagent (Viagen Biotech Inc.) and subjected to PCR amplification using specific sets of PCR primers for each genotype, including Pitx3-tTA knockin mice (Pitx3-F: GACTGGCTTGCCCTCGTCCCA and Pitx3-R: GTGCACCGAGGCCCCAGATCA), tetO-A53T transgenic mice (PrpEx2-F: TACTGCTCCATTTTGCGTGA and SNCA-R: TCCAGAATTCCTTCCTGTGG), Aldh1a1–/– mice (Aldh1a1muF: CTATCGCCTTCTTGACGAGTTCTT and Aldh1a1muR: CCTTGTACATCTTAACGGTGCACA), and Aldh1a1 wild-type mice (Aldh1a1wtF: TAAAGACCTGGATAAGGCCATCA and Aldh1a1wtR: ACGGTGCACAAAATAAACATCTG).
Immunohistochemistry and light microscopy
As described previously (10), mice were sacrificed and then perfused via cardiac infusion with 4% paraformaldehyde in cold PBS. To obtain frozen sections, brain tissues were removed and submerged in 30% sucrose for 24 hours and sectioned at 30-μm thickness using a cryostat (Leica CM1950). Antibodies specific to TH (rabbit polyclonal, 1:1,000, Pel-Freez; mouse monoclonal, TH-2, 1:500, Sigma-Aldrich), α-synuclein (4D6, 1:500, Santa Cruz Biotechnology), human α-synuclein (syn211, 1:500, Santa Cruz Biotechnology), ALDH1A1 (1:500, Sigma-Aldrich), and calbindin (C9848, 1:500, Sigma-Aldrich) were used as suggested by manufacturers. Alexa Fluor 488– or Alexa Fluor 546–conjugated secondary antibody (1:500, Invitrogen) was used to visualize the staining. Fluorescence images were captured using a laser scanning confocal microscope (LSM 510; Zeiss). The paired images in the figures were collected at the same gain and offset settings. After collection processing was applied uniformly to all paired images. The images were presented as either a single optic layer after acquisition in z-series stack scans at 0.8-μm intervals from individual fields or displayed as maximum intensity projections to represent confocal stacks.
PK digestion analysis
The frozen sections were pretreated with PK (50 μg/ml, Viagen Biotech Inc.) at 37°C for 30 minutes as described previously (19). Immunostaining and fluorescence images were performed as described above.
Image analysis
For the quantitative assessment of various marker protein accumulations and distributions, images were taken using identical settings and exported to ImageJ (NIH) for imaging analyses. Images were converted to an 8-bit color scale (fluorescence intensity from 0 to 255) using ImageJ. Areas of interest were first selected by Polygon or Freehand selection tools and then subjected to measurement by mean optical intensities. The mean intensity for the background area was subtracted from the selected area to determine the net mean intensity.
Stereology
According to the mouse brain in stereotaxic coordinates (41), a series of coronal sections across the midbrain (30 μm per section, every fourth section from bregma –2.54 mm to –4.24 mm) were chosen and processed for ALDH1A1 and TH staining, as described above, and visualized using a laser scanning confocal microscope (LSM 510; Zeiss). We examined 12–14 sections per brain. The images were captured as a projected layer at 20 μm (pinhole, 10 μm; interval, 10 μm, 2 layers) under ×10 magnification. The number of TH- and ALDH1A1-positive neurons was assessed using the Fractionator function of Stereo Investigator 10 (MicroBrightField Inc.). The sampling scheme was designed to have coefficient of error (CE) of less than 10% in order to get reliable results. A first count of samples was performed to achieve a total marking of >200 cells, which generally yields CE <10%. Once the initial cell count was completed, the CE was calculated. The counting parameters would be adjusted based on the CE value. To achieve CE <10%, normally 12 serial sections, with a total of 100 counting frames and, on average, 2 cells per frame would be counted. The final parameters for these studies were as follows: grid size, 200 × 200 μm; frame size, 50 × 50 μm. Three or more mice were used per genotype at each time point. Counters were blinded to the genotypes of the samples.
3D reconstruction of mouse midbrain DA neurons
According to the mouse brain in stereotaxic coordinates (41), a series of coronal sections across the midbrain (40 μm per section, every fourth section from bregma –2.54 mm to –4.24 mm, 10–12 sections per case) were processed for TH (1:1,000, Pel Freez) staining overnight and subsequently with ABC reagents (Vector Laboratories) for an additional hour. Visualization was performed using a DAB Kit (SK-4100, Vector Laboratories) for 5 minutes at room temperature. The serial sections were used for the 3D reconstruction of midbrain using Stereo Investigator 3D reconstruction software (MicroBrightField Inc.). Reconstruction was analyzed for stereology using the Neurolucida Explorer, which returned the nearest neighbor for the distance of DA neurons and the enclosed volume of SNpc.
Human brain tissues
Rapidly autopsied SNpc sections (within 3 hours of the postmortem interval) were obtained from the Brain and Body Donation Program of the Banner Sun Health Research Institute (42) as well as the brain bank of Johns Hopkins University School of Medicine (Supplemental Table 1). Subjects or their legal representatives provided informed consent, and studies were approved by the Banner Health and Johns Hopkins Institutional Review Boards. The diagnosis of PD was made based on clinicopathological criteria, including characteristic clinical features, and on the presence of Lewy bodies within pigmented neurons lost in the substantia nigra. Subjects with PD were divided into two groups on the basis of the presence or absence of clinically documented dementia. Nonpathological controls were selected based on the absence of cognitive impairment, Parkinsonism, and Lewy bodies. Tissues of patients with autosomal recessive juvenile Parkinsonism, a relatively rare syndrome that shares many features of Parkinsonism without the presence of Lewy bodies or Lewy neurites, were excluded from this study.
Human SNpc immunohistochemistry and 3D reconstruction
Human midbrain sections were from the John Hopkins University Brain Resource Center and Banner Sun Health Research Institute and are described in Supplemental Table 1. According to the Atlas of the Human Brain (43), transverse SNpc sections (50 μm per section, every 20th section from bregma 16.0 mm to 27.8 mm; 10 sections per case) from 9 control subjects and 10 patients with PD (between 71 and 90 years old) were deparaffinized and rehydrated in ethanol series. Antigen retrieval techniques were then used (sections were microwaved at full power in TBS buffer [pH 8.8] for 15 minutes twice) before H2O2 treatment. Sections were incubated with rabbit anti-ALDH1A1 polyclonal antibody (1:500, Millipore) overnight and subsequently with the ABC reagents (Vector Laboratories) for an additional hour. Visualization was performed using a DAB Kit (SK-4100, Vector Laboratories) with nickel enhancement for 5 minutes at room temperature. After DAB, all sections were dehydrated and sealed with coverslips.
The 3D reconstruction of human SNpc was performed using Stereo investigator (MicroBrightField Inc.). Nigrostriatal DA neurons were marked by the presence of NM in the soma. The ALDH1A1+/NM+ and ALDH1A1–/NM+ DA neurons were designated as 2 different markers. Contours of dorsal, VL, VM, and PL parts were created according to the literature (3) (DL and DM were merged, as dorsal, VI, and VL were merged as VL in this study). Reconstruction was analyzed for stereology using the Neurolucida Explorer, which returned the markers-closed contours by section for the calculation of percentages of the two subpopulations of nigrostriatal DA neurons and their loss in dorsal, VL, VM, and PL parts of SNpc.
α-Synuclein oligomerization by DOPAL
DOPAL (Santa Cruz Biotechnology) was dissolved in 1% benzyl alcohol and then diluted to a final concentration of 1 mM. Recombinant α-synuclein protein (2 μM, rPeptide) was incubated at 37°C in 50 μl 20 mM Tris-HCl buffer (pH 7.4) with DOPAL for 0, 10, 30, 60, and 120 minutes. The reaction was stopped by heating at 70°C for 3 minutes in 1× SDS loading buffer (Invitrogen). Mixtures (20 μl) were loaded on the 4% to 12% NuPAGE Bis-Tris gel for electrophoresis (Invitrogen) using MES running buffer. After transfer to nitrocellulose membranes, the membranes were immunoblotted with the appropriate dilutions of the primary antibody: α-synuclein (C20, 1:1,000; Santa Cruz Biotechnology) at 4°C. Signals were visualized by enhanced chemiluminescence development (Thermo Fisher Scientific).
Identifying DOPAL-modified α-synuclein via an APBA resin
APBA resin has been used previously to isolate a DOPAL-modified protein (44). The boronic acid complex of the APBA resin can bind to the catechol moiety of the DOPAL-modified protein. A53T/Aldh1a–/– and A53T/Aldh1a1+/+ striatum tissues were homogenized with 10 volumes of sucrose buffer (0.32 M sucrose, 1 mM NaHCO3, 1 mM MgCl2, and 0.5 mM CaCl2 plus protease and phosphatase inhibitor cocktails) and centrifuged at 10,000 g for 10 minutes. Protein concentrations in supernatant were measured using a BCA Kit (Thermo Fisher Scientific). To determine whether more DOPAL-modified α-synuclein proteins were in A53T/ Aldh1a–/– mice than in A53T/Aldh1a1+/+ mice, the striatum samples (50 μl, 1 mg/ml) were incubated with APBA resin (50 μl) on a shaker (4°C) overnight. After incubation, the samples were centrifuged (7,317 g, 2 minutes), and the supernatant was removed. The resin was washed subsequently with 1:1 acetonitrile/50 mM PBS (pH 7.4) (twice, 50 μl each) and distilled water (twice, 50 μl each), centrifuging after each wash fraction (7,317 g, 2 minutes). The resin was added subsequently to 20 μl 1× loading buffer and heated at 90°C for 5 minutes. The samples were centrifuged (10,621 g, 5 minutes), and the supernatants were loaded on the 4% to 12% NuPAGE Bis-Tris gel for electrophoresis (Invitrogen) using MES running buffer. After transfer to nitrocellulose membranes, the membranes were immunoblotted with the appropriate dilutions of the primary antibody α-synuclein (C20, 1:1,000) at 4°C overnight. Signals were visualized by enhanced chemiluminescence development (Thermo Fisher Scientific) and quantified with ImageJ.
Tissue fractionation and Western blot
Striatum tissues were homogenized with 10 volumes of sucrose buffer (0.32 M sucrose, 1 mM NaHCO3, 1 mM MgCl2, and 0.5 mM CaCl2 plus protease and phosphatase inhibitor cocktails) and centrifuged at 10,000 g for 10 minutes. Protein concentrations in supernatant were measured by BCA (Thermo Fisher Scientific). Proteins were size fractioned by 4% to 12% NuPAGE Bis-Tris gel electrophoresis (Invitrogen) using MES running buffer (Invitrogen). After transfer to nitrocellulose membranes, the membranes were immunoblotted with the appropriate dilutions of the primary antibodies α-synuclein (C20, 1:1,000; Santa Cruz Biotechnology), ALDH1A1 (1:1,000; Sigma-Aldrich), and DARPP32 (Cell Signaling Technology) at 4°C. Signals were visualized by enhanced chemiluminescence development and quantified with ImageJ.
Behavior tests
Open-field test.
As described previously (45), the ambulatory and rearing activities of mice were measured by the Flex-Field activity system (San Diego Instruments). Flex-Field software was used to trace and quantify mouse movement in the unit as the number of beam breaks per 30 minutes.
Rotarod test.
As described previously (45), mice were placed onto a rotating rod with autoacceleration from 0 to 40 rpm for 5 minutes (San Diego Instruments). The length of time the mouse stayed on the rotating rod was recorded across 10 trials.
Generation of AAV-overexpressing ALDH1A1
Human ALDH1A1 full-length cDNA (Addgene plasmid 11610) was subcloned into pAAV-NMCS vector (46), which was provided by Joanna Jankowsky from College of Medicine, Houston, Texas, USA. The Gene Core Laboratory at Baylor College of Medicine prepared the AAV-ALDH1A1 and AAV-GFP viruses for the infection experiments.
Midbrain neuronal culture; AAV viral infection; MPP+, glutamate, camptothecin treatment; and immunocytochemistry
Midbrain neuron-enriched cultures were prepared from postnatal day 0 pups from tetO-A53T and Pitx3-tTA crossbreeding (10). The pups were under the DOX treatment to inhibit the expression of transgenic α-synuclein during embryonic stages. Midbrain tissues containing SNpc and VTA, without the meninges and blood vessels, were subjected to papain (5 U/ml, Worthington) digestion for 40 minutes at 37°C. The digested tissue was carefully triturated into single cells using increasingly smaller pipette tips. Cells were centrifuged at 250 g for 5 minutes and resuspended in warm Basal Medium Eagle (Sigma-Aldrich) supplemented with 5% heat-inactivated fetal bovine serum, 1× B27, 1× N2, 1× GlutaMAX (Invitrogen), 0.45% d-glucose (Sigma-Aldrich), 10 U/ml penicillin, and 10 μg/ml streptomycin (Invitrogen). Dissociated cells from each midbrain were equally divided (~2.5 × 105 cells were plated on each coverslip) and plated onto five 12-mm round coverslips precoated with poly-d-lysine and laminin (BD Bioscience), and maintained at 37°C in a 95% O2- and 5% CO2-humidified incubator. 24 hours after seeding, the cultures were switched to serum-free medium supplemented with 5 μM cytosine β-D-arabinofuranoside (Sigma-Aldrich), which was used to suppress the proliferation of glia. Cells in one sister coverslip were maintained in the presence of 2.5 mg/ml DOX after plating. For AAV viral infection, after 3 days in vitro (DIV3), cells without DOX treatment were infected with AAV-expressing GFP and ALDH1A1 at the multiplicity of infection of 107. AAV-infected cells were then labeled with primary antibodies against TH (Santa Cruz Biotechnology, 1:500) and/or ALDH1A1 (Sigma-Aldrich, 1:1,000) overnight at 4°C in a humidified chamber.
For ALDH1A1 activator SIB1757 (Sigma-Aldrich) treatment, after DIV5, cells without DOX treatment were treated with DMSO or 10 μM SIB1757. On DIV8, cells were fixed with 4% paraformaldehyde and 4% sucrose in PBS for 15 minutes, permeabilized by 0.1% Triton X-100 for 5 minutes, and blocked in 10% nonimmune donkey serum (Invitrogen) for 1 hour at room temperature. SIB1757-treated cells were stained with antibody against cleaved caspase-3 (Cell Signaling Technology, 1:100), TH (Abcam, 1: 200), and human α-synuclein (Santa Cruz Biotechnology, 1:500). After 3 washes with PBS, donkey-derived secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 546, and/or Alexa Fluor 647 (Invitrogen; 1:1,000) were applied and incubated for 1 hour at room temperature in the dark. After extensive washes, coverslips were mounted on glass slides with Prolong Gold antifade reagent containing DAPI (Invitrogen), and fluorescence signals were detected using a laser scanning confocal microscope (LSM 510; Zeiss). The total number of TH-positive neurons on each sister coverslip was counted under a ×25 objective.
For MPP+, glutamate, and camptothecin treatment, midbrain neurons derived from Aldh1a1+/+ and Aldh1a1–/– P0 pups were treated with these cell death inducers on DIV12 at dosages ranging from 0 to 1,000 μM. After 24 or 48 hours of treatment, neurons were fixed with 4% PFA and immunostained with TH antibody and secondary antibody. The total number of all TH-positive neurons on each of the sister coverslips was counted under a ×25 objective.
The survival rate of TH-positive neurons was calculated by dividing the number of TH-positive neurons on each coverslip by the number of TH-positive neurons on the corresponding nTg coverslip (with an average number of TH+ neuron per coverslip around 225) infected with AAV-GFP (for viral infection) or treated with vehicle (for SIB1757, MPP+, glutamate, and camptothecin treatment).
Cortical neuronal culture, AAV viral infection, and MTT assay
Primary cortical neurons were cultured from P0 CaMKII-tTA/tetO-A53T pups (30). 1 × 105 neurons were seeded per well in 96-well plate for MTT assay or 1.5 × 106 neurons were seeded per well in 6-well plate for Western blot analysis. Neurons were either treated with 2.5 μg/ml DOX to inhibit the expression of transgenic α-synuclein from DIV0 or infected with 1 × 1010 AAV8-GFP or AAV8-ALDH1A1 on DIV1. On DIV7, the neuron loss in the 96-well plate was measured by MTT assay (Sigma-Aldrich). A53T overexpression level was confirmed by Western blot in neurons in a 6-well plate with or without DOX treatment (Supplemental Figure 5).
LCM and quantitative reverse transcriptase–PCR assay
Brains of Pitx3-tTA/tetO-H2BjGFP control double-transgenic and Pitx3-tTA/tetO-H2BjGFP/tetO-A53T triple-transgenic mice (10) were quickly dissected out, and the frozen brains were sectioned at 20-μm thickness by a cryostat onto a PAN membrane frame slide (Applied Biosystems) and stored at –80°C until LCM. The GFP-positive cells in the SNpc were selected and captured by an ArturusXT micro-dissection system with fluorescent illumination (Applied Biosystems) onto LCM Macro Caps (Applied Biosystems) separately at the following working parameters: spot size, 7–25 μm; power, 50–70 mW; duration, 20–40 μs. The total RNA was extracted with the PicoPure Isolation Kit (Applied Biosystems) using the protocol provided by the manufacturer. The cDNA was synthesized from 50 ng RNA by the First-Strand Kit (QIAGEN after genomic DNA elimination. The SYBR green real-time PCR detection method was used to quantitate the ALDH1A1 expression levels in the control and A53T transgenic SNpc DA neurons, which were normalized by β-actin (Actb) expression. The Aldh1a1 and Actb primers used were from QIAGEN and tested by the manufacturer.
Statistics
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software Inc.). Data are presented as mean ± SEM. Statistical significance was determined by comparing means of different groups and conditions using unpaired 2-tailed Student’s t test, 1-way ANOVA with post-hoc Tukey test, and 2-way ANOVA with post-hoc Bonferroni test.
Supplementary Material
Acknowledgments
This work was supported by the intramural research programs of National Institute on Aging (AG000959-07 and AG000945-03) and by extramural NIH funding of the Johns Hopkins University Alzheimer’s Disease Research Center (P50AG05146) and Morris K. Udall Centers of Excellence in Parkinson’s Disease Research. We thank David Goldstein of National Institute of Neurological Disorders and Stroke Intramural Research Program for advice on ALDH1A1 in dopamine metabolism, Thomas Beach of Brain and Body Donation Program for help in obtaining postmortem human brain sections, Xing-Long Gu for mouse breeding and genotyping, Joanna Jankowsky of Baylor College of Medicine for providing pAAV-NMCS vector, and Kazuhiro Oka of Baylor College of Medicine for packaging recombinant AAV viruses. We also thank the NIH Fellows Editorial Board for editorial assistance.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(7):3032–3046. doi:10.1172/JCI72176.
References
- 1.Schapira AH. Pathogenesis of Parkinson’s disease. Baillieres Clin Neurol. 1997;6(1):15–36. [PubMed] [Google Scholar]
- 2.Hirsch EC, Graybiel AM, Agid Y. Selective vulnerability of pigmented dopaminergic neurons in Parkinson’s disease. Acta Neurol Scand Suppl. 1989;126:19–22. doi: 10.1111/j.1600-0404.1989.tb01778.x. [DOI] [PubMed] [Google Scholar]
- 3.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(pt 5):2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 4.Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain. 1999;122(pt 8):1437–1448. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- 5.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 6.Polymeropoulos MH, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 7.Singleton AB, et al. α-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 8.Simon-Sanchez J, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satake W, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41(12):1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 10.Lin X, et al. Conditional expression of Parkinson’s disease-related mutant alpha-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci. 2012;32(27):9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCaffery P, Drager UC. High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proc Natl Acad Sci U S A. 1994;91(16):7772–7776. doi: 10.1073/pnas.91.16.7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchitti SA, Deitrich RA, Vasiliou V. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol Rev. 2007;59(2):125–150. doi: 10.1124/pr.59.2.1. [DOI] [PubMed] [Google Scholar]
- 13.Rees JN, Florang VR, Eckert LL, Doorn JA. Protein reactivity of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem Res Toxicol. 2009;22(7):1256–1263. doi: 10.1021/tx9000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fitzmaurice AG, et al. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc Natl Acad Sci U S A. 2013;110(2):636–641. doi: 10.1073/pnas.1220399110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson DW, Schray RC, Duester G, Schneider JS. Functional significance of aldehyde dehydrogenase ALDH1A1 to the nigrostriatal dopamine system. Brain Res. 2011;1408:81–87. doi: 10.1016/j.brainres.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wey MC, Fernandez E, Martinez PA, Sullivan P, Goldstein DS, Strong R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson’s disease. PLoS One. 2012;7(2):e31522. doi: 10.1371/journal.pone.0031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conway KA, Rochet JC, Bieganski RM, Lansbury PT, Lansbury PT., Jr Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science. 2001;294(5545):1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 18.Burke WJ, et al. Aggregation of α-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115(2):193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- 19.Neumann M, et al. Misfolded proteinase K-resistant hyperphosphorylated α-synuclein in aged transgenic mice with locomotor deterioration and in human α-synucleinopathies. J Clin Invest. 2002;110(10):1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated α-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci. 2007;27(12):3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cremades N, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149(5):1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis. 2003;14(3):637–647. doi: 10.1016/j.nbd.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Ouimet CC, Miller PE, Hemmings HC, Hemmings HC, Jr, Walaas SI, Greengard P. DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. III. Immunocytochemical localization. J Neurosci. 1984;4(1):111–124. doi: 10.1523/JNEUROSCI.04-01-00111.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan X, et al. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol Cell Biol. 2003;23(13):4637–4648. doi: 10.1128/MCB.23.13.4637-4648.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jinsmaa Y, et al. Dopamine-derived biological reactive intermediates and protein modifications: Implications for Parkinson’s disease. Chem Biol Interact. 2011;192(1–2):118–121. doi: 10.1016/j.cbi.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong D, Kotraiah V. Modulation of aldehyde dehydrogenase activity affects (+/–)-4-hydroxy-2E-nonenal (HNE) toxicity and HNE-protein adduct levels in PC12 cells. J Mol Neurosci. 2012;47(3):595–603. doi: 10.1007/s12031-011-9688-y. [DOI] [PubMed] [Google Scholar]
- 27.Bilsland J, et al. Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J Neurosci. 2002;22(7):2637–2649. doi: 10.1523/JNEUROSCI.22-07-02637.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15(5 pt 1):3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol. 1996;134(3):757–770. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin X, et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64(6):807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sulzer D, Surmeier DJ. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov Disord. 2013;28(1):41–50. doi: 10.1002/mds.25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Surmeier DJ. Calcium, ageing, and neuronal vulnerability in Parkinson’s disease. Lancet Neurol. 2007;6(10):933–938. doi: 10.1016/S1474-4422(07)70246-6. [DOI] [PubMed] [Google Scholar]
- 33.Surmeier DJ, Schumacker PT. Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J Biol Chem. 2013;288(15):10736–10741. doi: 10.1074/jbc.R112.410530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sulzer D. α-Synuclein and cytosolic dopamine: stabilizing a bad situation. Nat Med. 2001;7(12):1280–1282. doi: 10.1038/nm1201-1280. [DOI] [PubMed] [Google Scholar]
- 35.Goldstein DS, et al. Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J Neurochem. 2013;126(5):591–603. doi: 10.1111/jnc.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurz A, et al. A53T-α-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS One. 2010;5(7):e11464. doi: 10.1371/journal.pone.0011464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lam HA, et al. Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human α-synuclein. J Neurosci Res. 2011;89(7):1091–1102. doi: 10.1002/jnr.22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacobs FM, et al. Retinoic acid counteracts developmental defects in the substantia nigra caused by Pitx3 deficiency. Development. 2007;134(14):2673–2684. doi: 10.1242/dev.02865. [DOI] [PubMed] [Google Scholar]
- 39.Luk KC, et al. The transcription factor Pitx3 is expressed selectively in midbrain dopaminergic neurons susceptible to neurodegenerative stress. J Neurochem. 2013;125(6):932–943. doi: 10.1111/jnc.12160. [DOI] [PubMed] [Google Scholar]
- 40.Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274(5293):1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 41. Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. 3rd ed. Waltham, Massachusetts, USA: Academic Press; 2008. [Google Scholar]
- 42.Beach TG, et al. The Sun Health Research Institute Brain Donation Program: description and experience, 1987–2007. Cell Tissue Bank. 2008;9(3):229–245. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mai JK, Paxinos G, Voss T. Atlas Of The Human Brain. 3rd ed. Waltham, Massachusetts, USA: Academic Press; 2007. [Google Scholar]
- 44.LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11(11):1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- 45.Chandran JS, et al. Progressive behavioral deficits in DJ-1-deficient mice are associated with normal nigrostriatal function. Neurobiol Dis. 2008;29(3):505–514. doi: 10.1016/j.nbd.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levites Y, et al. Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid β, amyloid β40, and amyloid β42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J Neurosci. 2006;26(46):11923–11928. doi: 10.1523/JNEUROSCI.2795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.