

Abstract
Microvillus inclusion disease (MVID) is a severe form of congenital diarrhea that arises from inactivating mutations in the gene encoding myosin Vb (MYO5B). We have examined the association of mutations in MYO5B and disruption of microvillar assembly and polarity in enterocytes. Stable MYO5B knockdown (MYO5B-KD) in CaCo2-BBE cells elicited loss of microvilli, alterations in junctional claudins, and disruption of apical and basolateral trafficking; however, no microvillus inclusions were observed in MYO5B-KD cells. Expression of WT MYO5B in MYO5B-KD cells restored microvilli; however, expression of MYO5B-P660L, a MVID-associated mutation found within Navajo populations, did not rescue the MYO5B-KD phenotype but induced formation of microvillus inclusions. Microvilli establishment required interaction between RAB8A and MYO5B, while loss of the interaction between RAB11A and MYO5B induced microvillus inclusions. Using surface biotinylation and dual immunofluorescence staining in MYO5B-KD cells expressing mutant forms of MYO5B, we observed that early microvillus inclusions were positive for the sorting marker SNX18 and derived from apical membrane internalization. In patients with MVID, MYO5B-P660L results in global changes in polarity at the villus tips that could account for deficits in apical absorption, loss of microvilli, aberrant junctions, and losses in transcellular ion transport pathways, likely leading to the MVID clinical phenotype of neonatal secretory diarrhea.
Introduction
Microvillus inclusion disease (MVID) is a rare neonatal diarrheal disorder of the small intestine that arises mainly in European, Middle Eastern, and Navajo Native American cohorts (1, 2). Recent investigations have identified putative inactivating mutations in myosin Vb (MYO5B) in patients with MVID. In the Navajo population, MVID has an incidence of 1 case per 12,000 live births, with a single homozygous MYO5B-P660L (1979C>T p.Pro660Leu, exon 16) mutation responsible for all of these cases, inherited in an autosomal recessive pattern (2, 3). Of the 41 published mutations in the MYO5B gene that contribute to MVID, 16 mutations were homozygous, 9 mutations were heterozygous, and 16 further mutations were found in patients who have compound heterozygous mutations.
MYO5B is a highly processive motor, which functions as a homodimer and is activated by calmodulin binding. Structurally, MYO5B has 3 distinct domains: an N-terminal motor domain, a central light chain binding domain, and the C-terminal cargo-binding tail domain. The motor domain generates force, and the central calmodulin light chain binding domain acts as the lever arm. Together, these domains generate motion that allows MYO5B to function as a dynamic tether for cargo bound to the tail domain by treadmilling in place on F-actin (11–16). MYO5B has binding regions in its tail domain for RAB8A via exon C (exon 30), RAB11 via the globular tail, and RAB10 through its alternatively spliced exon D (exon 31). It should be noted the predominant splice variant of MYO5B in enterocytes lacks exon D (17). Mutations that disrupt the function of the motor, lever arm, calmodulin binding, or RAB binding or induce premature termination before the tail domain can lead to MVID (1, 4, 6, 9).
MVID was first characterized in 1978 in newborns with chronic, unremitting diarrhea (18, 19). The underlying cause of the diarrhea remains to be determined definitively, but biopsy data point to a decrease in sodium absorption that could be the result of absent microvilli or leaky tight junctions in the enterocytes of the patients’ small intestine (20, 21). In these biopsy samples, pathognomonic microvillus inclusions were observed in 10% of enterocytes. No studies until now have identified conclusively the origin or nature of these structures (22, 23). The accumulation of PAS-positive “granules” also occurs at higher frequency in samples from patients with MVID than in the enterocytes of normal duodenum (9). MVID is uniformly fatal and no current pharmacotherapy exists, but it can be treated with total parenteral nutrition or an intestinal transplant (24).
While recent studies have identified MYO5B mutations in MVID, the pathophysiology remains less clear. Previous investigations have established that microvillus inclusions contain apical enterocyte proteins such as sucrase isomaltase, alkaline phosphatase, and sodium hydrogen exchanger 3 (NHE3) (25). In patients with MVID, normally apically trafficked proteins are mislocalized subapically, while sodium potassium ATPase (Na/K-ATPase) basolateral localization was unaffected in the biopsy samples (25). In another study, CD10, which is normally associated with the brush border, accumulated on the subapical surface of the enterocytes in patients with MVID (26). Since the initial discovery that MYO5B mutations were responsible for MVID, much speculation has focused on the role of MYO5B interacting proteins in the manifestations of this disease. In previous studies, we have demonstrated that MYO5B regulates intracellular trafficking and endocytic recycling by localizing with specific RAB small GTPases (RAB8A, RAB10, and RAB11) in subapical vesicle populations (27). Of interest, Rab8a knockout mice display a deficit in intestinal microvilli with prominent intracellular vacuoles that has similarities to some of the features of MVID, but no mutations in Rab8a have been associated with the disease in humans (28). All of these previous investigations suggest that MVID represents a pathophysiologic window into this apical trafficking process, because it arises as a result of inactivating mutations in MYO5B predicted to elicit aberrant apical trafficking of proteins and loss of apical microvilli in intestinal enterocytes (2). Therefore, understanding how mutations in MYO5B lead to aberrant trafficking in enterocytes should provide novel insights into the fundamental mechanisms governing plasma membrane recycling system trafficking and lay the framework for producing a viable therapeutic alternative for patients with MVID.
In this study, we report that MYO5B regulates global intestinal enterocyte polarity. Using stable MYO5B knockdown (MYO5B-KD) in CaCo2-BBE cells, we demonstrate that MYO5B loss results in a decrease in microvilli, mistrafficking of apically and basolaterally trafficked proteins, a decrease in CDC42, and dispersal of RAB8A and RAB11A. In samples from Navajo patients with MVID, microvilli are decreased, apical membrane proteins are mistrafficked, and RAB8A and RAB11A are dispersed from their normal apical position. While microvillus inclusions were not observed in MYO5B-KD cells, the reexpression in MYO5B-KD CaCo2-BBE cells of MYO5B-P660L, a mutation found within Navajo populations that slows MYO5B movement, causes the formation of microvillus inclusions. Rescue of the microvilli loss in the MYO5B-KD cells required intact RAB8A binding with MYO5B, while expression of a MYO5B that cannot bind RAB11A elicited microvillus inclusions. Finally, we demonstrate in CaCo2-BBE cells that microvillus inclusions arise from internalization of the apical membrane and are likely the products of macropinocytosis. Together, these studies demonstrate that loss of apical microvilli in patients with MVID likely arises from a defect in RAB8A-dependent trafficking, while deficits in the RAB11A-dependent pathways account for the formation of microvillus inclusions. The broad changes in cell polarity in enterocytes at the microvillus tips likely account for the secretory diarrhea that dominates the clinical picture in patients with MVID.
Results
Loss or mutation of MYO5B causes a reduction in apical microvilli in enterocytes.
Previous work has demonstrated that both truncations and nonfunctional motor mutations in MYO5B are associated with the MVID phenotype in neonates, including the loss of microvilli and microvillus inclusion formation (6, 8). To examine the effects of loss of MYO5B, we stably knocked down MYO5B in CaCo2-BBE cells, a subclone from the CaCo2 parental cell line selected specifically for the extensive microvilli that develop when grown on permeable Transwell filters (29). For this study, 2 lentiviral shRNA vectors targeting MYO5B and a control shRNA were used for transduction of CaCo2-BBE cells. With both shRNA MYO5B constructs, MYO5B protein and mRNA were reduced by greater than 50% compared with the control cells (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI71651DS1). The MYO5B-KD shRNA2–expressing cell line was used for all the subsequent studies discussed below, unless stated otherwise.
Previous published work has demonstrated that CaCo2 cells polarize completely when grown on permeable filters for 14 days, and all CaCo2-BBE cell lines for this study were grown on Transwell filters for 15 days, unless stated otherwise (30). We examined the microvilli of these cells by staining both CaCo2-BBE cells expressing control and MYO5B-KD shRNA CaCo2-BBE cells with anti-ezrin antibodies and colabeling with fluorescent phalloidin (Figure 1, A–F, K, and L). In the control cells, we observed a normal apical brush border distribution of F-actin and ezrin (Figure 1, A–C and K). By scanning electron microscopy and transmission electron microscopy (TEM), normal densely packed microvilli were observed in the control cells with no microvillus inclusions (Figure 1, G and H). In the MYO5B-KD cells, phalloidin staining accumulated in the terminal web and apical ezrin decreased (Figure 1, D–F and L). Scanning electron microscopy and TEM studies of these cells showed sparse, shortened apical microvilli with no microvillus inclusions (Figure 1, I and J). Similar results were found with MYO5B-KD cell lines expressing both shRNA vectors. Quantitation of ezrin immunofluorescence revealed a reduction in CaCo2-BBE MYO5B-KD cells, and quantitation of ezrin by Western blot showed a significant decrease in total ezrin protein in the MYO5B-KD cells (Figure 1, M and N).
Figure 1. Confocal fluorescence imaging, scanning electron microscopy, and TEM of CaCo2-BBE cells demonstrating loss of microvilli with MYO5B-KD.

(A–F) x-y images are shown in the top panels with x-z images below each. (A and B) Immunostaining for ezrin (red) and phalloidin F-actin (green) in control cells showed prominent staining of microvilli. Triple overlay with DAPI nuclear staining (blue) is shown in C. (D and E) Immunostaining for ezrin (red) and phalloidin F-actin (green) in MYO5B-KD cells showed reduced apical ezrin and an accumulation of F-actin in the subapical region. Triple overlay with DAPI nuclear staining (blue) is shown in F. (G and H) Scanning electron microscopy and TEM of control cells showing normal densely packed microvilli. (I and J) Scanning electron microscopy and TEM of MYO5B-KD cells showing immature sparse microvilli. (K) Control cells x-z images from C magnified showing microvilli stained for both ezrin and F-actin. (L) CaCo2-BBE MYO5B-KD cells x-z images from F magnified showing loss of apical microvilli stained for both ezrin and F-actin and their accumulation in the terminal web. (M) Quantitation of ezrin mean fluorescence in maximum-intensity Z-stack projections showing a reduction in CaCo2-BBE MYO5B-KD cells. (N) Ezrin Western blot with quantitation showing a decrease in total ezrin in the MYO5B-KD cells. Scale bar: 10 μm (A–F, K, and L); 3 μm (G and I); 1 μm (G and I, insets); 500 nm (H and J). *P ≤ 0.05, **P ≤ 0.01, Mann-Whitney test. Error bars denote mean ± SEM.
To examine whether the loss of microvilli was due to improper formation or a delay in formation in the MYO5B-KD cells, we examined the stages of microvillar development in both control and MYO5B-KD CaCo2-BBE cell lines. In a time series of control cells grown on Transwell filters, microvilli formed in stages and established a mature brush border by 12 days after plating (Supplemental Figure 2A). In the control cells, microvilli began to bud from the plasma membrane after 2 days, initiated contact with their neighbors after 4 days, formed tepee-like structures after 6 days, and initiated the process of packing their microvilli after 8 days on Transwell filters. By Western blot analysis, villin-1 was upregulated throughout microvillar development (Supplemental Figure 2, B and C). In a time series for CaCo2-BBE MYO5B-KD cells, the initial stages of microvilli development were similar to those seen for the control cells. However, MYO5B-KD cells did not progress past the tepee-like stage and did not achieve microvillar packing, even when cultured for up to 20 days on Transwell filters (Supplemental Figure 3A). In MYO5B-KD cells, villin-1 was not upregulated during culture on Transwell filters, compared with the control cells, by Western blot analysis (Supplemental Figure 3, B and C).
A wide array of mutations in MYO5B are associated with MVID. As a result, it was unclear whether the loss of MYO5B or the effect of MYO5B mutation was responsible for the MVID phenotype. Notably, we did not observe evidence for microvillus inclusions in the MYO5B-KD cells. To examine the effects of the Navajo MYO5B mutation (referred to herein as MYO5B-P660L) on the MVID phenotype, we immunostained small intestine biopsy samples from Navajo patients with MVID for ezrin and MYO5B (Figure 2, A–F). In normal duodenum sections, MYO5B immunostaining was concentrated in the subapical region of enterocytes below the brush border, which stained strongly for ezrin (Figure 2, A–C). In contrast, samples from patients with MVID showed a marked decrease in ezrin, dispersal of MYO5B staining, and microvillus inclusion formation in a subset of enterocytes at the tips of intestinal villi, when compared with controls (Figure 2, D–F). Interestingly, the cells within the lower regions of the villi demonstrated normal ezrin staining of the apical brush border (Figure 2E). Structured illumination microscopy (SIM) of the samples from patients with MVID revealed an accumulation of β-actin in the terminal web and a dispersal of MYO5B from its normal subapical position above the terminal web (Figure 2, G and H). TEM of the samples from patients with MVID revealed a prominent decrease in microvilli and enlarged intracellular vesicular structures (Figure 2, I and J). Thus, while MYO5B-KD in CaCo2-BBE cells replicated losses in microvilli seen in patients with MVID, we did not observe microvillus inclusions.
Figure 2. Immunostaining of duodenum samples from Navajo patients with MVID reveals that mutation of MYO5B causes loss of apical microvilli, microvillus inclusion formation, and accumulation of F-actin into the terminal web.

(A–C) Normal duodenum immunostaining for MYO5B (green) and ezrin (magenta) showed subapical MYO5B with increased staining at the tips of villi and ezrin localization to the brush border. (D–F) MVID patient duodenum immunostaining for MYO5B and ezrin showed dispersal of MYO5B from the subapical region of enterocytes and ezrin localization to microvillus inclusions in cells at the villus tips. Note that cells near the crypts (white arrowhead) showed a normal apical ezrin distribution when compared with cells at the villus tips (red arrowhead). (G) SIM imaging of β-actin (red) and MYO5B (green) in normal duodenum showed actin staining in the apical brush border (yellow arrow), discrete staining of the terminal web (white arrow), and MYO5B localization above the terminal web and below the apical surface (blue arrow). (H) SIM imaging of the samples from patients with MVID revealed an accumulation of β-actin (red) in the terminal web and dispersal of MYO5B (green) from its normal subapical position above the terminal web. (I) TEM in normal duodenum showed normal apical microvilli. (J) TEM of the MVID patient duodenum revealed a decrease in microvilli and enlarged subapical vesicular structures. Scale bar: 50 μm (A–F); 10 μm (A–F, insets); 5 μm (G and H); 2 μm (I and J).
Alteration of tight junctions was observed in CaCo2-BBE MYO5B-KD cells and enterocytes from patients with MVID.
We evaluated the integrity of the tight junctions by immunostaining for claudin 1 (CLDN1), claudin 2 (CLDN2), claudin 4 (CLDN4), and ZO-1 (Supplemental Figure 4, A–H). Immunocytochemistry supported the loss of CLDN1 at the junctions as well as an increase in CLDN2 staining in the junctions of MYO5B-KD cells. Quantitation of CLDN1, CLDN2, CLDN4, and ZO-1 immunofluorescence showed a reduction in CLDN1 and ZO-1 and an increase in CLDN2 and CLDN4 in MYO5B-KD cells (Supplemental Figure 4I). Western blot analysis and quantitation showed that CLDN1 was downregulated and CLDN2 was upregulated in the MYO5B-KD cells compared with control cells (Supplemental Figure 4, J and K). The increase in CLDN1 and concomitant decrease in CLDN2 is indicative of a shift from a high resistance junctional complex to a leaky junction (31). In support of this concept, the transepithelial electrical resistance (TEER) in MYO5B-KD cells was reduced significantly compared with that in control cells (Supplemental Figure 4L). Interestingly, previous studies have noted that microvilli formation is correlated with TEER in CaCo2 cells (32).
We next examined the effects of the MYO5B-P660L mutation on components of the tight junction by immunostaining duodenal biopsy samples from Navajo patients for CLDN1 and CLDN4 (Supplemental Figure 5). Unfortunately, no CLDN2 antibody is presently available for reliable staining of human paraffin sections. CLDN1 in samples from patients with MVID was lost from junctions and mislocalized to the cytoplasm (Supplemental Figure 5, A and B). In contrast, CLDN4 maintained its normal localization in the samples from patients with MVID, although its expression was reduced (Supplemental Figure 5, C and D). These findings suggest that loss or mutation of MYO5B in both the CaCo2-BBE cell model and patients with MVID leads to alterations in junctional composition in enterocytes.
Loss of polarity in basolateral and apical compartments is observed in MYO5B-KD CaCo2-BBE cells and enterocytes from patients with MVID.
We next examined the effects of reducing MYO5B on components of the basolateral and apical compartments in CaCo2-BBE cells. To evaluate the integrity of the basolateral compartment, we immunostained for p120-catenin (p120), Na/K-ATPase, E-cadherin, and β-catenin (Figure 3). In the control CaCo2-BBE cells, p120, Na/K-ATPase, and β-catenin were distributed along the basolateral membranes, while E-cadherin was confined to the junctions (Figure 3, A, C, E, G, and I). In the MYO5B-KD cells, p120 and Na/K-ATPase staining were decreased at the lateral membrane by immunofluorescence (Figure 3, B and D). Remarkably, E-cadherin staining in the MYO5B-KD cells was redistributed to both apical and basolateral membranes as well as to vesicles underlying both surfaces (Figure 3, F and J). Nevertheless, β-catenin was not redistributed or decreased by immunofluorescence between these 2 cell lines (Figure 3, G and H). Quantitation of immunofluorescence for the basolateral proteins revealed that MYO5B-KD cells showed a reduction of p120, no change in Na/K-ATPase or β-catenin, and an increase in E-cadherin (Figure 3K). Interestingly, the apical-to-basolateral ratio in MYO5B-KD cells for p120 or β-catenin showed no change. However, Na/K-ATPase shifted to the apical surface, while E-cadherin was redistributed from the junction to both the apical and basolateral surfaces (Figure 3N). Western blot analysis of p120 in MYO5B-KD cells showed isoform switching when compared with the pattern observed in the control cells (Figure 3L), which is indicative of an alteration in polarity (33, 34). This change was not accompanied by a decrease in total p120 protein (Figure 3M). The apparent decrease, shown by immunofluorescence staining, was likely due to the restricted specificity of the immunostaining antibody for extended carboxyterminal isoforms (35). We observed no changes in Na/K-ATPase and β-catenin and an increase in total E-cadherin (Figure 3, L and M).
Figure 3. Loss of MYO5B in CaCo2-BBE cells causes the redistribution of basolateral markers.

(A–H) x-y confocal images are shown above z-axis reconstructions. (A) p120 staining in controls showed a lateral distribution, (B) while MYO5B-KD cells showed decreased p120 at the lateral membranes. (C) Na/K-ATPase staining in controls showed a lateral distribution, (D) while MYO5B-KD cells showed reduction in lateral membranes. (E) E-cadherin staining in controls showed a junctional distribution, (I) with the x-z image magnified in junctional E-cadherin (white arrows). (F) MYO5B-KD showed redistribution of E-cadherin along the apical and lateral membranes and with internal pools of E-cadherin distributed throughout the cells; x-z image magnified in J. (G and H) In control and MYO5B-KD cells, β-catenin stained lateral membranes. (K) Quantitation of basolateral mean fluorescence. MYO5B-KD cells showed a reduction of p120 and an increase in E-cadherin. (L and M) Western blot with quantitation of basolateral markers in MYO5B-KD showed an isoform switch of p120, with no decrease and an increase E-cadherin total protein. (N) Quantitation of apical-to-basolateral ratio in MYO5B-KD cells showed a redistribution of Na/K-ATPase to the apical surface, while E-cadherin was localized over both the apical and basolateral surfaces. (O) Surface biotinylation of either the apical or basolateral surfaces in control and MYO5B-KD cells with total protein, flow through from streptavidin beads (FT), and biotinylated-streptavidin-bound protein from control (Bound-Ctrl) or MYO5B-KD (Bound-MVBKD) cells showed an increase in apical and a decrease in basolateral E-cadherin in the MYO5B-KD cells. Scale bar: 10 μm. *P ≤ 0.05, **P ≤ 0.01, Mann-Whitney test. Error bars denote mean ± SEM.
To investigate the localization of E-cadherin using a biochemical approach, we performed cell surface biotinylation of both the apical and basolateral surfaces and analyzed the distribution of E-cadherin in proteins collected on streptavidin-coated beads (Figure 3O). Analysis of the relative distributions across biotinylated fractions demonstrated that E-cadherin in the apical-bound fraction was increased in MYO5B-KD cells compared with that in the control cells (17.93% ± 2.15% versus 9.76% ± 3.1%) and E-cadherin in the basolateral-bound fraction decreased in MYO5B-KD cells compared with that in control cells (12.43% ± 7.46% versus 22.8% ± 5.87%). All of these results suggest that loss of MYO5B caused aberrant trafficking of E-cadherin.
To evaluate the integrity of the basolateral compartment in samples from patients with MVID, we immunostained sections for p120, E-cadherin, Na/K-ATPase, and β-catenin (Figure 4). In the control duodenum biopsy samples, all 4 markers were distributed along the basolateral membranes (Figure 4, A, C, E, and G). In the samples from patients with MVID, p120 and β-catenin staining was decreased at the lateral membranes, as shown by immunofluorescence, but maintained a normal lateral distribution (Figure 4, B and H). E-cadherin staining in the samples from patients with MVID, however, was redistributed to both the apical and basolateral surfaces (Figure 4D). Na/K-ATPase staining in the samples from patients with MVID was maintained with dispersal in enterocytes at the villus tips (Figure 4F).
Figure 4. Immunostaining of MVID patient duodenum samples demonstrated alteration of the basolateral compartment.

(A) In normal duodenum, p120 staining showed a lateral distribution. (B) In MVID patient duodenum, p120 staining was reduced but maintained its lateral membrane localization. (C) In normal duodenum, E-cadherin staining demonstrated a lateral distribution. (D) In MVID patient duodenum, E-cadherin staining was present on both apical and lateral membranes. (E) In normal duodenum, Na/K-ATPase antibodies stained the lateral membranes. (F) In the MVID patient samples, Na/K-ATPase stained the lateral membranes with dispersal of staining in enterocytes at the villus tips. (G) In normal duodenum, β-catenin staining was present in the lateral membranes. (H) In MVID patient duodenum, β-catenin staining was decreased in the lateral membranes. Scale bar: 50 μm; 10 μm (insets).
To examine the effects of MYO5B-KD on a nonstructural component of the microvilli, we immunostained the CaCo2-BBE cells for dipeptidyl peptidase-4 (DPPIV). In control cells, DPPIV labeled the apical external surface of microvilli (Figure 5, A–F and M). However, in the MYO5B-KD cells, DPPIV staining was reduced in the apical membranes, consistent with the loss of microvilli, and we observed DPPIV staining in internal punctate vesicular structures. In the RAB8A knockout mouse, DPPIV was observed in enlarged lysosomes (28). To test whether DPPIV in MYO5B-KD cells was also diverted to the lysosomes, we dual immunostained the cells for DPPIV and LAMP2a. In the MYO5B-KD cells, DPPIV localized to both LAMP2a-negative and also LAMP2a-positive vesicles (Figure 5, G–L and N). To determine the origin of the LAMP2a-negative vesicles, we dual immunostained both the control and MYO5B-KD cell lines for RAB11A, which labels the apical recycling endosome, and DPPIV (Figure 5, O–R). In control cells, DPPIV labeled the apical surface and RAB11A labeled punctate vesicles below the apical surface (Figure 5O). In the MYO5B-KD cells, RAB11A localized to both DPPIV-positive vesicles and vesicles that contained no DPPIV (Figure 5, P–R). Quantitation of DPPIV immunofluorescence revealed a decrease in DPPIV in the MYO5B-KD cells, and, as shown by Western blot analysis, total protein expression of DPPIV was significantly reduced (Figure 5, S and T).
Figure 5. Coimmunostaining of DPPIV and LAMP2a in CaCo2-BBE cells with redistribution of DPPIV to large vesicles in MYO5B-KD cells.

(A–L) x-y confocal images are shown above z-axis reconstructions. Control and MYO5B-KD cells were immunostained for DPPIV (green) and LAMP2a (red). (A–F) Control cells showed a normal apical distribution of DPPIV and diffusely cytoplasmic LAMP2a. (G–L) In MYO5B-KD cells, DPPIV was seen in internal vesicles, some of which also stained for LAMP2a (white arrow). (M) Magnified x-z image from control cells in F. (N) Magnified x-z image from MYO5B-KD cells in L showing cytoplasmic distribution of DPPIV. (O) Control cells immunostained for DPPIV (green) and RAB11A (red) showed apical DPPIV and subapical RAB11A. (P–R) MYO5B-KD cells, immunostained for DPPIV (green) and RAB11A (red), showed dispersal of RAB11A from the subapical surface and localization in large DPPIV positive vesicles (white arrows). (S) DPPIV reduction in MYO5B-KD cells shown via mean fluorescence in maximum-intensity Z-stack projections. (T) Western blot comparing control and MYO5B-KD cell lines probed for DPPIV and α-tubulin demonstrating reduction of DPPIV expression. (U) Apical surface biotinylation in control and MYO5B-KD cells showing DPPIV immunoreactivity in total protein, flow through from streptavidin beads, and biotinylated streptavidin-bound protein, demonstrating an increase in the nonbiotinylated cytoplasmic pool and a decrease in DPPIV on the apical surface in the MYO5B-KD (Bound-MVBKD) versus control (Bound-Ctrl) cells. Scale bar: 10 μm. *P ≤ 0.05, Mann-Whitney test. Error bars denote mean ± SEM.
To investigate the localization of DPPIV using a biochemical approach, we performed cell surface biotinylation of the apical surface and analyzed proteins collected on streptavidin-coated beads for DPPIV (Figure 5U). In the MYO5B-KD cells, we observed a loss of DPPIV from the apical membranes (7.209% ± 3.63% versus 13.42% ± 2.98% in controls) and an increase in the nonbiotinylated pool of DPPIV (92.79% ± 3.63% versus 86.58 ± 2.98% in controls). All of these results suggest that DPPIV is lost from the apical membrane and localized intracellularly in part to lysosomes as well as to endosomes in MYO5B-KD cells.
Other proteins reported previously as mistrafficked in samples from patients with MVID, such as transferrin receptor (CD71) and CD10, also displayed an altered distribution in MYO5B-KD cells. CD10 was observed in subapical and lateral puncta in the MYO5B-KD cells, instead of in the more diffuse pattern observed in the control cells, and quantitated CD10 fluorescence was significantly decreased (Supplemental Figure 6, A, B, and F). CD71 was redistributed strongly to the subapical region throughout the cytoplasm, and total protein was increased in MYO5B-KD cells (Supplemental Figure 6, C–F). Finally, we also evaluated the pattern of transferrin trafficking in MYO5B-KD cells. Fluorescently labeled transferrin was endocytosed from the basolateral membranes and then chased with serum for 60 minutes in both control and MYO5B-KD cells. Fluorescently labeled transferrin was endocytosed appropriately from the basolateral membranes in both the control and MYO5B-KD cells (Supplemental Figure 6, G and H). In MYO5B-KD cells, fluorescently labeled transferrin accumulated in subapical vesicles, rather than recycling out of the cell following serum chase, as in the control cells (Supplemental Figure 6, I and J). Quantitation of transferrin trafficking showed no significant difference in uptake between the control and the MYO5B-KD cells, but transferrin was significantly retained in MYO5B-KD cells (Supplemental Figure 6K). All of these results suggested that MYO5B-KD caused alterations in apical and basolateral trafficking.
To examine the effects of the MYO5B-P660L mutation on apical nonstructural components of the microvilli, we immunostained the duodenal biopsy samples for CD10 and sodium glucose transporter 1 (SGLT1). In the duodenal biopsy control samples, CD10 and SGLT1 labeled the apical brush borders of enterocytes (Supplemental Figure 7, A and E). In contrast, transferrin receptor (CD71) was diffusely distributed throughout the enterocytes (Supplemental Figure 7C). However, in the samples from patients with MVID, CD10 and SGLT1 were redistributed away from the apical surface with a loss of SGLT1 (Supplemental Figure 7, B, D, and F). In contrast, CD71 staining was increased in both the apical and basolateral poles of enterocytes (Supplemental Figure 7D). Interestingly, when DPPIV was costained with villin-1, they colocalized in microvillus inclusions, but DPPIV was also observed in large internal punctate vesicular structures that did not stain for villin-1 (Figure 6). These findings confirmed that loss of MYO5B in CaCo2-BBE cells caused a disruption of normal apical and basolateral trafficking and localization similar to what is observed in Navajo patients with MVID enterocytes.
Figure 6. DPPIV is mistrafficked to intracellular vesicles and to microvillus inclusions in samples from patients with MVID.

(A–C) Normal duodenum villin-1 (red) and DPPIV (green) immunostaining showed localization of DPPIV and villin-1 to the brush border. Merged overlay images with DAPI nuclear staining (blue) are shown at right. (D–F) MVID patient duodenum villin-1 and DPPIV immunostaining showed mislocalization of DPPIV to microvillus inclusions (white arrows) and to large intracellular vesicles (red arrow). Merged overlay images with DAPI nuclear staining (blue) are shown at right. Scale bar: 50 μm; 10 μm (insets).
RAB8A- and RAB11A-positive vesicles are dispersed in CaCo2-BBE MYO5B-KD cells and enterocytes from Navajo patients with MVID.
To determine the underlying effects of MYO5B loss on trafficking to microvilli and polarity, we stained the MYO5B-KD cells for 2 RAB proteins that bind to MYO5B: RAB8A and RAB11A. Both RAB8A and RAB11A were concentrated in large vesicular complexes subapically in the control cells, with RAB11A showing a central subapical distribution and RAB8A having a subapical and lateral distribution. In the MYO5B-KD cells, both RAB8A and RAB11A were dispersed throughout the cytoplasm away from their normal subapical distributions (Figure 7, A–C). Maximum-intensity Z-stack projections of MYO5B-KD cells showed dispersal of both RAB8A and RAB11A into smaller vesicles located diffusely through the cell cytoplasm (Figure 7D). Quantitation of the mean total cellular fluorescence intensity demonstrated that RAB11A staining across the entire cell volume did not change for RAB11A but was decreased for RAB8A (Figure 7E). Total RAB8A and RAB11A protein, as shown by Western blot, showed no change between control and MYO5B-KD cells (Figure 7, F and G). All of these findings suggest that loss of MYO5B expression disrupts the normal defined submembrane localization of RAB8A and RAB11A.
Figure 7. RAB8A and RAB11A immunofluorescence staining in MYO5B-KD cells was dispersed from the apical surface.

(A and B) x-y confocal images are shown above z-axis reconstructions. (A) RAB8A (red) and p120 (green) costaining in CaCo2-BBE cells. Control cells showed apical-lateral RAB8A distribution. MYO5B-KD cells showed RAB8A dispersal throughout the cytoplasm. (B) RAB11A (red) and Na/K-ATPase (green) staining in CaCo2-BBE cells. In control cells, RAB11A showed a subapical distribution. In MYO5B-KD cells, RAB11A was dispersed throughout the cytoplasm. (C) Magnified RAB8A staining x-z images from control and MYO5B-KD cells in A, and magnified RAB11A staining x-z images from control and MYO5B-KD cells in B. (D) In control cells, z-axis projection showed the normal distribution of RAB8A-positive vesicles and RAB11A-positive vesicles. In MYO5B-KD cells, z-axis projection showed dispersal of RAB8A-positive vesicles and RAB11A-positive vesicles. (E) Quantitation of RAB8A and RAB11A mean fluorescence in maximum-intensity Z-stack projections showed a reduction in RAB8A but no change of RAB11A in MYO5B-KD cells. (F) Western blot with quantitation of RAB8A showed no change in RAB8A in MYO5B-KD cells. (G) Western blot with quantitation of RAB11A showed no change in RAB11A in MYO5B-KD cells. Scale bar: 10 μm. *P ≤ 0.05, Mann-Whitney test. Error bars denote mean ± SEM.
To examine the effects of loss of MYO5B on other markers of polarity, we also stained the CaCo2-BBE cells for CDC42. Activation of CDC42 is regulated by RAB8A via its guanine nucleotide exchange factor TUBA, and both CDC42- and RAB8A-deficient mice develop aspects of the MVID phenotype (36–38). In the control cells, we observed 2 distinct CDC42 populations. In one, CDC42 was dispersed throughout the cells, while in another, CDC42 localized to small puncta. In the MYO5B-KD cells, the punctate population was disrupted and total CDC42 was reduced, as assessed by immunofluorescence quantitation (Supplemental Figure 8, A and B). Total CDC42 protein was reduced in MYO5B-KD cells, and, furthermore, the active CDC42 population was also reduced (Supplemental Figure 8, B and C). Immunostaining in normal duodenum showed an apical distribution of CDC42; however, in samples from patients with MVID, total CDC42 was reduced and mislocalized away from the apical surface (Supplemental Figure 8D). It should be noted that atypical protein kinase C (aPKC) was also mislocalized in MVID patient tissue samples (Supplemental Figure 8D), consistent with regulation of the localization and activation of aPKC by CDC42 (39, 40). These findings, along with previous work using CDC42-deficient mice, suggest that loss of CDC42 contributes to the MVID phenotype.
To determine the underlying effects of the Navajo MYO5B mutation on trafficking to microvilli and polarity loss, we stained the samples from patients with MVID for RAB8A and RAB11A. Both RAB8A and RAB11A were distributed subapically in control duodenal biopsy samples, with RAB8A showing a subapical and lateral distribution and RAB11A showing a subapical vesicle distribution (Supplemental Figure 9, A and C). In the MVID patient sample, RAB8A was dispersed throughout the cytoplasm and accumulated in small puncta (Supplemental Figure 9B). Moreover, in the samples from patients with MVID, RAB11A was dispersed throughout the cytoplasm away from its normal distribution and accumulated with increased fluorescence intensity throughout the enterocytes (Supplemental Figure 9D). These findings demonstrate that loss or mutation of MYO5B leads to a disordered distribution of RAB8A and RAB11A both in vitro and in patients with MVID and suggest that MYO5B tethers these RABs and their associated vesicles in the apical domain.
MYO5B-P660L mutation causes the motor to move slowly along F-actin.
All of our preceding studies suggested that loss of MYO5B expression could replicate many aspects of the MVID phenotype, with the notable exception of the production of microvillus inclusions. The MYO5B-P660L mutation seen in Navajo patients with MVID lies outside the actin binding region, so the functional impact of this mutation required clarification. We expressed 3x-mCitrine-MYO5B-WT or 3x-mCitrine-MYO5B-P660L in HEK cells, and both proteins were precipitated by F-actin pull down (Supplemental Figure 10). It should be noted that MYO5B breaks down readily because of an internal PEST site, which can be appreciated by the second band visualized in the F-actin pull down. Previously published work has shown that MYO5B motors that move slowly along F-actin or cannot bind to F-actin are nonfunctional motors (6). Thus, to evaluate the effects of MYO5B-P660L mutation on MYO5B dynamics in live cells, we used single molecule TIRF-FRAP microscopy. We reasoned that fluorescent myosin motors with similar actin binding properties and motor function would demonstrate comparable photobleaching kinetics. For these studies, we tagged synthetic MYO5B wild-type (MYO5B-WT) motor domain (MYO5B-1016X) and MYO5B-P660L motor domain (the Navajo MVID mutation, MYO5B-P660L-1016X) with 3 tandem copies of mCitrine (3x-mCitrine) and expressed these constructs in BF16 cells (1). Previous studies have used this approach to measure average motor speed, the processivity of single kinesin molecules, and the lifetimes of myosin-1a tail/membrane interactions (41, 42). 3x-mCitrine-MYO5B-WT-1016X molecules demonstrated localization and movement toward the tips of filopodia, which indicated active forward movement along F-actin (Supplemental Figure 11A and Supplemental Video 1). In contrast, 3x-mCitrine-MYO5B-P660L-1016X localized with the F-actin binding protein espin but showed negligible movement toward the tips of filopodia, which is indicative of stalled forward movement along F-actin (Supplemental Figure 11A and Supplemental Video 2). Single molecule TIRF-FRAP imaging of 3x-mCitrine-MYO5B-WT-1016X showed that these motors were photobleached by 50% of the maximum intensity after 30 seconds of laser exposure in live BF16 cells (Supplemental Figure 11B). In contrast, 3x-mCitrine-MYO5B-P660L-1016X was photobleached to 20% of the maximum intensity after 30 seconds of laser exposure in live BF16 cells, consistent with a stalled motor (Supplemental Figure 11B). Together, these results suggest that, while MYO5B-P660L can bind F-actin, the motor movement is consistent with a rigor-type phenotype for a nonfunctioning slow moving motor.
Reexpression of MYO5B-WT rescues the phenotype in MYO5B-KD cells, but MYO5B-P660L reexpression does not rescue the knockdown phenotype and causes microvillus inclusion formation.
To determine the specificity of MYO5B-KD effects in CaCo2-BBE MYO5B-KD cells, we reexpressed synthetic full-length sequences for either mCherry-MYO5B-WT or mCherry-MYO5B-P660L. We stained both of these “rescued” cell lines for the microvillar markers ezrin and phalloidin (F-actin). The reexpression of mCherry-MYO5B-WT in MYO5B-KD cells restored apical microvilli, as visualized by both immunofluorescence and scanning electron microscopy (Figure 8, A, B, E, and G). In contrast, the expression of mCherry-MYO5B-P660L in CaCo2-BBE MYO5B-KD cells failed to restore microvilli and induced the formation of microvillus inclusions containing F-actin–based structures with inward facing microvilli, as visualized by both immunofluorescence and scanning electron microscopy (Figure 8, C, D, F, and G). Microvillus inclusions form normally in CaCo2 cells during their initial polarization on Transwell filters, but after 14 days on Transwell filters, unless induced by colchicine or nocodazole treatment, microvillus inclusions do not form (30). Quantitation of microvillus inclusions in CaCo2-BBE control, MYO5B-KD, MYO5B-WT rescue, and MYO5B-P660L reexpression cells showed a significant increase in microvillus inclusions associated with MYO5B-P660L reexpression over the other control or wild-type MYO5B-rescued cell lines (Figure 8H).
Figure 8. MYO5B-WT and MYO5B-YE/QR rescue the MYO5B-KD phenotype, while MYO5B-P660L and MYO5B-YE/QR promote microvillus inclusions.

(A) Ezrin and (B) phalloidin staining in microvilli was rescued in MYO5B-KD–expressing MYO5B-WT. Arrowhead, high expressers; –, low expressers. (C and D) Ezrin and phalloidin staining in microvilli was not rescued in MYO5B-KD–expressing MYO5B-P660L and induced microvillus inclusions (asterisks). (E and F) Scanning electron microscopy of (E) MYO5B-KD–expressing MYO5B-WT showed reestablished mature microvilli and (F) MYO5B-KD–expressing MYO5B-P660L showed immature microvilli in the tepee-like stage. (G) Western blot of rescue cell lines showed similar expression levels of mCherry. (H) Microvillus inclusions in MYO5B-KD reexpressing MYO5B-P660L and MYO5B-YE/QR were significantly higher compared with other cell lines. (I) Expression of MYO5B-YE/QR reestablished phalloidin staining of microvilli (yellow arrow) but also induced the formation of microvillus inclusions (white arrow). (J) Expression of MYO5B-QL/YC partially recovered microvilli. (K and M) Scanning electron microscopy and TEM of MYO5B-KD cells expressing MYO5B-YE/QR showed reestablished mature microvilli. (L and N) Scanning electron microscopy and TEM of MYO5B-KD cells expressing MYO5B-QL/YC showed partial rescue of nonuniform microvilli. (O) MYO5B-KD cells expressing MYO5B-YE/QR showed subapical microvillus inclusions (red arrows), with corresponding denuded microvilli (black arrow) and surrounding brush border (blue arrow). (P) MVID patient duodenum with pathognomonic microvillus inclusion (red arrows) and denuded microvilli (black arrow). Scale bar: 10 μm (A–D, I, and J); 2 μm (E and F); 500 nm (E and F, insets); 5 μm (K and L); 1 μm (K and L, insets), 2 μm (M and N); 500 nm (O and P). *P ≤ 0.05, ***P ≤ 0.001, Mann-Whitney test. Error bars denote mean ± SEM.
The mCherry-MYO5B-WT rescue of MYO5B-KD cells partially reestablished the normal distribution of CLDN1 and CLDN2 (Supplemental Figure 12A). In contrast, the expression of mCherry-MYO5B-P660L in MYO5B-KD cells caused CLDN1 to be mislocalized into the cytoplasm and an increase in CLDN2 at the lateral membrane (Supplemental Figure 12A). The mCherry-MYO5B-WT rescue of CaCo2-BBE MYO5B-KD cells also reestablished the normal distribution of DPPIV, while the expression of mCherry-MYO5B-P660L caused mislocalization of DPPIV to microvillus inclusions and large vesicles (Supplemental Figure 12B). Reexpression of mCherry-MYO5B-WT in MYO5B-KD cells restored the normal distributions of both RAB8A- and RAB11A-containing vesicles (Supplemental Figure 13A). In contrast, the expression of mCherry-MYO5B-P660L in CaCo2-BBE MYO5B-KD cells failed to rescue the dispersal of either RAB8A or RAB11A (Supplemental Figure 13A). The failure of MYO5B-P660L to rescue suggests that this motor has lost its ability to function as a tether, likely due to loss of the ability of MYO5B-P660L to treadmill on F-actin at the speed of actin polymerization (43–45). These findings suggest that MYO5B-P660L expression promotes microvillus inclusion formation and can recapitulate the MVID phenotype.
MYO5B mutant that selectively binds to RAB8A rescues the loss of microvilli in CaCo2-BBE MYO5B-KD cells.
In previous work, we have identified 2 mutants of MYO5B that have altered interactions with critical RAB proteins (27). MYO5B(Y1714E/Q1748R) (referred to herein as MYO5B-YE/QR) binds RAB8A but does not interact with RAB11A. In contrast, MYO5B(Q1300L/Y1307C) (referred to herein as MYO5B-QL/YC) can bind to RAB11A but does not interact with RAB8A. Thus, to determine whether aspects of MYO5B loss result from interactions with specific RAB proteins, we evaluated the MYO5B-KD cell lines with reexpression of either mCherry-MYO5B-YE/QR or mCherry-MYO5B-QL/YC. The expression of mCherry-MYO5B-YE/QR in MYO5B-KD cell lines reestablished microvilli, as visualized by phalloidin staining, scanning electron microscopy, and TEM (Figure 8, G, I, K, and M). However, we also observed the presence of microvillus inclusions in these cells by immunofluorescence and TEM (Figure 8, I and O). It should be noted that the microvillus inclusions that formed were consistent with the size seen in samples from patients with MVID (Figure 8, O and P). Reexpression of mCherry-MYO5B-QL/YC in MYO5B-KD cells elicited incomplete rescue of microvilli, but no microvillus inclusions formed (Figure 8, G, J, L, and N). Quantitation of microvillus inclusions in MYO5B-QL/YC– and MYO5B-YE/QR–rescued cells showed a significant increase in microvillus inclusions in the MYO5B-YE/QR–expressing cells compared with that in MYO5B-QL/YC–expressing cells (Figure 8H).
The expression of mCherry-MYO5B-YE/QR in MYO5B-KD cells reestablished the normal pattern for RAB8A, but not RAB11A, distribution (Supplemental Figure 13B). In contrast, the expression of mCherry-MYO5B-QL/YC in MYO5B-KD cells did not elicit the normal RAB8A distribution but was able to reestablish the normal distribution of RAB11A (Supplemental Figure 13B). Interestingly, mCherry-MYO5B-QL/YC was able to reestablish the proper distribution of both ezrin and DPPIV, while mCherry-MYO5B-YE/QR did not rescue either (data not shown). Thus, establishment of apical microvilli appeared to involve interactions of MYO5B with RAB8A, while microvillus inclusion formation was associated with a loss of RAB11A interactions with MYO5B.
Microvillus inclusions arise from internalization of the apical surface.
Microvillus inclusions could arise when either endocytosis or exocytosis is altered (30). Since microvillus inclusions do not contain acid phosphatase, a lysosomal marker, it was postulated that microvillus inclusions do not originate from autophagocytosis of the apical membrane (46). Previous investigations using organ culture of duodenal biopsies from patients with MVID incubated for 5 minutes in PBS containing cationized ferritin suggested that microvillus inclusions originate from engulfing brush border membrane (22). We postulated that, because of the large size of the microvillus inclusions, only macropinocytosis could facilitate such a large endocytic event. Macropinocytosis is an actin-based endocytic process, in which the plasma membrane is internalized to form large endocytic vesicles, >0.2 μm in diameter, designated macropinosomes (47). In MYO5B-YE/QR–reexpressing CaCo2-BBE cells, we observed microvillus inclusions forming in a series of stages. First, we observed cells with apical membrane invaginations into the cytoplasm (Figure 9A). Other cells showed invaginations with increased size as well as internalized inclusions in which the apical membrane was completely engulfed and separated from the apical surface (Figure 9, B and C). To determine the origin of these inclusions, we biotinylated the apical surface of the mCherry-MYO5B-YE/QR–expressing CaCo2-BBE MYO5B-KD cells, and we fixed and stained cells with phalloidin and fluorescent streptavidin after 24 hours (Figure 9, D–F). We observed the presence of fluorescent streptavidin in the microvillus inclusions in CaCo2-BBE cells expressing mCherry-MYO5B-YE/QR (Figure 9, D–F). Similar results were observed in cells expressing mCherry-MYO5B-P660L (data not shown). To examine whether microvillus inclusions in CaCo2-BBE cells represented products of macropinocytosis, we stained mCherry-MYO5B-YE/QR–expressing MYO5B-KD cells with phalloidin and antibodies against sorting nexin 18 (SNX18), a marker of early macropinosome formation (Figure 9, G–I, and ref. 47). SNX18 and phalloidin double labeling of these cells revealed that early stages of microvillus inclusion formation were marked by SNX18 staining, while more complete microvillus inclusions lost their SNX18 staining as they matured (Figure 9, G–I). Interestingly, some microvillus inclusions were detected in samples from patients with MVID triple labeled for SNX18, MYO5B, and DPPIV (Figure 10). As a whole, these results provide evidence that microvillus inclusions arise from macropinocytosis of the apical surface.
Figure 9. Microvillus inclusions in CaCo2-BBE cells arise from internalization of the apical surface.

x-y confocal images of MYO5B-YE/QR–expressing MYO5B-KD cells are shown above z-axis reconstructions. (A–C) CaCo2-BBE cells were stained with phalloidin (green). Images in A and B use corresponding x-y and different z-axis reconstructions from the same field of view. The z-axis reconstruction in A shows the development of an apical invagination, whereas that in B shows a nearly completed microvillus inclusion. (C) A separate field of view in which the z-axis reconstruction demonstrates a completely internalized microvillus inclusion. (D–F) The apical surface of CaCo2-BBE cells was biotinylated and fixed after 24 hours. These cells were then stained with phalloidin (green) and fluorescent streptavidin (red). Single asterisks indicate the position for the first z-axis reconstruction directly below the x-y confocal image. Double asterisks indicate the position for the second z-axis reconstruction directly below the first z-axis reconstruction. Fluorescent streptavidin was observed in microvillus inclusions inside the cells. (G–I) CaCo2-BBE cells from A and B double labeled with both phalloidin and SNX18 showed SNX18 localization at the bottom of a forming microvillus inclusion (white arrows), while more mature microvillus inclusions showed dispersal of SNX18 (red arrows). Scale bar: 10 μm.
Figure 10. Immunostaining of MVID patient duodenum shows early microvillus inclusions labeled with SNX18, MYO5B, and DPPIV.
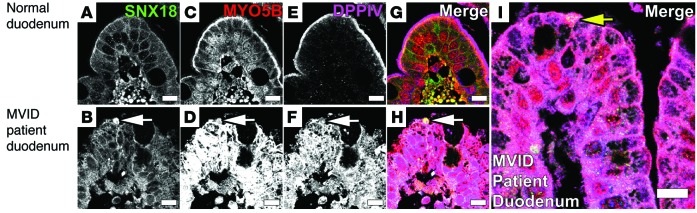
(A) Normal duodenum section stained for SNX18 showed subapical and lateral localization of SNX18. (B) Staining of MVID patient duodenum for SNX18 showed localization of SNX18 to early microvillus inclusion (white arrow). (C) Normal duodenum stained for MYO5B showed subapical localization of MYO5B. (D) Staining of MVID patient duodenum for MYO5B showed dispersal from the subapical region and accumulation and increase of MYO5B staining throughout the cytoplasm and localization of MYO5B to an early microvillus inclusion (white arrow). (E) Normal duodenum stained for DPPIV showed apical localization of DPPIV. (F) Staining of MVID patient duodenum for DPPIV showed localization of DPPIV to an early microvillus inclusion (white arrow). (G and H) Merged images of normal duodenum and samples from patients with MVID stained with SNX18, MYO5B, and DPPIV from A–F. (I) Merged image of MVID patient duodenum showed an early microvillus inclusion formation (yellow arrow) staining for SNX18, MYO5B, and DPPIV. Scale bar: 10 μm.
Discussion
In this study, we developed a cellular model of MVID using shRNA to stably knockdown MYO5B and then reexpressed wild-type MYO5B and MYO5B mutants in CaCo2-BBE cells. Using this model, we demonstrated that loss of MYO5B caused a decrease in apical microvilli and dispersal of RAB8A and RAB11A from the subapical surface. RAB8A and RAB11 family members act on recycling endosomes and are involved in the establishment of a variety of specialized apical processes that depend on apical trafficking. RAB8A and RAB11A interactions with MYO5B are required for proper establishment of apical polarity in MDCK cells, and phosphorylation of the RAB11 effector RAB11-FIP2 by MARK2/PAR1B also regulates the establishment of polarity and the proper assembly of junctional proteins (27, 48). Other studies have suggested that RAB8A and RAB11A are involved in the assembly and maintenance of apical primary cilia in kidney cells (49–51). However, enterocytes do not form primary cilia above the transition zone, and, in contrast, the mechanisms behind microvilli formation have not been well characterized. This study supports the concept that MYO5B functions as a dynamic tether for both RAB8A and RAB11A and normally maintains these proteins at their appropriate subapical membrane localization. The present investigation has also shown that uncoupling of MYO5B from RAB8A promotes loss of microvilli, since a MYO5B mutant that can bind RAB8A, but not RAB11A, reestablished microvilli in CaCo2-BBE MYO5B-KD cells. However, the RAB8A-binding mutant of MYO5B also induced microvillus inclusions. Thus, formation of microvillus inclusions resulted from a loss of interactions between RAB11A and MYO5B. Similar to expression of MYO5B-YE/QR, reexpression of MYO5B-P660L, the mutation found in the Navajo population, led to the formation of microvillus inclusions and exacerbation of the MYO5B-KD phenotype. Thus, MYO5B, in concert with RAB8A and RAB11A, regulates the polarity of intestinal epithelial cells, and uncoupling of these RABs from MYO5B causes gross changes in cellular polarity and mucosal integrity in patients with MVID. These results when paired with previous work demonstrate that MYO5B interacts with RAB8A and RAB11A to regulate enterocyte polarity, apical trafficking, and microvilli growth (Supplemental Figure 14). Both RAB8A and RAB11A bind to MYO5B directly on recycling vesicles (27). RAB11A also regulates actin dynamics in a MYO5B-dependent manner, and we have now implicated RAB11A in the recycling of apical brush border contents from macropinosomes (52). RAB8A also plays a role in apical recycling and trafficking. CDC42 associates with both RAB8A- and RAB11A-positive vesicles and is also activated via RAB8A through TUBA to facilitate regulation of cellular polarity and actin polymerization (36). All of these processes aid in the formation and maintenance of microvilli.
The pathognomonic characteristic of MVID is the presence of intracellular microvillus inclusions, although they are observed in only 10% of enterocytes from patients with MVID, mainly at the villus tips. Interestingly, knockdown of MYO5B expression in CaCo2-BBE cells did not induce microvillus inclusions. A previous investigation reported that transient knockdown of MYO5B using siRNA in CaCo2 cells caused an increase in microvillus inclusions (4). As this manuscript was being submitted, another group found structures reminiscent of microvillus inclusions with stable knockdown of MYO5B in parental CaCo2 cells (10). The differences observed in our studies likely accrue from our use of the highly polarized CaCo2-BBE cell line. Parental CaCo2 cell lines obtained from ATCC often show variable polarity. Nevertheless, reexpression of either MYO5B-YE/QR or MYO5B-P660L in CaCo2-BBE MYO5B-KD cells induced microvillus inclusions, which formed from internalization of segments of the apical membrane. Whether apical membrane macropinocytosis is a normal cellular process in polarized intestinal epithelial cells remains unclear, but colchicine treatment causes microvillar inclusions to form in normal rat small intestine (53). In CaCo2 cells, colchicine or nocodazole treatment can also elicit microvillus inclusion formation (30). Thus, the disruption of normal microtubule-dependent trafficking may cause microvillus inclusions to form. Since the normal apical recycling process is dependent on RAB11A and microtubule-dependent pathways, the microvillus inclusions may reflect the effects of mutant MYO5B molecules on the process of recycling. Thus, the nonfunctional MYO5B mutants may not cause the formation of microvillus inclusions but rather induce the phenotype by the inability of the cell to recycle internalized apical membrane through the RAB11A-dependent recycling system.
Currently, it is not understood why MVID mutations lead to an enterocyte-specific effect as opposed to more global effects, since other highly polarized epithelial tissues appear grossly unaffected by this disease. In addition to the chronic unremitting diarrhea, patients with MVID can also develop extraintestinal liver abnormalities, but it remains uncertain whether liver complications accrue from long-term total parenteral nutrition administration or the disease. In patients with MVID, kidney function generally appears normal, but these patients do have an increased rate of kidney stone development (54). In vertebrates, there are 3 isoforms of myosin V: MYO5A, MYO5B, and MYO5C (55). All 3 of these isoforms can interact with RAB10 and RAB8A, but only MYO5A and MYO5B can bind to RAB11 family members (17). One possible explanation for the severity of the intestinal phenotype in patients with MVID is that MYO5A may compensate for the loss of MYO5B in other polarized cell types and that low levels of MYO5A in enterocytes make enterocytes more vulnerable to loss or mutation of MYO5B (17). To address this possibility, we have attempted to rescue the MYO5B-KD of CaCo2-BBE cells with mCherry-MYO5A-WT, but reexpression of MYO5A was unable to rescue the MYO5B-KD phenotype in CaCo2-BBE cells (data not shown). It may be more likely that the mechanisms for establishing apical microvillar specializations differ in different polarized epithelial cells. Increasing evidence suggests that important parts of the trafficking machinery in one cell type might serve another function in others (56). Furthermore, it should be noted that even if mutant MYO5B were affecting primary cilia function in kidney cells, few patients with MVID have reached the age at which polycystic disease from ciliopathy would be detected.
The question remains why infants with MVID manifest such prominent secretory diarrhea. Loss of apical microvilli would be predicted to yield an absorptive diarrhea, and loss of intestinal villi in other intestinal diseases, such as celiac disease, usually occurs without secretory diarrhea (57). The results presented here in both samples from Navajo patients with MVID and in CaCo2-BBE cells indicate that loss of functional MYO5B causes not only a deficit in microvilli, but also a global loss in polarity, which leads to alterations in both intercellular junctions and ion transport pathways necessary for adaptation through transcellular pumps and channels. The results of these changes lead to a mucosa with decrements in absorptive pathways and a leaky epithelium at the villus tips. This scenario would account for the observed secretory diarrhea. Finally, it is important to note that we observed in the duodenal biopsies from Navajo patients with MVID that normal ezrin-containing brush borders were often present on the enterocytes in the proximal villus. Similar results have been reported recently in sporadic cases of MVID (10). The loss of apical brush border and formation of microvillus inclusions occurred in cells at the villus tips. Thus, it seems likely that the enterocyte microvilli can be formed initially and that stabilization of initial cell polarity may represent a future target for therapy in infants with MVID.
Methods
Cell lines, real-time PCR analysis, lentivirus-mediated MYO5B-KD, and rescue.
Human colonic epithelial CaCo2-BBE and human embryonic kidney 293 (HEK) cell lines were grown in DMEM. The mouse melanoma BF16 cells were grown in RPMI (Cellgro). All cell media was supplemented with L-glutamine (Cellgro), nonessential amino acids (Cellgro), penicillin/streptomycin (Cellgro), 10% fetal bovine serum (Hyclone), and glucose for RPMI. Cell culture dishes and Transwell filters were purchased from Costar. For CaCo2-BBE cells, 200,000 cells were plated and grown on 12-well Transwell filters for 15 days in culture, unless otherwise stated, and media was changed daily through the culture duration.
Two lentiviral shRNA vectors targeting MYO5B (Open Biosystems, V3LHS_323980 and V3LHS_323981) and a control shRNA (Open Biosystems, RHS4346) were used for transduction of Caco2-BBE cells. The CaCo2-BBE cells were transduced with lentiviral media produced in HEK cells and selected using puromycin (Cellgro). Rescue of CaCo2-BBE MYO5B-KD was performed using vectors that were synthesized from optimized MYO5B sequences (GeneArt) resistant to silencing by the above shRNAs, cloned into mCherry-C2 at the N terminus, and selected using both puromycin (shRNA vector) and G418 (mCherry vector) selection.
RNA was isolated from CaCo2-BBE control and MYO5B-KD cell lines using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions and treated with RQ1 RNase-Free DNase (Promega). cDNA was synthesized using a High-Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems) with both oligo-dT and random hexamer primers. Real-time PCR was performed using a StepOnePlus Real-Time PCR system with Express SYBR Green ER Supermix (Applied Biosystems) and the following primer pairs: MYO5B-forward 5′-CAGCAAGAAAGTCCAGGCGG-3′, MYO5B-reverse 5′-CCACGGTTTCCTCAGCTCA-3′, GAPDH-forward 5′-AGATCCCTCCAAAATCAAGTGG-3′, and GAPDH-reverse 5′-GGCAGAGATGATGACCCTTTT- 3′. These primers were validated for efficiency and melting temperature, and the results were analyzed by comparative CT method.
Human tissue preparation.
Samples from patients with MVID were obtained via endoscopic biopsy for 4 of the samples we used and via resection for 1 additional sample, for a total of 5 samples from patients with MVID. All tissue samples were obtained through protocols approved by the Phoenix Children’s Hospital IRB. All biopsy and resection samples were washed in PBS and fixed in 10% formalin. Samples were stored in 70% ethanol, embedded, and sectioned for immunostaining.
Western blot.
The CaCo2-BBE MYO5B-KD and CaCo2-BBE control cell lines were plated on 6-well Transwell filters for 15 days, washed with PBS, and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) for 10 minutes at 4°C. The lysates were sonicated for 25 seconds and centrifuged at 16,000 g for 10 minutes. The protein concentration was determined by the bicinchoninic acid (BCA) method using the Pierce BCA protein assay reagent (Pierce). A 20 μg total of protein was suspended in 1× SDS sample buffer, heated for 10 minutes at 70°C, resolved on a 6% to 12% SDS-PAGE gel, and transferred to Protran Nitrocellulose Transfer Membrane (Whatman). The blots were blocked in 5% DMP/TBS-T (5% dry milk powder in Tris-buffered saline, 0.01% Tween-20). The blots were incubated for 1 hour at room temperature or overnight at 4°C with primary antibody diluted in 1% DMP/TBS-T. The blots were washed 5 times for 10 minutes at room temperature with TBS-T (0.1% Tween-20) and incubated for 1 hour at room temperature with horseradish peroxidase–conjugated secondary antibodies (Jackson ImmunoResearch) and washed 5 times for 10 minutes at room temperature with TBS-T (0.1% Tween-20), followed by 1 wash with TBS. Specific tagging was identified by chemiluminescence reagent (Pierce), with detection using BioMax ML film (Kodak). Western blot quantitation was performed using scanned tiffs placed into Image Studio (LI-COR Biosciences), which was used to determine the signal. Using Excel, the normalized signal was determined relative to α-tubulin or β-actin. Significance was determined using the Mann-Whitney 1-tailed test, and these measurements were graphed using Prism GraphPad. All of these experiments were performed in triplicate for each protein of interest.
Confocal immunofluorescence and SIM.
The CaCo2-BBE MYO5B-KD and CaCo2-BBE control cell lines were plated on 12-well Transwell filters and cultured for 15 days. Cells were washed with PBS and fixed. For tight and adherens junction staining, cells were fixed with 100% methanol for 5 minutes at –20°C. For all other staining, the cells were fixed in 4% PFA for 15 minutes at room temperature. These cells were washed 3 times with PBS and blocked/extracted for 30 minutes at room temperature in 10% normal donkey serum (Jackson ImmunoResearch) and 0.3% Triton X-100 in PBS. Cells were incubated overnight at 4°C with primary antibodies diluted in 1% normal donkey serum and 0.01% Tween-20 in PBS. Cells were washed 3 times for 15 minutes at room temperature with 0.01% Tween-20 in PBS (PBS-T). Appropriate secondary antibodies were conjugated for immunofluorescence with Alexa Fluor 488, Alexa Fluor 568, Alexa Fluor 647, Cy3, or Cy5 (1-hour incubation at room temperature). Detailed information on primary antibodies and their dilutions is provided in Supplemental Table 1. The cells were washed 3 times in PBS-T and washed once in PBS and then washed in water and mounted with ProLong Gold plus DAPI (Invitrogen).
All images were captured with an Olympus FV-1000 confocal microscope (Olympus), using a ×60 oil immersion objective with a numerical aperture of 1.42 and using the microscope’s software at a ×3 optical zoom. The individual images were converted to tiff files with the FV-1000, and Photoshop (Adobe) or ImageJ was used to create the final figures.
Samples for SIM were prepared as for confocal immunofluorescence (see above). SIM imaging was performed on a DeltaVision OMX microscope (Applied Precision Inc.) using 488 nm, 568 nm, and 642 nm lasers. Reconstruction and alignment of SIM images was performed using softWoRx version 5.0 (Applied Precision Inc.). These corrections were applied back into the acquired images, and Photoshop (Adobe) was used to produce the final images.
TEER measurement.
The cell lines were plated on 12-well Transwell filters, and media was changed every day. The TEER was measured using a Millicell-ERS (Millipore) for each Transwell filter, the numbers were recorded, and the results are expressed in Ω × cm2. All of these experiments were done in triplicate for each time point of interest. Significance was determined using the Mann-Whitney 1-tailed test, and these measurements were graphed using Prism GraphPad.
Scanning electron microscopy.
CaCo2-BBE cell lines were washed 3 times with PBS. The cells were fixed with 3% glutaraldehyde in scanning electron microscopy buffer (0.1 M sodium phosphate buffer, pH 7.4, 0.1 M sucrose) at 4°C overnight. Cells were washed with scanning electron microscopy buffer twice, treated with 1% osmium tetroxide (in scanning electron microscopy buffer) for 1 hour on ice, and washed 3 times with scanning electron microscopy buffer. The cells were dehydrated with a serial dilution of ethanol (35%, 50%, 70%, 95%, and 100%) for 15 minutes each and incubated with hexamethyldisilazane (HMDS). After the HMDS treatment, samples were mounted on stubs and coated with gold in a sputter coater and analyzed using an FEI Quanta 250 scanning electron microscopy (Vanderbilt Cell Imaging Shared Resource, Vanderbilt University).
Immunofluorescence quantitation.
All images were captured with an Olympus FV-1000 confocal microscope (Olympus) using a ×60 oil immersion objective, as detailed above. The Z-stacks were exported as individual images and converted to tiff files using FV-1000 software. These image sequences were opened in ImageJ, and the fluorescence mean intensity was determined for each Z-stack (maximum intensity projections), placed in Excel, and means, standard deviation, and standard error were calculated. Significance was determined using the Mann-Whitney 1-tailed test, and these measurements were graphed using Prism GraphPad. All of these experiments were done in triplicate for each selected protein of interest.
Histology and TEM.
Normal human duodenum sections and those from patients with MVID were deparaffinized and were submitted to antigen retrieval in a pressure cooker using the target retrieval solution (Dako North America Inc.). Serum-free protein block (Dako North America Inc.) was used for blocking the duodenum, and immunofluorescence staining was performed as stated above for confocal immunofluorescence for all proteins of interest except for RAB8A and RAB11A. Duodenum sections stained for both RAB8A and RAB11A were placed in serum-free block for 8 hours at room temperature or for 48 hours at 4°C. All other aspects of immunofluorescence staining were performed as stated above. All imaging was performed using the Olympus FV-1000. TEM samples from patients with MVID were prepared and analyzed by the Vanderbilt Cell Imaging Shared Resource, Vanderbilt University.
Transferrin trafficking assay.
CaCo2-BBE cells were serum starved for 60 minutes in serum-free media at 37°C. Transferrin–Alexa Fluor 633 (Molecular Probes) was added to the basolateral surface for 1 hour at 37°C. The transferrin was chased out with normal media for 60 minutes, and cells were washed 2 times in PBS and fixed and prepared for imaging as described above.
CDC42 activity assay.
CDC42 activity was assayed using the CDC42 activity assay kit (BK034, Cytoskeleton). Cells were plated on 6-well Transwell filters for 15 days, as described above for Western blot protein isolation. Protein isolation and CDC42 activity were performed as described in the manufacturer’s data sheet (BK034, Cytoskeleton).
F-actin pull down of MYO5B.
0.4 mg/ml actin was polymerized in actin polymerization buffer (500 mM KCl and 20 mM MgCl2) and 2 mM ATP and 1 mM DTT were added to the mixture and incubated for 1 hour at room temperature. The polymerized F-actin was stabilized using 6.6 μM phalloidin (Molecular Probes) and incubated for 20 minutes at room temperature. HEK cells transfected with either 3x-mCitrine-MYO5B-WT-1016x or 3x-mCitrine-MYO5B-P660L-1016x were lysed and spun down at 150,000 g. 1 μM actin was added to the cell lysate supernatant and incubated for 1 hour. The lysates were spun down at 150,000 g, and samples from the supernatant and actin pellet were collected and analyzed by Western blot using an affinity-purified chicken anti-MYO5B antibody (39).
Live cell single molecule total internal reflection fluorescence–fluorescence recovery after photobleaching microscopy.
MYO5B-WT-1016x and MYO5B-P660L-1016x were tagged with 3x-mCitrine and verified by sequencing. BF16 cells expressing 3x-mCitrine-MYO5B-WT-1016x or 3x-mCitrine-MYO5B-P660L-1016x were plated sparsely in glass bottom imaging dishes. Cells were gently washed twice in PBS and placed in CO2-independent media (Gibco) for imaging experiments. Total internal reflection fluorescence–fluorescence recovery after photobleaching microscopy (TIRF-FRAP) and TIRF microscopy was performed on a Nikon TiE inverted light microscope equipped with a Nikon TIRF illuminator, a 100×/1.49 numerical aperture TIRF objective (used in combination with a 1.5× optivar), and a Hamamatsu ImagEM-CCD camera. Rectified pixel size was 75 nm per pixel. Fluorescence was excited using a 50-milliwatt 491-nm diode laser working between 20% to 30% of full power. Single particle events appeared as bright diffraction restricted spots on the surface of the cell. Cells expressing exceedingly high levels of each construct were chosen for analysis to improve the signal/noise ratio of single molecule detection; image stacks consisting of 500 frames (30 seconds) were captured at 16.667 frames per second and subjected to contrast enhancement using ImageJ. Using ImageJ, fluorescence maximum intensity for each video was measured, placed in Excel, normalized to 1, and graphed as a percentage. All of these experiments were done in triplicate for each construct.
Microvillus inclusion scoring.
Microvillus inclusions in the cell lines presented in this study were measured by assembling 3D reconstructions of Z-stacks from 8 randomly chosen fields of view of each cell line stained with DPPIV, ezrin, or phalloidin. The absolute number of microvillus inclusions in each 3D reconstruction was counted. Significance was determined using the Mann-Whitney 1-tailed test, and these measurements were graphed using Prism GraphPad.
Biotin labeling and cell surface biotinylation.
For biotin labeling, CaCo2-BBE cell lines on Transwell filters were washed 3 times with PBS containing 0.1 mM CaCl2 and 1.0 mM MgCl2 (PBS-CM) on ice. Biotin working solution (0.5 mg/ml in PBS-CM from a 200 mg/ml stock dissolved in anhydrous DMSO) was added to the apical side of the filters for 20 minutes at 4°C. The biotin was quenched with 5 washes of PBS-CM containing 0.2% BSA and 100 mM glycine at 4°C. The cells were incubated for 24 hours at 37°C. CaCo2-BBE cells were fixed with PFA as described above. Streptavidin 647 (Molecular Probes) and Phalloidin 405 (PromoKine) were added in PBS-T. The cells were washed 3 times in PBS-T, washed once in PBS, and washed in water and mounted with ProLong Gold (Invitrogen).
For cell surface biotinylation assays, CaCo2-BBE cell lines on Transwell filters were washed as specified above, and biotin (0.5 mg/ml in PBS-CM from a 200 mg/ml stock dissolved in anhydrous DMSO) was added to the apical or basolateral side of the filters for 20 minutes at 4°C. This process was subsequently repeated for an additional 20 minutes at 4°C. The biotin was quenched with 5 washes of PBS-CM containing 0.2% BSA and 100 mM glycine and washed 2 times with PBS-CM at 4°C. The Transwell filters were cut out of the Transwell inserts and placed in Eppendorf tubes containing 800 μl lysis buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10% glycerol, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate, 10 mM sodium fluoride, 1 mM PMSF, 5 μg/ml leupeptin, 5 μg/ml pepstatin, and 10 μg/ml aprotinin added fresh) (58). The tubes were gently rotated for 1 hour at 4 °C, filters were removed from the tubes, and lysates were processed for biotinylated protein isolation on streptavidin beads. Protein concentration was determined as described above, and 800 μg total protein was resuspended in isolation dilution buffer (200 mM HEPES [pH 7.5], 600 mM NaCl, 40% glycerol, and 0.4% Triton X-100) brought up to 700 μl with mammalian protease inhibitors (Sigma-Aldrich). 50 μl of the total protein was obtained and dissociated in 1× SDS sample buffer and heated for 10 minutes at 70°C. Streptavidin agarose beads (Novagen 69203) were added to the above lysates and incubated overnight at 4 °C while gently rotating. The samples were centrifuged at 1,600 g for 3 minutes, and 50 μl of the flow through was obtained and dissociated in 1× SDS sample buffer and heated for 10 minutes at 70°C. The beads were washed 3 times with 500 μl of the isolation dilution buffer and centrifuged at 1,600 g for 3 minutes each time. The beads were resuspended in 1× SDS sample buffer, heated for 10 minutes at 70°C, and resolved on a 10% SDS-PAGE gel, and Western blots were performed for DPPIV and E-cadherin, as described above. Band densities were quantified using digitally scanned tiffs placed into Image Studio (LI-COR Biosciences). Using Excel, the percentage of total protein in the unbound and streptavidin-bound pools was determined by adding the relative unbound pools to the streptavidin-bound protein pools.
Statistics.
Reported values represent the mean ± SEM of 3 or more independent experiments, unless otherwise indicated. Statistical significance for all experiments was determined using a Mann-Whitney 1-tailed test with significance at P ≤ 0.05 using Prism GraphPad.
Study approval.
The studies in these investigations used archival paraffin tissue duodenal specimens from healthy children and patients with MVID. All procedures and studies were performed according to protocols approved by IRBs at both Phoenix Children’s Hospital and Vanderbilt University School of Medicine. IRB-approved informed consent was obtained from all families of living subjects prior to research, and IRBs approved decedent research on tissue blocks from patients who had died.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant RO1 DK70856 to J.R. Goldenring and Research Allocation Committee (RAC) Awards to M. Shub from Phoenix Children’s Hospital for initial support of this project. Confocal and structured illumination fluorescence microscopy, TEM, and scanning electron microscopy imaging were performed through the use of the Vanderbilt University Medical Center Cell Imaging Shared Resource and histological sectioning was performed by Translational Pathology Shared Resource, both supported by NIH grants CA68485, DK20593, DK58404, and HD15052. Fluorescence slide imaging was performed on an Ariol SL-50 digitizing scanner in the Vanderbilt University Medical Center Digital Histology Shared Resource. We thank Cathy Caldwell for help with cell culture, Kenya Avant-Mitchell for help with Western blots, Z. Bradshaw and Y. Tu for help with actin pull downs of MYO5B constructs, J. Higginbotham for assistance with CaCo2-BBE cell sorting, J. Mazerik for help with TIRF imaging, R. Nambiar and J. Williams for help with scanning electron microscopy and TEM imaging, A. Reynolds and N. Markham for p120 antibody, F. Ghishan for NHE3 antibodies, I. Macara and R. Guyer for aPKC antibody, and the Vanderbilt University School of Medicine Medical Scientist Training Program for continued support. We thank G. Silber, K. Ingebo, and D. Ursea for the outstanding care that they have provided over the years for the Navajo patients with MVID. We thank all of the families of our Navajo patients, who consented to allow archived tissue samples from their children to be used in this study.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(7):2947–2962. doi:10.1172/JCI71651.
References
- 1.Muller T, et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet. 2008;40(10):1163–1165. doi: 10.1038/ng.225. [DOI] [PubMed] [Google Scholar]
- 2.Erickson RP, Larson-Thome K, Valenzuela RK, Whitaker SE, Shub MD. Navajo microvillous inclusion disease is due to a mutation in MYO5B. Am J Med Genet A. 2008;146A(24):3117–3119. doi: 10.1002/ajmg.a.32605. [DOI] [PubMed] [Google Scholar]
- 3.Pohl JF, et al. A cluster of microvillous inclusion disease in the Navajo population. J Pediatr. 1999;134(1):103–106. doi: 10.1016/S0022-3476(99)70380-X. [DOI] [PubMed] [Google Scholar]
- 4.Ruemmele FM, et al. Loss-of-function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo-2 RNAi cell model. Hum Mutat. 2010;31(5):544–551. doi: 10.1002/humu.21224. [DOI] [PubMed] [Google Scholar]
- 5.Chen CP, et al. Microvillus inclusion disease: prenatal ultrasound findings, molecular diagnosis and genetic counseling of congenital diarrhea. Taiwan J Obstet Gynecol. 2010;49(4):487–494. doi: 10.1016/S1028-4559(10)60102-7. [DOI] [PubMed] [Google Scholar]
- 6.Szperl AM, et al. Functional characterization of mutations in the myosin Vb gene associated with microvillus inclusion disease. J Pediatr Gastroenterol Nutr. 2011;52(3):307–313. doi: 10.1097/MPG.0b013e3181eea177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golachowska MR, et al. MYO5B mutations in patients with microvillus inclusion disease presenting with transient renal Fanconi syndrome. J Pediatr Gastroenterol Nutr. 2012;54(4):491–498. doi: 10.1097/MPG.0b013e3182353773. [DOI] [PubMed] [Google Scholar]
- 8.Chiang MC, et al. Bowel “Dissection” in microvillus inclusion disease. Pediatr Neonatol. 2013;pii:S1875-9572(13)00039-9 doi: 10.1016/j.pedneo.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 9.van der Velde KJ, et al. An overview and online registry of microvillus inclusion disease patients and their MYO5B mutations. Hum Mutat. 2013;34(12):1597–1605. doi: 10.1002/humu.22440. [DOI] [PubMed] [Google Scholar]
- 10.Thoeni CE, et al. Microvillus inclusion disease: loss of myosin Vb disrupts intracellular traffic and cell polarity. Traffic. 2014;15(1):22–42. doi: 10.1111/tra.12131. [DOI] [PubMed] [Google Scholar]
- 11.Rief M, Rock RS, Mehta AD, Mooseker MS, Cheney RE, Spudich JA. Myosin-V stepping kinetics: a molecular model for processivity. Proc Natl Acad Sci U S A. 2000;97(17):9482–9486. doi: 10.1073/pnas.97.17.9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehta AD, Rock RS, Rief M, Spudich JA, Mooseker MS, Cheney RE. Myosin-V is a processive actin-based motor. Nature. 1999;400(6744):590–593. doi: 10.1038/23072. [DOI] [PubMed] [Google Scholar]
- 13.De La Cruz EM, Wells AL, Rosenfeld SS, Ostap EM, Sweeney HL. The kinetic mechanism of myosin V. Proc Natl Acad Sci U S A. 1999;96(24):13726–13731. doi: 10.1073/pnas.96.24.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pierobon P, et al. Velocity, processivity, and individual steps of single myosin V molecules in live cells. Biophys J. 2009;96(10):4268–4275. doi: 10.1016/j.bpj.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Provance DW, Addison E, Wood P, Chen D, Silan C, Mercer J. Myosin-Vb functions as a dynamic tether for peripheral endocytic compartments during transferrin trafficking. BMC Cell Biology. 2008;9:44. doi: 10.1186/1471-2121-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schafer JC, Baetz NW, Lapierre LA, McRae RE, Roland JT, Goldenring JR. Rab11-FIP2 interaction with MYO5B regulates movement of Rab11a-containing recycling vesicles. Traffic. 2014;15(3):292–308. doi: 10.1111/tra.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roland JT, Lapierre LA, Goldenring JR. Alternative splicing in class V myosins determines association with Rab10. J Biol Chem. 2009;284(2):1213–1223. doi: 10.1074/jbc.M805957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chehade MS. Infantile food protein-induced enterocolitis syndrome. In: David TJ, ed. Recent Advances in Paediatrics. London, United Kingdom: Royal Society of Medicine Press; 2005:140. [Google Scholar]
- 19.Davidson GP, Cutz E, Hamilton JR, Gall DG. Familial enteropathy: a syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villus atrophy. Gastroenterology. 1978;75(5):783–790. [PubMed] [Google Scholar]
- 20. Arpin MC, Crepaldi T, Louvard D. Cross-talk between apical and basolateral domains of epithelial cells regulates microvillus assembly. In: Birchmeier WB, Birchmeier C, eds. Epithelial Morphogenesis in Development and Disease. Amsterdam, The Netherlands: Harwood Academic; 1999:95–116. [Google Scholar]
- 21.Rhoads JM, et al. Microvillus inclusion disease. In vitro jejunal electrolyte transport. Gastroenterology. 1991;100(3):811–817. doi: 10.1016/0016-5085(91)80031-4. [DOI] [PubMed] [Google Scholar]
- 22.Reinshagen K, Naim HY, Zimmer KP. Autophagocytosis of the apical membrane in microvillus inclusion disease. Gut. 2002;51(4):514–521. doi: 10.1136/gut.51.4.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cutz E, Rhoads JM, Drumm B, Sherman PM, Durie PR, Forstner GG. Microvillus inclusion disease: an inherited defect of brush-border assembly and differentiation. N Engl J Med. 1989;320(10):646–651. doi: 10.1056/NEJM198903093201006. [DOI] [PubMed] [Google Scholar]
- 24.Oliva MM, Perman JA, Saavedra JM, Young-Ramsaran J, Schwarz KB. Successful intestinal transplantation for microvillus inclusion disease. Gastroenterology. 1994;106(3):771–774. doi: 10.1016/0016-5085(94)90714-5. [DOI] [PubMed] [Google Scholar]
- 25.Ameen NA, Salas PJ. Microvillus inclusion disease: a genetic defect affecting apical membrane protein traffic in intestinal epithelium. Traffic. 2000;1(1):76–83. doi: 10.1034/j.1600-0854.2000.010111.x. [DOI] [PubMed] [Google Scholar]
- 26.Groisman GM, Amar M, Livne E. CD10: a valuable tool for the light microscopic diagnosis of microvillous inclusion disease (familial microvillous atrophy). Am J Surg Pathol. 2002;26(7):902–907. doi: 10.1097/00000478-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Roland JT, Bryant DM, Datta A, Itzen A, Mostov KE, Goldenring JR. Rab GTPase-Myo5B complexes control membrane recycling and epithelial polarization. Proc Natl Acad Sci U S A. 2011;108(7):2789–2794. doi: 10.1073/pnas.1010754108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato T, et al. The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature. 2007;448(7151):366–369. doi: 10.1038/nature05929. [DOI] [PubMed] [Google Scholar]
- 29.Peterson MD, Mooseker MS. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci. 1992;102(pt 3):581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- 30.Gilbert T, Rodriguez-Boulan E. Induction of vacuolar apical compartments in the Caco-2 intestinal epithelial cell line. J Cell Sci. 1991;100(pt 3):451–458. doi: 10.1242/jcs.100.3.451. [DOI] [PubMed] [Google Scholar]
- 31.Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol. 2006;68:403–429. doi: 10.1146/annurev.physiol.68.040104.131404. [DOI] [PubMed] [Google Scholar]
- 32.Vandrangi P, Lo DD, Kozaka R, Ozaki N, Carvajal N, Rodgers VG. Electrostatic properties of confluent Caco-2 cell layer correlates to their microvilli growth and determines underlying transcellular flow. Biotechnol Bioeng. 2013;110(10):2742–2748. doi: 10.1002/bit.24939. [DOI] [PubMed] [Google Scholar]
- 33.Yanagisawa M, et al. A p120 catenin isoform switch affects Rho activity, induces tumor cell invasion, and predicts metastatic disease. J Biol Chem. 2008;283(26):18344–18354. doi: 10.1074/jbc.M801192200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukumoto Y, Shintani Y, Reynolds AB, Johnson KR, Wheelock MJ. The regulatory or phosphorylation domain of p120 catenin controls E-cadherin dynamics at the plasma membrane. Exp Cell Res. 2008;314(1):52–67. doi: 10.1016/j.yexcr.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J, Mariner DJ, Thoreson MA, Reynolds AB. Production and characterization of monoclonal antibodies to the catenin p120ctn. Hybridoma. 1998;17(2):175–183. doi: 10.1089/hyb.1998.17.175. [DOI] [PubMed] [Google Scholar]
- 36.Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F, Mostov KE. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol. 2010;12(11):1035–1045. doi: 10.1038/ncb2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakamori R, et al. Cdc42 and Rab8a are critical for intestinal stem cell division, survival, and differentiation in mice. J Clin Invest. 2012;122(3):1052–1065. doi: 10.1172/JCI60282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melendez J, et al. Cdc42 coordinates proliferation, polarity, migration, and differentiation of small intestinal epithelial cells in mice. Gastroenterology. 2013;145(4):808–819. doi: 10.1053/j.gastro.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin Y, Meisen WH, Hao Y, Macara IG. Tuba, a Cdc42 GEF, is required for polarized spindle orientation during epithelial cyst formation. J Cell Biol. 2010;189(4):661–669. doi: 10.1083/jcb.201002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atwood SX, Chabu C, Penkert RR, Doe CQ, Prehoda KE. Cdc42 acts downstream of Bazooka to regulate neuroblast polarity through Par-6 aPKC. J Cell Sci. 2007;120(pt 18):3200–3206. doi: 10.1242/jcs.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai D, McEwen DP, Martens JR, Meyhofer E, Verhey KJ. Single molecule imaging reveals differences in microtubule track selection between Kinesin motors. PLoS Biol. 2009;7(10):e1000216. doi: 10.1371/journal.pbio.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mazerik JN, Tyska MJ. Myosin-1A targets to microvilli using multiple membrane binding motifs in the tail homology 1 (TH1) domain. J Biol Chem. 2012;287(16):13104–13115. doi: 10.1074/jbc.M111.336313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kodera N, Yamamoto D, Ishikawa R, Ando T. Video imaging of walking myosin V by high-speed atomic force microscopy. Nature. 2010;468(7320):72–76. doi: 10.1038/nature09450. [DOI] [PubMed] [Google Scholar]
- 44.Pierobon P, et al. Velocity, processivity, and individual steps of single myosin V molecules in live cells. Biophys J. 2009;96(10):4268–4275. doi: 10.1016/j.bpj.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rafelski SM, Theriot JA. Bacterial shape and ActA distribution affect initiation of Listeria monocytogenes actin-based motility. Biophys J. 2005;89(3):2146–2158. doi: 10.1529/biophysj.105.061168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lake BD. Microvillus inclusion disease: specific diagnostic features shown by alkaline phosphatase histochemistry. J Clin Pathol. 1988;41(8):880–882. doi: 10.1136/jcp.41.8.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang JT, Kerr MC, Karunaratne S, Jeanes A, Yap AS, Teasdale RD. The SNX-PX-BAR family in macropinocytosis: the regulation of macropinosome formation by SNX-PX-BAR proteins. PLoS One. 2010;5(10):e13763. doi: 10.1371/journal.pone.0013763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lapierre LA, et al. Phosphorylation of Rab11-FIP2 regulates polarity in MDCK cells. Mol Biol Cell. 2012;23(12):2302–2318. doi: 10.1091/mbc.E11-08-0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaplan OI, et al. The AP-1 clathrin adaptor facilitates cilium formation and functions with RAB-8 in C. elegans ciliary membrane transport. J Cell Sci. 2010;123(pt 22):3966–3977. doi: 10.1242/jcs.073908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim YS, Chua CE, Tang BL. Rabs and other small GTPases in ciliary transport. Biol Cell. 2011;103(5):209–221. doi: 10.1042/BC20100150. [DOI] [PubMed] [Google Scholar]
- 51.Knodler A, et al. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci U S A. 2010;107(14):6346–6351. doi: 10.1073/pnas.1002401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holubcova Z, Howard G, Schuh M. Vesicles modulate an actin network for asymmetric spindle positioning. Nat Cell Biol. 2013;15(8):937–947. doi: 10.1038/ncb2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Achler C, Filmer D, Merte C, Drenckhahn D. Role of microtubules in polarized delivery of apical membrane proteins to the brush border of the intestinal epithelium. J Cell Biol. 1989;109(1):179–189. doi: 10.1083/jcb.109.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halac U, et al. Microvillous inclusion disease: how to improve the prognosis of a severe congenital enterocyte disorder. J Pediatr Gastroenterol Nutr. 2011;52(4):460–465. doi: 10.1097/MPG.0b013e3181fb4559. [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez OC, Cheney RE. Human myosin-Vc is a novel class V myosin expressed in epithelial cells. J Cell Sci. 2002;115(pt 5):991–1004. doi: 10.1242/jcs.115.5.991. [DOI] [PubMed] [Google Scholar]
- 56.Weisz OA, Rodriguez-Boulan E. Apical trafficking in epithelial cells: signals, clusters and motors. J Cell Sci. 2009;122(pt 23):4253–4266. doi: 10.1242/jcs.032615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guarino A, Lo Vecchio A, Berni Canani R. Chronic diarrhoea in children. Best Pract Res Clin Gastroenterol. 2012;26(5):649–661. doi: 10.1016/j.bpg.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 58.Singh B, Bogatcheva G, Washington MK, Coffey RJ. Transformation of polarized epithelial cells by apical mistrafficking of epiregulin. Proc Natl Acad Sci U S A. 2013;110(22):8960–8965. doi: 10.1073/pnas.1305508110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.