

Abstract
α–Intercalated cells (A-ICs) within the collecting duct of the kidney are critical for acid-base homeostasis. Here, we have shown that A-ICs also serve as both sentinels and effectors in the defense against urinary infections. In a murine urinary tract infection model, A-ICs bound uropathogenic E. coli and responded by acidifying the urine and secreting the bacteriostatic protein lipocalin 2 (LCN2; also known as NGAL). A-IC–dependent LCN2 secretion required TLR4, as mice expressing an LPS-insensitive form of TLR4 expressed reduced levels of LCN2. The presence of LCN2 in urine was both necessary and sufficient to control the urinary tract infection through iron sequestration, even in the harsh condition of urine acidification. In mice lacking A-ICs, both urinary LCN2 and urinary acidification were reduced, and consequently bacterial clearance was limited. Together these results indicate that A-ICs, which are known to regulate acid-base metabolism, are also critical for urinary defense against pathogenic bacteria. They respond to both cystitis and pyelonephritis by delivering bacteriostatic chemical agents to the lower urinary system.
Introduction
Urinary tract infections (UTIs) are among the most prevalent and resource-taxing diseases in the United States. Each year, 13.3% of women, 2.3% of men (1), and 3.4% of children (2) are infected, most commonly with Gram– uropathogenic E. coli (UPEC) (3). 2 billion dollars per year are spent caring for infected patients (4). It is well known that the kidney constitutively synthesizes a number of antimicrobial peptides with broad specificity, for example RNAse7 (5) and the cathelicidins (6), but less is known about the kidney’s acute responses to UTIs. We found that the bacteriostatic protein lipocalin 2 (LCN2; also known as NGAL, 24p3, and SCN) was secreted in response to Gram– UTIs in both human cohorts and in a variety of mouse models. LCN2 was expressed by specialized cells located in distal segments of the nephron.
Lcn2 expression was first observed in embryonic and adult collecting duct (CD) cells (7, 8), but its message was expressed at low levels. However, Lcn2 message was intensively upregulated (10- to 1,000-fold increase; refs. 8, 9), and its protein appeared in the urine and in the serum (5- to 500-fold increase) within hours of stimuli that typically cause acute kidney injury (AKI). Inductive stimuli included sepsis (10–14), ischemia-reperfusion injury (IR) (9, 15–27), obstructive uropathy (28), nephrotoxins (15, 29–33), trauma-rhabdomyolysis (34), kidney graft injury (35, 36), or a mixture of these maladies (37, 38). Sepsis was a particularly strong stimulus for LCN2 expression. To model our findings, we created a bioluminescent-fluorescent LCN2-Luc2-mCherry reporter mouse to continuously monitor LCN2 expression in real time in vivo (15). IR of the kidney induced bioluminescent LCN2 reporter expression in the kidney medulla in proportion to the intensity of the stimulus and identical in timing with the appearance and disappearance of urinary LCN2 protein (uLCN2) over 2 days. In situ hybridization revealed that LCN2 was expressed after IR (15) by the thick ascending limbs (TALH) and by A-ICs, but not by B-ICs or principal cells of the CDs. Similar data were obtained using different models of sepsis. In all of these cases, A-ICs consistently and prominently expressed LCN2, whereas the contribution of the TALH was variable. These findings raised questions with respect to the role of the A-IC in different acute injuries. They also questioned the function of LCN2 in the urinary system.
LCN2 is a member of a broad group of proteins known as the lipocalins. Structurally, all lipocalins have a calyx created by an 8-stranded antiparallel β-sheet that is closed back on itself to form a β-barrel (39), which binds a variety of ligands. During the cloning of recombinant LCN2, the protein bound a bacterial molecule composed of 3 catechol rings held together by a triserine lactone. The bacterial molecule is known as enterochelin (Ent) (40), a siderophore synthesized by Gram– bacteria. Ent can remove Fe from transferrin with high affinity (Kd, 10–49 M), but in the presence of LCN2, Ent:Fe3+ binds to LCN2 (Kd, 10–10 M), rerouting Ent:Fe3+ for degradation and preventing Fe transfer to bacteria (40, 41). In fact, the LCN2:Ent:Fe3+ complex is stable at pH 4.0, which implies that LCN2 can sequester Ent:Fe3+ in most biological fluids, even acidified urine (8). Fe can only be released from the LCN2:Ent:Fe3+ complex in acidified lysosomes, following the reduction of Fe and the proteolytic cleavage of LCN2 (42). Hence, LCN2 arrests extracellular bacterial growth by chelating Ent:Fe3+, limiting its bioavailability in solution (40, 43, 44). Conversely, Lcn2–/– mice succumb to systemic (peritoneal) infection with Ent-expressing laboratory strains, due to unchecked growth (43, 45), which suggests that chelation of the siderophore is critical for innate defense. Because we have previously observed high levels of LCN2 in the circulation and urine of both human (11, 45) and mouse models of sepsis (including cecal ligation and puncture, which produced a 531-fold increase in kidney Lcn2 mRNA; N. Paragas and J. Barasch, unpublished observations), we speculated that kidney LCN2 suppresses the growth of Ent-producing bacteria, not only in circulation, but also in the urinary system.
The dominant mechanism of bacterial clearance in the urogenital tract includes urinary flow and regular bladder emptying, as well as shedding of infected cells (46). In addition, the urogenital system constitutively expresses antimicrobial proteins (AMPs) that are released into the urine, most strikingly by loop of Henle and CDs (HBD1, refs. 47, 48; THP, refs. 49–52; and lactoferrin, ref. 53), as well as by ICs (RNase7, ref. 5). Some of these molecules can be directly upregulated by urinary infections (e.g., cathelicidin by 3- to 8-fold, ref. 48) or indirectly by cytokines that are stimulated by the urinary infection (e.g., HBD1, ref. 48), while other AMPs are constitutively expressed (54, 55). The AMPs typically disrupt the phospholipid membranes of different types of microorganisms, including both bacteria and fungi, rather than target a specific species (56, 57). RNase7, for example, inhibits Gram– E. coli, Pseudomonas, and Proteus as well as Gram+ Enterococcus and Staphyloccocus (58). Lactoferrin and THP also inhibit both Gram+ and Gram– organisms (59). LCN2 differs from these defense mechanisms not only because of the specificity of its target (Ent siderophores produced by Gram– organisms), but also because it is rapidly upregulated orders of magnitude by both bacterial and aseptic causes of AKI.
Here we provide evidence that the A-IC plays a role in immune defense by secreting LCN2 and H+ in order to suppress urinary tract pathogens and ascending pyelonephritic infections during the course of tissue damage. To test these ideas, we examined human urine samples and created different models of UTI using a variety of genetically modified mice.
Results
LCN2 expression in human and murine UTI.
To identify the expression pattern of uLCN2 in humans with UTI, we analyzed data collected from patients admitted to the hospital from the Emergency Department (60). In this cohort, we found a number of patients with acute UTIs. In infected patients (urinary leukocyte esterase– and urine culture–positive [LE+Cx+]; n = 43) who were selected because they did not have additional renal diseases, we found that supernatant uLCN2 was upregulated on average 10-fold compared with uninfected patients (LE–Cx–; n = 514) (P < 0.001; Figure 1A) (60). Where quantitative bacteriologic data were available in the hospital record, we found that uLCN2 correlated with urinary CFUs (uCFUs; n = 141; Figure 1B) in LE+ patients (>30 wbc per high-powered field). In addition, when clinic patients presented with dysuria, frequency, urgency, and urinary findings including hematuria and LE+ urine (>30 wbc per high-powered field) and were treated with antibiotics, uLCN2 expression was suppressed within 3 days (n = 4; P < 0.01; Figure 1C). These data demonstrated that supernatant uLCN2 was associated in a dose-dependent, reversible fashion with uCFUs, although the wide variation in uLCN2 suggested that a multistep process was required to induce Lcn2 expression.
Figure 1. LCN2 is markedly upregulated in human and mouse UTI.

(A) uLCN2 expression in LE+Cx+ (100 ng/ml; IQR 30–300 ng/ml; n = 43) versus LE–Cx– patients (14.2 ng/ml; IQR 7.4–35.3 ng/ml; n = 514) in an Emergency Department cohort lacking other forms of kidney disease. (B) Relationship between uLCN2 and uCFU in all LE+ Emergency Department patients (LE+ and <104 uCFU [limit of detection], 45 ng/ml uLCN2, IQR 20–150 ng/ml, n = 75; LE+ and 104–105 uCFU, 250 ng/ml, IQR 40–425 ng/ml, n = 21; LE+ and >105 uCFU, 520.2 ng/ml, IQR 236.7–1,126 ng/ml, n = 45). (C) Suppression of uLCN2 by antibiotics (Pre, 600 ng/ml, IQR 533–700 ng/ml; Post, 83.0 ng/ml, IQR 21–129 ng/ml) in patients presenting to clinic with dysuria, frequency, urgency and LE+ (>30 wbc per high-powered field), hematuria, and mucus threads (n = 4). Values represent median and IQR.
To model the UTI, we challenged C57BL/6 mice with small volumes of the UPEC strain CFT073 (20 μl of 5 × 108 CFU/ml; refs. 61–63) by transurethral catheterization (TU) (62, 64). We then serially assayed the urinary supernatant to monitor both uLCN2 and uCFUs longitudinally over the course of 7 days. Similar to our patient data, supernatant uLCN2 grossly mirrored uCFUs at both peak (days 1–3) and resolution (days 4–5) phases of infection (n = 7; Figure 2, A and B). uLCN2 expression levels reached 10 μM, but then declined rapidly. Hence, UPECs acutely and reversibly induced the expression of a graded amount of uLCN2 in mice and in humans.
Figure 2. LCN2 is necessary and sufficient to suppress UTI.

(A) Longitudinally sampled WT C57BL/6 mouse urine (n = 7 per time point) demonstrated faster clearance of Ent+ uCFUs than that of sibling-matched Lcn2–/– mice (n = 6 per time point). *P < 0.05, Mann-Whitney test. (B) Longitudinal measurements of uLCN2 by immunoblot, in the same urine samples as in A, correlated with uCFUs. Lanes for 0–3 days and for 4–7 days were run on different gels, but with the same in-gel LCN2 standards. (C) In contrast, there was no significant difference in uCFUs in Lcn2–/– versus WT mice inoculated with Ent-null UPEC. (D) Bladder CFUs in Lcn2–/– mice (n = 9) exceeded those in WT mice (n = 5) and mirrored the difference in uCFUs at 1 day after infection with Ent+ UPEC. (E) LCN2 (5 μM; n = 3) suppressed the growth of UPEC in urine (pH 5.8). P < 0.0001.
LCN2 is necessary and sufficient to suppress UPECs.
LCN2 acts as a narrow-spectrum antimicrobial by chelating the Ent siderophore (8). To determine its importance in our model of UTI, we deleted murine Lcn2 by BAC recombineering (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI71630DS1). Lcn2loxP/loxP mice were generated and mated with EIIa-Cre mice (65) to generate Lcn2–/–, a global knockout in a C57BL/6 background (see Methods and Supplemental Figure 1). These mice developed normally and were fertile. However, Lcn2–/– mice (n = 6) took a significantly longer time than littermates to resolve a small inoculum of UPEC (20 μl of 5 × 108 CFU/ml); elevated uCFUs were identified in Lcn2–/– mice on days 1–5 after TU (P < 0.05, Figure 2A). In contrast, WT Lcn2+/+ littermates (n = 7) cleared the infection over 4 days, in parallel with their expression of uLCN2 (Figure 2, A and B).
LCN2 suppresses bacterial growth by binding Ent (40). To test whether the LCN2-Ent interaction was essential for the suppression of urinary bacterial growth in vivo, we challenged Lcn2-deleted mice and WT controls with a CFT073 UPEC mutant lacking entF (66). Unlike infection with WT CFT073 UPEC (Figure 2A), we did not find a significant difference in urine, bladder, or kidney cultures 1 day after TU with Ent-null CFT073 UPEC (Figure 2C), which indicated that LCN2 activity depends on the expression of bacterial Ent.
uCFUs have been found to parallel bladder and kidney CFUs in different mouse models of UTI (67). To examine whether this correlation also applied to Lcn2–/– and Lcn2+/+ littermates, we measured CFUs in bladder homogenates obtained 24 hours after TU. Similar to our findings in urine samples, colonization of the bladder was generally higher in Lcn2–/– versus Lcn2+/+ littermates (Figure 2D). Hence, serial measurements of uCFUs, as well as bladder CFUs, suggested that LCN2 was required to suppress the growth of UPEC in acute UTIs.
Next, we tested whether LCN2 was sufficient to inhibit urinary bacterial growth in vitro. We used UPEC grown to log phase in a Fe-restricted minimal media (M9), and then subsequently transferred the bacteria into urine or into a new batch of M9. Growth in urine (pH <6.0) was similar to growth in acidified M9 (pH <6.0). Addition of LCN2 (5 μM, a 5-fold molar excess compared with urine Fe, and an amount consistent with uLCN2 in UTI; Figure 2B) inhibited bacterial growth in urine (n = 3; P < 0.0001; Figure 2E). Conversely, the addition of excess Fe (1 mM) to M9 cultures restored UPEC growth (data not shown), which indicated that LCN2 restricts Fe availability.
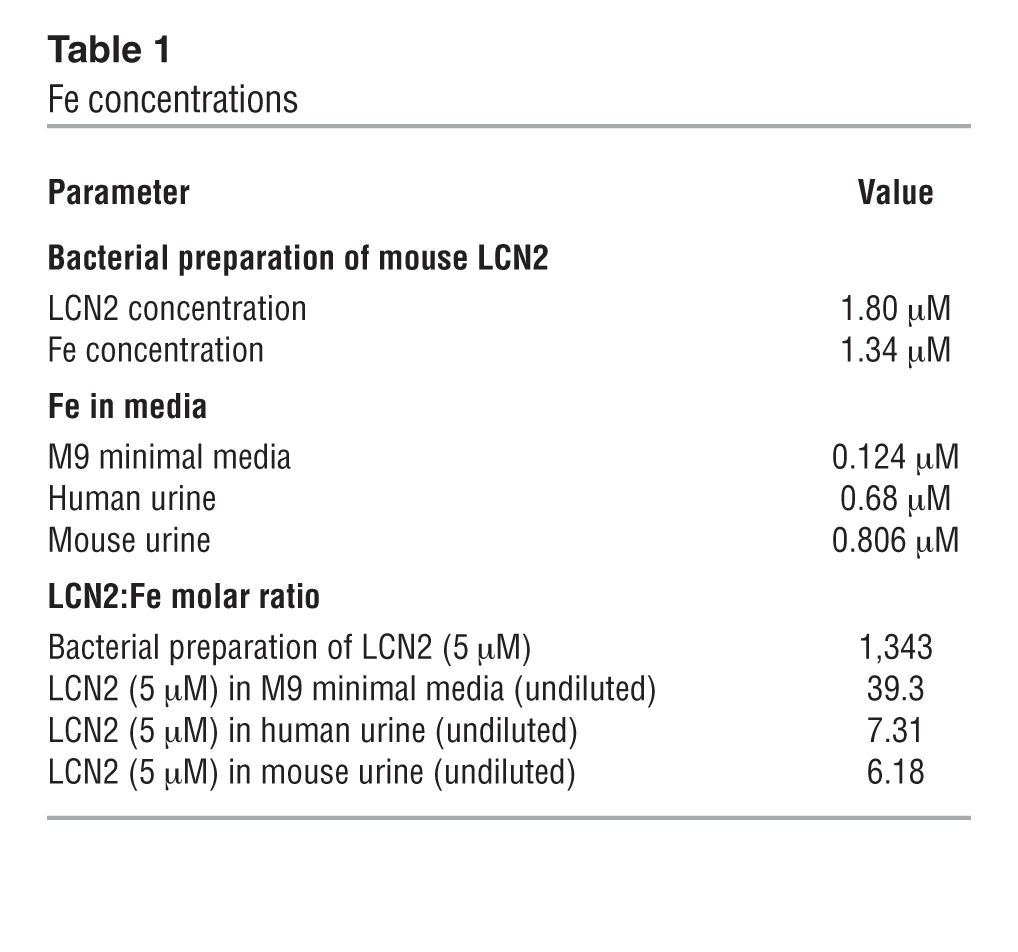
To further examine whether LCN2 induces bacterial Fe starvation, we measured a series of Fe acquisition systems known to be regulated by Fe load via fur (63, 68), including the catecholates Ent (ent genes) and salmochelin (iro genes), the hydroxamate aerobactin (iuc genes) (69, 70), and their respective receptors, fepA, iroN (71–73), and iutA as well as the heme-scavenging chu receptors (73). We found that the addition of LCN2 (5 μM; n = 5) for 30 minutes to UPEC in M9 media upregulated genes in the fur regulon, including synthetic enzymes (e.g., entA and entF, 396.2- and 36,294.5-fold, respectively; ref. 74) and receptors (e.g., fepA and iroN, 18.0- and 2.94-fold, respectively; ref. 75); genes in the aerobactin pathway, including synthetic enzymes (e.g., iucA and iucD, 12,568.5- and 19.0-fold, respectively) and receptors (e.g., iutA, 13.4-fold); and genes in the heme pathway (e.g., chuS, 26.9-fold) (Figure 3A), which suggested that LCN2 induces the widespread activation of compensatory pathways due to Fe starvation. In order to confirm the physiologic relevance of the growth conditions, we added trace amounts of Fe to M9 to match the urinary concentration (806 nM; see Methods and Table 1) and found that LCN2 still activated Ent and aerobactin genes. To test whether the upregulation of bacterial siderophores by LCN2 was typical of Fe chelation, we tested the medicinal Fe chelator desferoxamine (DFO). Fe chelation with 50 μM DFO produced gene expression changes similar to those produced by LCN2 (n = 5; Figure 3B), which confirmed the notion that LCN2 induces Fe starvation. Hence, while UPEC express many types of siderophores (68), these data demonstrated that LCN2 was both necessary and sufficient to acutely limit UPEC growth in an Fe-dependent fashion, despite selectively recognizing Ent (43, 76).
Figure 3. Response of UPEC to LCN2 and DFO.

UPEC were grown in M9 medium with (A) LCN2 (5 μM; n = 5) or (B) DFO (50 μM; n = 5) for 30 minutes. Note the upregulation of Ent synthetic enzymes (entA and entF) and receptors (fepA and iroN), aerobactin synthetic enzymes (iucA and iucD) and receptors (iutA), and heme receptors (chuS), which indicates that LCN2 induced Fe starvation and widespread activation of compensatory pathways, similar to Fe restriction by DFO.
Table 1.
Fe concentrations
The kidney medulla expresses LCN2.
To localize sites of expression of LCN2 in acute UTI in C57BL/6 mice, we used our bioluminescent Lcn2-Luc2 (C57BL/6 background) reporter mice (15). The reporters were constructed by the placement of Luc2 at the start codon of the Lcn2 gene so that expression of Luc2 faithfully and quantitatively paralleled the expression of Lcn2 message in the kidney in different types of kidney damage (15). When reporter mice were exposed to a UTI, we found that LCN2 bioluminescence originated from the kidney medulla (peak, 4.88 ± 0.95–fold at day 1; n = 11; P < 0.0001; Figure 4, A and B), which was most clearly evident when we explanted the urinary system and hemisected the kidney (Figure 4C). We confirmed the induction of LCN2 in the kidney by performing quantitative PCR and measuring copy number, which showed log-order increases in Lcn2 message (n = 5; Figure 4D). Hence, similar to aseptic kidney injuries (15, 60), the C57BL/6 model of UTI induced LCN2 in the medulla of the kidney.
Figure 4. Temporal and spatial expression of LCN2 in the kidney.

(A–D and G) Cystitis model. (A and B) Lcn2-Luc2 (C57BL/6 background) reporter mice were inoculated with UPEC, and luciferase was quantified (n = 11). (C) Excised urogenital tracts (1 day after inoculation) verified that LCN2 luminescence (arrowheads) originated from the kidney medulla. (D) Lcn2 copy number in C57BL/6 kidney and bladder before and 1 day after inoculation. (E and F) C57BL/6 and C3H/HeN mice inoculated with bioluminescent CFT073 UPEC-lux (20 μl of 5 × 108 CFU/ml; n = 4 each) were imaged on the dorsal and ventral sides for 3 days. UPEC-lux were detected 1–3 days after inoculation in C3H kidney (cystitis and pyelonephritis), but not in C57BL/6 kidney (cystitis). (G) Cytokine activation in C57BL/6 kidney (cystitis model; n = 10). *P < 0.05; #P < 0.001. Scale bar: 1 cm. Kd, kidney; g, gonad; Blad, bladder.
C57BL/6 mice are known to be resistant to kidney colonization by bacteria (64). Moreover, our inoculum volume (20 μl) was not likely to generate gross vesicular-urinary reflux in the C57BL/6 background (77). Trials with bioluminescent CFT073 UPEC (CFT073 UPEC-lux; 20 μl of 5 × 108 CFU/ml; n = 4; Supplemental Figure 2 and Figure 4, E and F) and measurement of CFUs (n = 4; Supplemental Figure 3) also failed to reveal obvious infection of the kidney (limit of detection, 102 CFU), which indicated that LCN2 message and medullary bioluminescence may have been triggered by a small number of bacteria reaching the kidney. Alternatively, bacterial components may have reached the kidney and induced LCN2. These included LPS (lipid A), which we previously found to powerfully induce high levels of LCN2 in the kidney medulla (15), and Ent, which we found to induce uLCN2 and kidney Lcn2 message (2.8 ± 0.68–fold; n = 5; P = 0.037 at 6 hours; Supplemental Figure 4), consistent with cytokine activation by Ent in other models (78). The UPEC inoculum in C57BL/6 mice also induced kidney cytokines (e.g., Il1a, Il18, and Cxcl1; n = 10; Figure 4G), some of which are known to amplify LCN2 expression (79–81). These data indicated that urinary bacteria might induce uLCN2 with a variety of ligands.
In addition to the kidney medulla, uLCN2 might have also originated from bladder cells. However, while GFP-labeled UPECs (Supplemental Figure 5) bound to the thin layer of bladder uroepithelia (Figure 5A) and induced both Lcn2 and cytokines (n = 5; Figure 5, B and C), the vast majority of Lcn2 transcripts were still expressed by kidney, rather than by bladder (P < 0.05, kidney vs. bladder transcripts; Figure 4D). Additionally, bladders explanted from LPS-treated C57BL/6 mice (1 mg/kg i.p.; n = 3) secreted only 10 ng/ml LCN2 per 12 hours (Supplemental Figure 6), representing about 0.004% of the uLCN2 induced by the same dose of LPS in matched mice (∼250 μg/ml). LCN2 may have also originated from neutrophils, but uLCN2 was expressed in urine supernatants even after neutrophil ablation with RB6-8C5 (150 μg), compared with treatment with control rat IgG2b (150 μg) (15, 82). In summary, whereas bladder epithelia and neutrophils undoubtedly contributed to soluble uLCN2, it appears that the kidney medulla is also an important source of uLCN2 in the first 24–48 hours after inoculation of UPEC.
Figure 5. Temporal and spatial expression of LCN2 in bladder.

(A) GFP-labeled UPEC in C3H bladder wall. (B) Lcn2 expression by uroepithelium (1 day after inoculation). (C) Cytokine activation in C3H/HeN Lpsn bladder (pyelonephritis model; n = 6). Note the consistent expression of Il1a and Il1b in bladder and kidney (see Figure 4G). Scale bars: 100 μm.
The A-IC expresses LCN2.
Since there was little evidence of colonization of the C57BL/6 kidney with UPECs in our model of UTI, we turned to another model of urinary infection: pyelonephritis in the C3H/HeN mouse. These mice are susceptible to UPEC-mediated pyelonephritis because they are naturally prone to vesiculoureteral reflux due to the Vurm1 locus (83, 84). We confirmed the reflux using bioluminescent CFT073 UPEC-lux, which rapidly ascended to the kidney (Figure 4, E and F). To locate the bacteria in the kidney, we inoculated GFP-transfected UPEC (20 μl of 5 × 108 CFU/ml) under control of the E. coli Lac promoter (Supplemental Figure 5). GFP-labeled UPECs associated with a subset of CD cells, which expressed luminal v-ATPase (Figure 6, A and B), characteristic of A-ICs (15). In addition, in situ hybridization showed that Lcn2 message was induced in scattered epithelia after inoculation (Figure 6C), typical of ICs. To further examine the interaction between UPEC and CD epithelia, we used an IC line (85, 86) and applied heat-killed UPECs (5 × 106 CFU; P < 0.005; Figure 6D). The bacteria induced LCN2 expression in an NF-κB–sensitive fashion in the ICs (85, 86). In addition, when we explanted epithelia directly from the medulla of Lcn2-Luc2–expressing kidneys (15, 43, 45) and treated the cells with living UPEC (P < 0.01; Figure 6E), we visualized the activated LCN2-Luc2 reporter. LCN2-Luc2 activation reflected the bacterial burden, because treatment with antibiotics reversed the activation. These data implied that CD cells (ICs) can directly sense and respond to bacteria or their components in a cell-autonomous fashion.
Figure 6. UPEC bound to A-ICs.

(A) In the C3H/HeN model of pyelonephritis, GFP-labeled UPEC (green) specifically bound A-ICs (marked by apical v-ATPase; red; n = 4). Original magnification, ×40. (B) High-resolution image of UPEC-GFP bound to A-ICs. Original magnification, ×100. (C) UPEC induced Lcn2 (arrowheads) in C3H medullary cells/ICs in kidney (paraffin in situ hybridization). (D) Treatment of an IC line with killed UPEC (24 hours; n = 4) induced Lcn2 message. NF-κB inhibitor (5 μM) reversed the LCN2 signal. (E) Coculture of Lcn2-Luc2 kidney medullary cells with living UPECs (3 hours) induced LCN2-Luc2 reporters. Gentamicin reversed the LCN2 signal (n = 3). Note the luminescent wells (blue, baseline; green/red, activated). Scale bars: 50 μm (A and C); 10 μm (B).
The IC is required for bacteriostasis.
To test the role of the ICs in UTI in vivo, we developed a method for deleting these cells from CDs. We sought transcription factors active in IC development by converting the metanephric mesenchyme into epithelia with growth factors (LIF plus FGF), and then we identified transcription factors expressed with repetitive microarray analyses during tubulogenesis (87). The transcription factor Tcfcp2l1 was expressed in late stages of both mesenchymal tubulogenesis in vitro and kidney development in vivo (E18–P5). Deletion of this transcription factor (Tcfcp2l1fl/fl;Ksp-Cre mice; M. Werth, unpublished observations) abolished the IC lineage in toto, as demonstrated by v-ATPase staining (Figure 7, A and B). All other segments of the nephron developed normally, as indicated by the normal serum creatinine and urinary protein profiles (Supplemental Figure 7) as well as the normal distribution and density of segment-specific marker proteins for CDs (Krt8 and calbindin; Figure 7B and data not shown), principal cells (Aqp2), and TALH (Tamm-Horsfall; M. Werth, unpublished observations). To determine whether knockout of ICs affects bacteriostasis, we examined the responses to UPEC and LPS using the same doses and protocols as in Figures 2 and 4–6. While WT mice responded by expressing uLCN2 (n = 6, Figure 7C) and by acidifying the urine (n = 15, P < 0.05, Figure 7D; n = 6, P < 0.05, Figure 7E), IC-knockout Tcfcp2l1fl/fl;Ksp-Cre littermates suppressed both responses. Consistently, Tcfcp2l1fl/fl;Ksp-Cre mice demonstrated higher CFU levels in both bladder and urine (n = 8, P < 0.05, Figure 7F), which indicated that they were less fit to defend the urinary tract than their littermates.
Figure 7. A-ICs regulate uLCN2 and urinary pH.

(A) ICs in sibling-matched Tcfcp2l1fl/fl and (B) IC-knockout Tcfcp2l1fl/fl;Ksp-Cre mice were visualized by immunostaining with Troma1-Krt8 (red), v-H+ATP6v1b1 (blue), and Tcfcp2l1 (green). (C and D) Deletion of IC Tcfcp2l1 suppressed (C) uLCN2 (n = 6) and (D) acidification in response to LPS (n = 15) compared with Tcfcp2l1fl/fl littermates. (E) UPEC also induced urinary acidification in C57BL/6 mice (n = 6). (F) IC-knockout Tcfcp2l1fl/fl;Ksp-Cre mice were significantly less able to clear bladder and urinary bacteria 1 day after TU inoculation than Tcfcp2l1fl/fl mice (n = 8). *P < 0.05. Scale bars: 10 μm.
ICs classically regulate acid-base balance by secreting H+. To evaluate whether acidification also reduced bacterial growth, we treated UPEC in M9 minimal media or human urine with increasing H+ concentrations and found that bacterial growth was reproducibly suppressed in a dose-dependent fashion in both media (n = 4 each; Figure 8, A and B). Because acidification inhibited the growth of UPEC and did not disrupt the chelation of Ent:Fe by LCN2 (8), these data suggested that A-ICs respond to infection by coordinately expressing 2 compatible bacteriostatic agents. Indeed, deletion of Tcfcp2l1 resulted in reduced urinary acidification in mice stimulated by LPS or by UPEC (Figure 7, D and E).
Figure 8. Acidification inhibits bacterial growth.
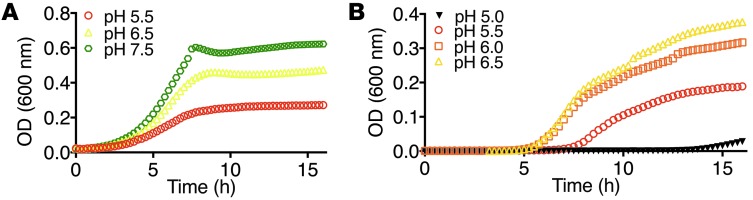
UPEC growth was significantly suppressed by acidification in both (A) M9 media (n = 4; P < 0.0001) and (B) acidified urine (n = 4; P < 0.0001, pH 5.0 vs. pH 5.5, pH 5.5 vs. pH 6.0, and pH 6.0 vs. pH 6.5).
Activation of LCN2 by TLR4.
Human data demonstrated that uLCN2 was particularly elevated in patients with Gram– rather than Gram+ UTI (P < 0.05, Figure 1C and Figure 9A), suggesting a role for the LPS receptor TLR4 (88). To examine the physiological role of TLR4 in the regulation of LCN2 expression, we used TLR4-defective C3H mice. Lpsd (C3H/HeJ) mice carry a mutation in TLR4 that renders them insensitive to LPS (88). The Lpsd mouse could not effectively suppress the growth of UPEC, as shown by higher uCFUs (n = 15, P < 0.01, Figure 9B), nor could they express as much LCN2 (n = 15, P < 0.05, Figure 9C) as WT Lpsn mice (C3H/HeN or HeOuJ, n = 9). To determine whether TLR4 regulates additional genes, we measured the same cytokines that UPEC induced in the C57BL/6 kidney (e.g., Il1a and Il1b, Figure 5C). C57BL/6 and C3H Lpsn kidneys expressed a similar profile of cytokine responses to UTI, but the Lpsd mouse severely blunted their expression (n = 15 and 9, respectively, P < 0.05, Figure 9D) (79–81). To determine whether the Lpsd phenotype could be ascribed to TLR4 expression in the kidney parenchyma, we transplanted Lpsn kidneys into nephrectomized Lpsd hosts and vice versa, and treated the recipients with LPS (1 mg/kg i.p.). LPS is a powerful inducer of LCN2 in many different tissues (15), including Lpsn kidneys, which demonstrated a 15.6 ± 2.3–fold increase in Lcn2 message when transplanted into Lpsd hosts (n = 3); conversely, the Lpsd kidney (which is not a null mutation) generated only a 4.5 ± 2.6–fold increase when transplanted into Lpsn hosts (P < 0.05, Figure 9E). In fact, administration of LPS (1 mg/kg) specifically activated LCN2 in ICs (Figure 9F), consistent with their local expression of TLR4 (Figure 9G and ref. 89). Hence, despite many potential systemic effects of LPS, these data demonstrated that the expression of kidney TLR4 critically regulates both LCN2 and IL expression in response to UPEC and LPS. The IC appears to be a critical target of TLR4 signaling.
Figure 9. TLR4 is required to induce LCN2 in UTI.

(A) Gram– infections, but not Gram+ infections, induced high levels of uLCN2 in the subset of the Emergency Department patients with documented speciation data, CFU counts, and no other renal disease (Gram+, 200.0 ng/ml, IQR 30.0–425.0 ng/ml, n = 9; Gram–, 400.0 ng/ml, IQR 135.0–775.0 ng/ml, n = 77). Both Gram+ and Gram– patients had >100,000 CFU/ml urine. (B) uCFUs were significantly lower, and (C) kidney Lcn2 and (D) kidney Il1 levels were significantly higher, in C3H Lpsn (n = 9) versus Lpsd (n = 15) mice after inoculation (pyelonephritis model). (E) Lpsn kidneys transplanted into Lpsd hosts demonstrated a 3-fold greater response to LPS (as assessed by kidney Lcn2 levels) than the reciprocal transplantation (n = 3 each). (F) In situ hybridization showing Lcn2 signal in medullary cells and ICs in LPS-challenged mice (paraffin in situ hybridization). Lcn2 RNA signal (dark purple stain) is denoted by arrowheads. (G) UPEC-GFP (green) bound TLR4+ (blue) cells in the CDs of C3H mice 1 day after inoculation (original magnification, ×100). *P < 0.05; **P < 0.001. Scale bars: 10 μm.
Discussion
A-ICs regulate acid-base homeostasis by secreting H+ into the urine. The process involves the reiterative insertion of H+ATPase containing vesicles into the apical membrane, particularly in response to metabolic and respiratory acidoses (90, 91). The H+ titrates urinary NH3+ and HPO42–, producing acidified urine in the range of pH 4.5–pH 6.5. It has been known for nearly a century that acidified urine inhibits the growth of E. coli (92) and other urinary organisms; however, A-ICs, which mediate acid-base balance, have not previously been assigned a dedicated role in antimicrobial defense. Here, we demonstrated that A-ICs sense Gram– UTIs ranging in severity from cystitis to pyelonephritis and respond with innate immune defenses.
A-ICs were strategically located in the CDs, where they bound UPEC ascending from the bladder in the pyelonephritic C3H mouse (Figure 4E, Figure 5, and ref. 89). A-ICs may use their microvilli and microplicae to contact bacterial FimH, PapG, or Dr (93), each of which has been shown to ligate different epithelial receptors (94). Moreover, A-ICs used TLR4 (Figure 9G) to sense UPEC and to trigger a program that included expression of cytokines (Figure 9D), expression of LCN2 (Figure 6E and Figure 9, C and F), and acidification of the urine (Figure 7, D and E). Even isolated and cultured kidney medullary epithelia and ICs responded to UPEC by expressing LCN2 in an NF-κB–dependent fashion (Figure 6D). Constitutively expressed antimicrobials, including the nonselective RNase7 (5, 58); cathelicidins, such as LL-37 (6, 95); and defensins (96), such as HBD1 (48), HBD2, and HD5 (54, 58) — as well as the inflammatory cytokine IL-18 (97) — may also derive from A-ICs. These data demonstrated that A-ICs bind, sense, and respond to Gram– pathogenic bacteria in a cell-autonomous fashion.
To determine the physiological importance of the IC in the defense of the urinary tract, we used a number of approaches, including data from our in vitro tubulogenesis assays (98), which demonstrated a role for the transcription factor Tcfcp2l1 in epithelial morphogenesis (87). We created conditional mice (M. Werth, unpublished observations) and deleted Tcfcp2l1 in TALH and CDs, but not in other parts of the kidney, adrenal, spinal cord, liver, pancreas, intestine, or bladder, using the Ksp-Cre driver (99). In Tcfcp2l1 knockout kidneys, principal cells (shown by Troma staining in Figure 7, A and B), proximal tubule cells (shown by surrounding tubules [background green] in Figure 7), CDs and connecting segment (calbindin), TALH (Tamm-Horsfall), and glomeruli (podocalyxin) stainings were unaffected (M. Werth, unpublished observations), and excess proteinuria was not apparent (Supplemental Figure 7). Only ICs were deleted from the kidney, and only subtle renal tubular acidosis was evident in knockout mice fed normal diets (M. Werth, unpublished observations), in agreement with a prior report (100). Consequently, the reduced clearance from urine and from bladder homogenates in Tcfcp2l1 knockouts (the kidneys were uninfected in this predominant C57BL/6 background) was most likely attributable to the wholesale deletion of ICs and their constitutive (i.e., RNAse7, cathelicidins, defensins, and acidification) and regulated (i.e., LCN2) antimicrobial defenses.
An antimicrobial role for A-ICs was also suggested by their prominent transcriptional and translational expression of Lcn2 and the Lcn2-Luc2 reporter in different injuries, including infection (Figure 6, C–E), LPS treatment (Figure 9F), and aseptic damage to the kidney (15). The expression level of uLCN2 in fact depended on the IC, because Tcfcp2l1 deletion reduced uLCN2 (Figure 7C). In addition, both deletion of Tcfcp2l1 (Figure 7, A and B) and deletion of Lcn2 (Figure 2A) resulted in excess uCFUs. Even after neutrophil ablation (15, 82), inoculation with UPEC still resulted in Lcn2 gene expression in the kidney and LCN2 protein in the urine (Figure 9E), demonstrating a contribution of kidney parenchyma to LCN2 expression. In summary, A-ICs can contribute to immune defense not only by urinary acidification (Figure 8), but also by prominently expressing LCN2 in different settings. Consequently, ICs are necessary for bacteriostasis.
The LCN2 protein is an unusual defense mechanism because of its specificity for a single virulence pathway. The bacteriostatic activity of LCN2 depended on the expression of Ent, because unlike WT bacteria (Figure 2A), Ent-null bacteria grew equally well in Lcn2–/– and Lcn2+/+ littermates. These data suggested that control of UPEC growth depended on the relative expression levels of bacterial Ent and mammalian LCN2, because prominent LCN2 expression would limit the contribution of Ent in favor of alternative bacterial Fe carriers (Figure 3A and ref. 69). In this light, the contribution of Ent to UPEC fitness in the setting of additional siderophores may have been underestimated, because the level of the Ent inhibitor LCN2 was unknown in previous studies (3, 43, 81, 101, 102). The LCN2-Ent interaction is also likely to dominate LCN2 function, because the LCN2:Ent:Fe3+ complex was stable in acidified urine (Figure 2E), even at pH 4.5. Hence, coexpression of H+ and LCN2 by A-ICs might activate the LCN2-Ent interaction because other ligands, such as urinary catechols, would dissociate from LCN2 below pH 6.0 (103). In summary, LCN2 regulates bacterial growth in acidified urine by binding to Ent, although non-Ent siderophore pathways must partially compensate. Moreover, LCN2 may have non–siderophore-dependent activities, such as chemotactic activities, which we have not evaluated, but remain compatible with our IC-LCN2 model (102).
It should also be noted that measurements of tissue homogenates and longitudinal measurements of the urine provided similar data in our assays, consistent with publications demonstrating that uCFUs parallel bladder and kidney CFUs (67). However, LCN2 activity has only been tested in the soluble phase (40, 45), rather than within tissues (104). The focus on extracellular LCN2 is due in part to the rapid accumulation of LCN2 protein (10 μM) in the media or in the urine or plasma after an inductive stimulus (e.g., LPS, ischemia, and UPEC inoculation; Figure 2B and refs. 40, 45), despite only weak intracellular staining. Consequently, LCN2 most likely acts in the extracellular fluid. Hence, sampling the urine not only demonstrated the bacteriostatic pool of LCN2, but it also allowed us to simultaneously follow the time course of uCFU and uLCN2 expression in individual mice, which provided internally controlled, longitudinal data. uCFU assays are also a well-known component of the definition of human UTI (e.g., >50,000 uCFUs in children; ref. 105).
While our present data demonstrated a role for A-ICs and kidney LCN2 in the control of urinary infections, the question of how kidney cells sense infection was still not resolved. To approach this problem, we capitalized on the natural differences between C57BL/6 mice, which are resistant to kidney colonization, and C3H mice, which are readily infected due to the Vurm1 locus (84). Hence, low-volume inocula (20 μl) resulted in few, if any, bacteria in the C57BL/6 kidney, while the same inocula resulted in infection of the C3H kidney (Figure 4, E and F). In both lines, however, LCN2 was consistently and abundantly expressed (Figure 4, A and C, and Figure 9C), as were a number of cytokines (Figure 4G and Figure 5C), despite obvious differences in bacterial colonization. Moreover, knockout of ICs in the C57BL/6 background (Figure 7, A and B) reduced uLCN2 expression (Figure 7C), confirming the contribution of the kidney, even in a model of cystitis. In short, regardless of the severity of the UTI, both C57BL/6 and C3H mice activated Lcn2 expression, suggestive of a shared sensing mechanism in both lines.
The most important sensing mechanism is likely to be mediated by TLR4 expression. TLR4 was not only expressed by IC (Figure 9G and ref. 89), but was apparently required for LCN2 expression and for the suppression of infection. Lpsd defective mice had 5-fold less kidney Lcn2, but a 4-log-order increase in uCFUs, compared with Lpsn WT mice. These data are consistent with Flo et al., who found that Lcn2 is TLR4 dependent (45), and the groups of Hagberg (106) and Schilling (107), who demonstrated log-order increases in CFUs in Lpsd compared with Lpsn mice. In humans, Karoly et al. suggested that polymorphisms in human TLR4 increased the risk of UTI in children (108). In short, the most parsimonious explanation for LCN2 expression is the activation of TLR4 in CDs. In the case of C3H mice, prominent infection of the kidney induced the expression of Lcn2 and cytokines in a TLR4-dependent manner (Figure 9, B–D). In the case of C57BL/6 mice, we surmise that a qualitatively similar mechanism is active, despite the failure to demonstrate kidney colonization. In other words, C57BL/6 Lcn2 expression could be activated by the circulation or the ascent of a small number of bacteria or LPS or fragments containing LPS. Non-TLR4 ligands may also play a role, because while the TLR4 mutation in the C3H mouse line (HeJ strain; refs. 83, 109, 110), markedly limited the expression of Lcn2, it did not abolish its expression. Exciting preliminary observations indicated that Ent can stimulate cytokine expression in a cell line (111) and LCN2 expression in mice (Supplemental Figure 4). This is an intriguing finding, because it would suggest that a specific defense (LCN2) is activated by its own cognate bacterial ligand (Ent). Finally, it is also possible that sensing is caused by cytokine signaling to the kidney (e.g., Il8; refs. 112, 113). Notably, Il1 members were consistently induced in the bladder and kidneys of C3H and C57BL/6 mice (Figure 4G and Figure 5C). Regardless of mechanism, however, our data suggested that kidney A-ICs play a part in the innate immune response to UTIs, even when few, if any, bacteria can be cultured from the kidney itself.
Earlier work of Safirstein, Heyman, Rosen, and others showed that the distal nephron can respond to a variety of injuries by expressing cytokines (114) and growth factors (115), activating ERK (116) and HIF (117), and redistributing corticomedullary circulation (118). Using mouse models, we showed that various injuries induce a cell-specific response in the CD, namely the activation of A-ICs (15). Here we show that stimulated A-ICs in turn limit bacterial growth by secreting LCN2 and H+ and other antimicrobials. Human diseases of A-ICs, such as chronic distal renal tubular acidosis, are characterized by recurrent UTI and pyelonephritis (119) as well as by kidney stones, making it difficult to determine whether the recurrent UTI is primarily due to a failure of A-IC function or secondarily due to the presence of kidney stones. Here, we showed that a defect in ICs can be a primary factor for the increased susceptibility of the urinary tract to infection.
It is well known that the gastrointestinal tract (Paneth cells), the frog skin, and the epididymis (clear cells) are populated by scattered epithelia (120), which secrete acid-base, antimicrobial peptides, or both types of bacteriostatic agents. In this context, we conclude that A-ICs represent another example of the innate cellular defense, because they not only regulate acid-base homeostasis (90), but also serve as a critical sentinels and effectors in UTIs, including both cystitis and pyelonephritis (Figure 10).
Figure 10. LCN2 induction.

(A) CD (boxed region) and connecting segments contain A-ICs. (B) Systemic sepsis, cystitis, and pyelonephritis (UTI) activate A-ICs. Kidney TLR4 expression is critical for the secretion of uLCN2 and cytokines and for the suppression of UTI. LCN2 is secreted into the urine (apically) and is potentially also secreted into the circulation (basolaterally). Note that LCN2 and H+ secretion are both activated in UTI.
Methods
Mouse husbandry.
Lcn2loxP/loxP, Lcn2–/–, C57BL/6, C3H/HeJ, C3H/HeOuJ, C3H/HeN, Tcfcp2l1loxP/loxP, Tcfcp2l1–/–, EIIa-flpe, Ksp-Cre, and Lcn2-Luc2/mC mice were generated and analyzed by protocols approved by the IACUC at Columbia University.
Generation of Lcn2loxP/loxP mice.
We created a targeting vector to delete exons 2–5 (a span of 2.1 kb) because this region contains important caliceal amino acids that bind Ent. Using a C57BL/6J library (RPCI-23; CHORI) and bacterial recombineering, a single loxP was inserted into intron 1, and frt-loxP-neo-frt-loxP was inserted into intron 5. The targeting construct was 14.2 kb consisting of a (5′) 9.9-kb-long homology arm, a loxP in intron 1, exons 2–5, a 2-kb pGK-neo cassette flanked by frt-loxP-neo-frt-loxP, and finally a (3′) 2.3-kb short homology arm. A third loxP site provided a backup in case flp was inefficient. The targeting vector was electroporated, and ES clones were selected with neo and validated by PCR. 13 heterozygous F1 pups carrying targeted alleles (Lcn2+/loxP-flp) were generated from F0 mice and crossed with the flp deleter (Actb promoter–FLP B6; SJLTg [ACTFLPe] 9205 Dym/J; JAX Mice stock no. 003800) that had been backcrossed to C57BL/6 for 5 generations to reduce genetic heterogeneity. The offspring Actb-flp;Lcn2loxP/+ mice were mated with C57BL/6 mice to eliminate Actb-flp, and then brother-sister mating produced Lcn2loxP/loxP mice. Lcn2 was deleted by breeding Lcn2loxP/loxP to EIIa-Cre mice (B6.FVB-Tg[EIIa-cre] C5379Lmgd/J; JAX Mice stock no. 003724).
Generation of Tcfcp2l1loxP/loxP mice.
We generated a knockout of Tcfp2l1 by inserting loxP sites flanking exons 3, 4, and 5 (∼1.8 kb) using BAC recombineering (BAC clone no. RP24-291G6; CHORI). This strategy was chosen in order to delete the CP2 conserved functional domain and to create an open reading frame shift. For cloning and ES cell generation, in brief, the endogenous loxP site in the BAC DNA was removed using a spectinomycin cassette from Columbia University Transgenic Facility, and loxP sites were subsequently introduced using loxP-neo-loxP (LNL) and frt-neo-frt-loxP (FNFL) cassettes. The vector was introduced into mouse KV1 ES cells (generated by Columbia University Transgenic Facility); neo cassettes were eventually removed in ES cells with Cre recombinase and in the mouse by mating with the EIIa-flpe deleter. Tcfp2l1 was deleted using Ksp-Cre (Jackson Labs), and the deletion was confirmed by PCR and immunostaining for Tcfcp2l1 (AF5726; R&D Systems). The absence of ICs was detected by immunostaining for H+ATPase (ATP6v1b1; Santa Cruz). TROMA1 (Krt8; Developmental Studies Hybridoma Bank) was used as a pan-CD marker.
Imaging of live Lcn2-Luc2 reporter mice.
Lcn2-Luc2 reporter mice (15) were injected i.p. with 150 mg/kg D-luciferin (Caliper Life Sciences) in PBS (pH 7.0), anesthetized (2.5% isofluorane), and then imaged for 30 s using the PhotonIMAGER optical imaging system (Biospace Labs) with open filter position for luminescence. Regions of interest (ROIs) were quantified using bundled photo acquisition software (BioSpace Labs). A CCD camera digitizer measured the ROIs, and counts were converted to physical units of radiance in photons/s/cm2/steradian.
Kidney cross-transplantation.
To evaluate kidney TLR4 expression, we cross-transplanted Lpsd kidneys into Lpsn controls and vice versa. The surgical cross-transplants (15) were monitored for 2 weeks until serum creatinine stabilized at 0.2 mg/dl and uLCN2 was undetectable prior to challenge with LPS (1 mg/kg i.p.).
UTIs.
We used 8- to 16-week-old female C57BL/6, Lcn2–/–, Lcn2-Luc2 reporter, C3H/HeJ, C3H/HeN, and C3H/HeOuJ mice. Bacteria (20 μl; containing 1 × 107 CFUs or heat killed) were introduced into the bladders of anesthetized mice through a soft polyethylene catheter (0.61 mm outer diameter; Intramedic). CFUs in kidney homogenates or in urine (collected directly from mice) were quantified by serial dilution on LB agar plates. Mutant entF-CFT073 UPEC was obtained from S. Payne (University of Texas at Austin, Austin, Texas, USA; ref. 66). The mutant was constructed by insertion of the Kan cassette and confirmed by PCR (entF-1792 forward, 5′-GCTTATATCATCTTCACCTCAGGTTCCACC-3′; entF-3115 reverse, 5′-GCTGTCTTCTTCACCATCAATAATCGTTGC-3′) (66).
Inhibition of bacterial growth in vitro.
A single colony of CFT073 UPEC was grown in M9 or in urine to log phase. Bacteria were pelleted and resuspended in either M9 or urine and monitored in a 96-well plate (Tecan Infinite 200 Pro). Notably, CFT073 UPEC grown to log phase in LB carry over Fe into downstream assays.
Generation of a luxCDABE-labeled CFT073 UPEC strain.
The mini-Tn5-luxCDABE transposon was delivered to CFT073 UPEC (ATCC 700928) by conjugation with the donor strain E. coli S17-1 λ-pir (pUTmini-Tn5-luxCDABE-km2). The donor and CFT073 UPEC recipient cells were grown overnight in 5 ml LB broth at 37°C prior to biparental mating. Harvested cells were gently washed twice in LB and resuspended in one-tenth the original culture volume. A mixture of donor and recipient cells (1:5) was gently mixed, spot-inoculated onto the surface of nitrocellulose filters (0.13 mm; catalog no. VMWP01300; Millipore), and incubated at 37°C for 6–8 hours. To select for CFT073 UPEC transconjugants, cells were scraped off the filters, washed twice in LB, and plated onto LB agar plates containing 50 μg/ml kanamycin (Km2) for 24–48 hours. Highly bioluminescent transconjugants were identified in an IVIS Spectrum (PerkinElmer) and further purified by subculturing twice on LB containing 50 μg/ml kanamycin. The CFT073 UPEC-lux strains were further differentiated from donor strains by negative growth on LB agar with 100 μg/ml ampicillin.
LCN2 protein production.
Recombinant protein was produced in BL21 E. coli transformed with mouse or human LCN2 cDNA lacking 29–amino acid signal sequence (pGEX-4T-3-vector) and grown for 16 hours at 37°C in Modified Terrific Broth supplemented with 150 μM Fe to inhibit the endogenous production of Ent. IPTG (0.2 mM final concentration) was added for 5 hours. Bacterial pellets were lysed by sonication in lysis buffer, followed by Triton-X 100 (0.5%) treatment for 30 minutes on ice. Supernatants were collected after high-speed centrifugation and filter sterilized (0.45 μm). LCN2-GST was purified by binding to glutathione sepharose beads, followed by cleavage of the GST tag with thrombin. Released protein was fractionated by a Sephacryl S100HR column. Ent (EMC Microcollections gmbh) capture by LCN2 was tested at a 1:1:1 Ent:Fe:LCN2 ratio. The complex was washed 5 times on a 10-k micron, and binding was detected by its red coloration or by retention of 55Fe.
Isolation and culture of cells.
Lcn2-Luc2 reporter mice (8–12 weeks of age) were perfused with PBS, and kidney cells were isolated with collagenase (2 mg/ml; Sigma-Aldrich) and cultured (1 × 105 cells/well in 24-well plates; Falcon) in DMEM/F12 medium supplemented with 10% FBS, 1% penicillin-streptomycin, and 46 mg/l L-valine for 24 hours. Cells were treated for 24 hours with either living or heat-killed (30 minutes boiling) CFT073 UPEC (5 × 106 CFUs) or with LPS (4 μg/ml) and NF-κB inhibitor, analog 31 (5 μM) (15). Luciferase substrate (Luciferase Assay System; Promega) was added, and luminescence from Luc2 was imaged in a IVIS Spectrum optical imaging system (PerkinElmer).
In vitro ICs.
Rabbit ICs Clone C were obtained from S. Vijayakumar (University of Rochester, Rochester, New York, USA), maintained at 32°C, and then seeded on Corning Transwell no. 3412 at a density of 5 × 105 cells/cm2 (high density) in DMEM/F12 50:50 (MT10090CV; Mediatech Cellgro) with 10% heat-inactivated FBS (Invitrogen), 1% penicillin-streptomycin, 20 mg/l hydrocortisone, and an insulin, transferrin, and selenium supplement (Lonza) at 40°C (to inactivate the T antigen). Cells were serum starved prior to treatment, and RNA was extracted using an Ambion kit (AM1560) with DNAse digestion.
Fe quantification.
Pooled C3H/HeN urine (n = 15) was collected by clean catch and centrifuged for 10 minutes at 10,000 g. Urine, protein, and media Fe concentrations were measured by a Graphite Furnace Atomic Absorption Spectrophotometer (GFAAS), model Analyst 800 (PerkinElmer) by the Trace Metals Core Facility at the Columbia University Mailman School of Public Health.
Western blot.
Human urines were analyzed using nonreducing 4%–15% tris-HCL gels (Bio-Rad Laboratories) and monoclonal human LCN2 (1:1,000; BPD-HYB-211-01-02; Enzo Lifesciences). LCN2 was reproducibly detected to 0.4 ng/lane. Mouse urine LCN2 was detected by the polyclonal antibody (1:1,000; AF1857; R&D Systems), which has lower sensitivity than the anti-human antibodies. LCN2 protein was semiquantified by comparison with mouse or human LCN2 protein standards using Image J (NIH).
In situ hybridization and immunohistochemistry.
Lcn2 RNA was detected using digoxigenin-labeled antisense riboprobes (Roche Applied Biosystems) from cDNAs encoding LCN2 (exon 1–7, 566 bp) by linearization with XhoI followed by T7 RNA polymerase as previously described.
Frozen and paraffin-embedded sections were used for in situ hybridization. Frozen sections were used for immunohistochemical analyses with vATPase B1/2 (1:50; Santa Cruz Biotechnology) and TLR4 (1:50; Abcam) antibodies. DAPI and TOTO3 (1:1,000) were used for nuclear staining.
Real-time PCR analysis.
Total RNA was isolated with mirVANA (for eukaryotic cells) or ribopure (for bacteria) RNA extraction kits (Ambion). First-strand cDNA was synthesized with Superscript III (Invitrogen). Real-time PCR was performed in a 7500 Fast (Applied Biosystems) with SYBR green supermix reagent (Fisher) or Gene Expression Master Mix (Invitrogen) and primers (Supplemental Table 1) using Actb (eukaryotic cells) or gapA (bacteria) as internal controls. Fold amplification of transcripts was measured by the ΔΔCt method.
Ent induction of Lcn2.
Ent (30 μg) in 0.5 ml saline buffer (Sigma-Aldrich) was introduced in mice i.p. Kidneys were harvested 4–6, 8, or 12 hours after treatment. Total RNA was isolated from kidneys using TRIzol reagent (Invitrogen) according to the manufacturer’s instruction. First-strand cDNA was synthesized using the High Capacity Reverse Transcription Kit (Applied Biosystems). Real-time PCR was performed in a LightCycler 96 (Roche) with Taqman gene expression master mix reagent (Applied Biosystems) and primers using Actb (eukaryotic cells) as internal control. Fold amplification of transcripts was measured by the ΔΔCt method.
Statistics.
We evaluated our entire cohort of Emergency Department patients, as well as a subset with normal lab indices (serum creatinine and proteinuria), over a 7-day hospital course. UTI was defined by LE 1+ or 2+ and urinary Cx containing known urinary pathogens. Analyses used SPSS version 16.0. Continuous data were log-transformed prior to analysis, but presented as non–log-transformed values. 2-tailed t test for unequal variances was used for comparisons (Welsh’s t test). A P value less than 0.05 was considered significant. All box plots represent median and interquartile range (IQR), and whiskers represent minimum to maximum values. Mean and SEM was utilized with all fold change data. Prism 5 was used for graphing all data (GraphPad Software).
Study approval.
Patient datasets were obtained from the Charité–Universitätsmedizin and Helios Clinic, Berlin, Germany (60) and Columbia University Medical Center according to IRB protocols, with written informed consent prior to study inclusion. All animal experiments followed protocols approved by the IACUC at Columbia University.
Supplementary Material
Acknowledgments
We are grateful to Shelley Payne for providing mutant UPEC. N. Paragas is supported by a Transition to Independence grant by the National Institute of Diabetes and Digestive and Kidney Diseases (5K99DK094873-02). R. Kulkarni is supported by a Flight Attendant Medical Research Institute (FAMRI) Young Clinical Scientist Award (YCSA_092417). N. Paragas, J. Barasch, and A.J. Ratner are supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (2R01DK073462-06) and by a March of Dimes Research Grant.
Footnotes
Conflict of interest: Columbia University has licensed the use of NGAL (LCN2) to detect intrinsic AKI (ATN) to Abbott and Alere.
Citation for this article:J Clin Invest. 2014;124(7):2963–2976. doi:10.1172/JCI71630.
See the related Commentary beginning on page 2844.
References
- 1.Litwin MS, et al. Urologic diseases in America Project: analytical methods and principal findings. J Urol. 2005;173(3):933–937. doi: 10.1097/01.ju.0000152365.43125.3b. [DOI] [PubMed] [Google Scholar]
- 2.Freedman AL, et al. Urologic diseases in North America Project: trends in resource utilization for urinary tract infections in children. J Urol. 2005;173(3):949–954. doi: 10.1097/01.ju.0000152092.03931.9a. [DOI] [PubMed] [Google Scholar]
- 3.Brzuszkiewicz E, et al. How to become a uropathogen: comparative genomic analysis of extraintestinal pathogenic Escherichia coli strains. Proc Natl Acad Sci U S A. 2006;103(34):12879–12884. doi: 10.1073/pnas.0603038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Am J Med. 2002;113(suppl 1A):5S–13S. doi: 10.1016/s0002-9343(02)01054-9. [DOI] [PubMed] [Google Scholar]
- 5.Spencer JD, et al. Ribonuclease 7 is a potent antimicrobial peptide within the human urinary tract. Kidney Int. 2011;80(2):174–180. doi: 10.1038/ki.2011.109. [DOI] [PubMed] [Google Scholar]
- 6.Chromek M, et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med. 2006;12(6):636–641. doi: 10.1038/nm1407. [DOI] [PubMed] [Google Scholar]
- 7.Yang J, et al. An iron delivery pathway mediated by a lipocalin. Mol Cell. 2002;10(5):1045–1056. doi: 10.1016/S1097-2765(02)00710-4. [DOI] [PubMed] [Google Scholar]
- 8.Mori K, et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest. 2005;115(3):610–621. doi: 10.1172/JCI23056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishra J, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14(10):2534–2543. doi: 10.1097/01.ASN.0000088027.54400.C6. [DOI] [PubMed] [Google Scholar]
- 10.Parravicini E, Lorenz JM, Nemerofsky SL, O’Rourke M, Barasch J, Bateman D. Reference range of urinary neutrophil gelatinase-associated lipocalin in very low-birth-weight infants: preliminary data. Am J Perinatol. 2009;26(6):437–440. doi: 10.1055/s-0029-1214242. [DOI] [PubMed] [Google Scholar]
- 11.Bagshaw SM, et al. Plasma and urine neutrophil gelatinase-associated lipocalin in septic versus non-septic acute kidney injury in critical illness. Intensive Care Med. 2010;36(3):452–461. doi: 10.1007/s00134-009-1724-9. [DOI] [PubMed] [Google Scholar]
- 12.Kümpers P, et al. Serum neutrophil gelatinase-associated lipocalin at inception of renal replacement therapy predicts survival in critically ill patients with acute kidney injury. Crit Care. 2010;14(1):R9. doi: 10.1186/cc8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruz DN, et al. Plasma neutrophil gelatinase-associated lipocalin is an early biomarker for acute kidney injury in an adult ICU population. Intensive Care Med. 2010;36(3):444–451. doi: 10.1007/s00134-009-1711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wheeler DS, et al. Serum neutrophil gelatinase-associated lipocalin (NGAL) as a marker of acute kidney injury in critically ill children with septic shock. Crit Care Med. 2008;36(4):1297–1303. doi: 10.1097/CCM.0b013e318169245a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paragas N, et al. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med. 2011;17(2):216–222. doi: 10.1038/nm.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett M, et al. Urine NGAL predicts severity of acute kidney injury after cardiac surgery: a prospective study. Clin J Am Soc Nephrol. 2008;3(3):665–673. doi: 10.2215/CJN.04010907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McIlroy DR, Wagener G, Lee HT. Neutrophil gelatinase-associated lipocalin and acute kidney injury after cardiac surgery: the effect of baseline renal function on diagnostic performance. Clin J Am Soc Nephrol. 2010;5(2):211–219. doi: 10.2215/CJN.04240609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mishra J, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365(9466):1231–1238. doi: 10.1016/S0140-6736(05)74811-X. [DOI] [PubMed] [Google Scholar]
- 19.Niemann CU, et al. Acute kidney injury during liver transplantation as determined by neutrophil gelatinase-associated lipocalin. Liver Transpl. 2009;15(12):1852–1860. doi: 10.1002/lt.21938. [DOI] [PubMed] [Google Scholar]
- 20.Capuano F, et al. Neutrophil gelatinase-associated lipocalin levels after use of mini-cardiopulmonary bypass system. Interact Cardiovasc Thorac Surg. 2009;9(5):797–801. doi: 10.1510/icvts.2009.212266. [DOI] [PubMed] [Google Scholar]
- 21.Haase-Fielitz A, et al. The predictive performance of plasma neutrophil gelatinase-associated lipocalin (NGAL) increases with grade of acute kidney injury. Nephrol Dial Transplant. 2009;24(11):3349–3354. doi: 10.1093/ndt/gfp234. [DOI] [PubMed] [Google Scholar]
- 22.Tuladhar SM, Puntmann VO, Soni M, Punjabi PP, Bogle RG. Rapid detection of acute kidney injury by plasma and urinary neutrophil gelatinase-associated lipocalin after cardiopulmonary bypass. J Cardiovasc Pharmacol. 2009;53(3):261–266. doi: 10.1097/FJC.0b013e31819d6139. [DOI] [PubMed] [Google Scholar]
- 23.Wan X, et al. [Association between urinary neutrophil gelatinase-associated lipocalin and acute kidney injury after cardiac surgery]. Zhonghua Yi Xue Za Zhi. 2008;88(19):1318–1322. [PubMed] [Google Scholar]
- 24.Xin C, Yulong X, Yu C, Changchun C, Feng Z, Xinwei M. Urine neutrophil gelatinase-associated lipocalin and interleukin-18 predict acute kidney injury after cardiac surgery. Ren Fail. 2008;30(9):904–913. doi: 10.1080/08860220802359089. [DOI] [PubMed] [Google Scholar]
- 25.Koyner JL, et al. Urinary cystatin C as an early biomarker of acute kidney injury following adult cardiothoracic surgery. Kidney Int. 2008;74(8):1059–1069. doi: 10.1038/ki.2008.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagener G, Gubitosa G, Wang S, Borregaard N, Kim M, Lee HT. Increased incidence of acute kidney injury with aprotinin use during cardiac surgery detected with urinary NGAL. Am J Nephrol. 2008;28(4):576–582. doi: 10.1159/000115973. [DOI] [PubMed] [Google Scholar]
- 27.Dent CL, et al. Plasma neutrophil gelatinase-associated lipocalin predicts acute kidney injury, morbidity and mortality after pediatric cardiac surgery: a prospective uncontrolled cohort study. Crit Care. 2007;11(6):R127. doi: 10.1186/cc6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuwabara T, et al. Urinary neutrophil gelatinase-associated lipocalin levels reflect damage to glomeruli, proximal tubules, and distal nephrons. Kidney Int. 2009;75(3):285–294. doi: 10.1038/ki.2008.499. [DOI] [PubMed] [Google Scholar]
- 29.Mishra J, Mori K, Ma Q, Kelly C, Barasch J, Devarajan P. Neutrophil gelatinase-associated lipocalin: a novel early urinary biomarker for cisplatin nephrotoxicity. Am J Nephrol. 2004;24(3):307–315. doi: 10.1159/000078452. [DOI] [PubMed] [Google Scholar]
- 30.Malyszko J, Bachorzewska-Gajewska H, Poniatowski B, Malyszko JS, Dobrzycki S. Urinary and serum biomarkers after cardiac catheterization in diabetic patients with stable angina and without severe chronic kidney disease. Ren Fail. 2009;31(10):910–919. doi: 10.3109/08860220903216113. [DOI] [PubMed] [Google Scholar]
- 31.Vaidya VS, et al. Urinary biomarkers for sensitive and specific detection of acute kidney injury in humans. Clin Transl Sci. 2008;1(3):200–208. doi: 10.1111/j.1752-8062.2008.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirsch R, et al. NGAL is an early predictive biomarker of contrast-induced nephropathy in children. Pediatr Nephrol. 2007;22(12):2089–2095. doi: 10.1007/s00467-007-0601-4. [DOI] [PubMed] [Google Scholar]
- 33.Ling W, et al. Urinary IL-18 and NGAL as early predictive biomarkers in contrast-induced nephropathy after coronary angiography. Nephron Clin Pract. 2008;108(3):c176–c181. doi: 10.1159/000117814. [DOI] [PubMed] [Google Scholar]
- 34.Makris K, et al. Urinary neutrophil gelatinase-associated lipocalin (NGAL) as an early marker of acute kidney injury in critically ill multiple trauma patients. Clin Chem Lab Med. 2009;47(1):79–82. doi: 10.1515/CCLM.2009.004. [DOI] [PubMed] [Google Scholar]
- 35.Lebkowska U, et al. Neutrophil gelatinase-associated lipocalin and cystatin C could predict renal outcome in patients undergoing kidney allograft transplantation: a prospective study. Transplant Proc. 2009;41(1):154–157. doi: 10.1016/j.transproceed.2008.10.092. [DOI] [PubMed] [Google Scholar]
- 36.Parikh CR, et al. Urine NGAL and IL-18 are predictive biomarkers for delayed graft function following kidney transplantation. Am J Transplant. 2006;6(7):1639–1645. doi: 10.1111/j.1600-6143.2006.01352.x. [DOI] [PubMed] [Google Scholar]
- 37.Ferguson MA, et al. Urinary liver-type fatty acid-binding protein predicts adverse outcomes in acute kidney injury. Kidney Int. 2010;77(8):708–714. doi: 10.1038/ki.2009.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nickolas TL, et al. Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase-associated lipocalin for diagnosing acute kidney injury. Ann Intern Med. 2008;148(11):810–819. doi: 10.7326/0003-4819-148-11-200806030-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flower DR, North AC, Sansom CE. The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta. 2000;1482(1–2):9–24. doi: 10.1016/s0167-4838(00)00148-5. [DOI] [PubMed] [Google Scholar]
- 40.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10(5):1033–1043. doi: 10.1016/S1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 41.Barasch J, Mori K. Cell biology: iron thievery. Nature. 2004;432(7019):811–813. doi: 10.1038/432811a. [DOI] [PubMed] [Google Scholar]
- 42.Abergel RJ, et al. The siderocalin/enterobactin interaction: a link between mammalian immunity and bacterial iron transport. J Am Chem Soc. 2008;130(34):11524–11534. doi: 10.1021/ja803524w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berger T, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103(6):1834–1839. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holmes MA, Paulsene W, Jide X, Ratledge C, Strong RK. Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure. 2005;13(1):29–41. doi: 10.1016/j.str.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 45.Flo TH, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432(7019):917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 46.Zhou G, et al. Uroplakin Ia is the urothelial receptor for uropathogenic Escherichia coli: evidence from in vitro FimH binding. J Cell Sci. 2001;114(pt 22):4095–4103. doi: 10.1242/jcs.114.22.4095. [DOI] [PubMed] [Google Scholar]
- 47.Hiratsuka T, et al. Structural analysis of human β-defensin-1 and its significance in urinary tract infection. Nephron. 2000;85(1):34–40. doi: 10.1159/000045627. [DOI] [PubMed] [Google Scholar]
- 48.Valore EV, Park CH, Quayle AJ, Wiles KR, McCray PB, Ganz T. Human β-defensin-1: an antimicrobial peptide of urogenital tissues. J Clin Invest. 1998;101(8):1633–1642. doi: 10.1172/JCI1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Serafini-Cessi F, Malagolini N, Cavallone D. Tamm-Horsfall glycoprotein: biology and clinical relevance. Am J Kidney Dis. 2003;42(4):658–676. doi: 10.1016/S0272-6386(03)00829-1. [DOI] [PubMed] [Google Scholar]
- 50.Bates JM, et al. Tamm-Horsfall protein knockout mice are more prone to urinary tract infection: rapid communication. Kidney Int. 2004;65(3):791–797. doi: 10.1111/j.1523-1755.2004.00452.x. [DOI] [PubMed] [Google Scholar]
- 51.Mulvey MA, Schilling JD, Martinez JJ, Hultgren SJ. Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proc Natl Acad Sci U S A. 2000;97(16):8829–8835. doi: 10.1073/pnas.97.16.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wright KJ, Seed PC, Hultgren SJ. Uropathogenic Escherichia coli flagella aid in efficient urinary tract colonization. Infect Immun. 2005;73(11):7657–7668. doi: 10.1128/IAI.73.11.7657-7668.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abrink M, Larsson E, Gobl A, Hellman L. Expression of lactoferrin in the kidney: implications for innate immunity and iron metabolism. Kidney Int. 2000;57(5):2004–2010. doi: 10.1046/j.1523-1755.2000.00050.x. [DOI] [PubMed] [Google Scholar]
- 54.Zasloff M. Antimicrobial peptides, innate immunity, and the normally sterile urinary tract. J Am Soc Nephrol. 2007;18(11):2810–2816. doi: 10.1681/ASN.2007050611. [DOI] [PubMed] [Google Scholar]
- 55.Reinhart H, Obedeanu N, Hooton T, Stamm W, Sobel J. Urinary excretion of Tamm-Horsfall protein in women with recurrent urinary tract infections. J Urol. 1990;144(5):1185–1187. doi: 10.1016/s0022-5347(17)39687-8. [DOI] [PubMed] [Google Scholar]
- 56.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 57.Perron GG, Zasloff M, Bell G. Experimental evolution of resistance to an antimicrobial peptide. Proc Biol Sci. 2006;273(1583):251–256. doi: 10.1098/rspb.2005.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spencer JD, et al. An endogenous ribonuclease inhibitor regulates the antimicrobial activity of ribonuclease 7 in the human urinary tract. Kidney Int. 2013;85(5):1179–1191. doi: 10.1038/ki.2013.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raffi HS, Bates JJM, Laszik Z, Kumar S. Tamm-Horsfall protein acts as a general host-defense factor against bacterial cystitis. Am J Nephrol. 2005;25(6):570–578. doi: 10.1159/000088990. [DOI] [PubMed] [Google Scholar]
- 60.Nickolas TL, et al. Diagnostic and prognostic stratification in the emergency department using urinary biomarkers of nephron damage: a multicenter prospective cohort study. J Am Coll Cardiol. 2012;59(3):246–255. doi: 10.1016/j.jacc.2011.10.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gupta K, et al. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the infectious diseases society of america and the European society for microbiology and infectious diseases. Clin Infect Dis. 2011;52(5):e103–e120. doi: 10.1093/cid/ciq257. [DOI] [PubMed] [Google Scholar]
- 62.Mobley HL, et al. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect Immun. 1990;58(5):1281–1289. doi: 10.1128/iai.58.5.1281-1289.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Welch RA, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2002;99(26):17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hopkins WJ, Gendron-Fitzpatrick A, Balish E, Uehling DT. Time course and host responses to Escherichia coli urinary tract infection in genetically distinct mouse strains. Infect Immun. 1998;66(6):2798–2802. doi: 10.1128/iai.66.6.2798-2802.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lakso M, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93(12):5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Torres AG, Redford P, Welch RA, Payne SM. TonB-dependent systems of uropathogenic Escherichia coli: aerobactin and heme transport and TonB are required for virulence in the mouse. Infect Immun. 2001;69(10):6179–6185. doi: 10.1128/IAI.69.10.6179-6185.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hvidberg H, Struve C, Krogfelt KA, Christensen N, Rasmussen SN, Frimodt-Moller N. Development of a long-term ascending urinary tract infection mouse model for antibiotic treatment studies. Antimicrob Agents Chemother. 2000;44(1):156–163. doi: 10.1128/AAC.44.1.156-163.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia EC, Brumbaugh AR, Mobley HL. Redundancy specificity of Escherichia coli iron acquisition systems during urinary tract infection. Infect Immun. 2011;79(3):1225–1235. doi: 10.1128/IAI.01222-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carbonetti NH, Boonchai S, Parry SH, Vaisanen-Rhen V, Korhonen TK, Williams PH. Aerobactin-mediated iron uptake by Escherichia coli isolates from human extraintestinal infections. Infect Immun. 1986;51(3):966–968. doi: 10.1128/iai.51.3.966-968.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnson JR. Virulence factors in Escherichia coli urinary tract infection. Clin Microbiol Rev. 1991;4(1):80–128. doi: 10.1128/cmr.4.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buchanan SK, et al. Crystal structure of the outer membrane active transporter FepA from Escherichia coli. Nat Struct Biol. 1999;6(1):56–63. doi: 10.1038/4931. [DOI] [PubMed] [Google Scholar]
- 72.Russo TA, McFadden CD, Carlino-MacDonald UB, Beanan JM, Olson R, Wilding GE. The Siderophore receptor IroN of extraintestinal pathogenic Escherichia coli is a potential vaccine candidate. Infect Immun. 2003;71(12):7164–7169. doi: 10.1128/IAI.71.12.7164-7169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alteri CJ, Hagan EC, Sivick KE, Smith SN, Mobley HL. Mucosal immunization with iron receptor antigens protects against urinary tract infection. PLoS Pathog. 2009;5(9):e1000586. doi: 10.1371/journal.ppat.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ehmann DE, Shaw-Reid CA, Losey HC, Walsh CT. The EntF and EntE adenylation domains of Escherichia coli enterobactin synthetase: Sequestration and selectivity in acyl-AMP transfers to thiolation domain cosubstrates. Proc Natl Acad Sci U S A. 2000;97(6):2509–2514. doi: 10.1073/pnas.040572897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Russo TA, McFadden CD, Carlino-MacDonald UB, Beanan JM, Barnard TJ, Johnson JR. IroN functions as a siderophore receptor and is a urovirulence factor in an extraintestinal pathogenic isolate of Escherichia coli. Infect Immun. 2002;70(12):7156–7160. doi: 10.1128/IAI.70.12.7156-7160.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Strong RK. Siderocalins. In: Åkerström B, Borregard N, Flower DR, Salier JP, eds. Lipocalins. Georgetown, Texas, USA: Landes Bioscience; 2006:83–98. [Google Scholar]
- 77.Hung C-S, Dodson KW, Hultgren SJ. A murine model of urinary tract infection. Nat Protoc. 2009;4(8):1230–1243. doi: 10.1038/nprot.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nelson AL, Barasch JM, Bunte RM, Weiser JN. Bacterial colonization of nasal mucosa induces expression of siderocalin, an iron-sequestering component of innate immunity. Cell Microbiol. 2005;7(10):1404–1417. doi: 10.1111/j.1462-5822.2005.00566.x. [DOI] [PubMed] [Google Scholar]
- 79.Cowland JB, Sorensen OE, Sehested M, Borregaard N. Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1β, but not by TNF-α. J Immunol. 2003;171(12):6630–6639. doi: 10.4049/jimmunol.171.12.6630. [DOI] [PubMed] [Google Scholar]
- 80.Bando M, et al. Interleukin-1α regulates antimicrobial peptide expression in human keratinocytes. Immunol Cell Biol. 2007;85(7):532–537. doi: 10.1038/sj.icb.7100078. [DOI] [PubMed] [Google Scholar]
- 81.Chan YR, et al. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol. 2009;182(8):4947–4956. doi: 10.4049/jimmunol.0803282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lysenko ES, Ratner AJ, Nelson AL, Weiser JN. The role of innate immune responses in the outcome of interspecies competition for colonization of mucosal surfaces. PLoS Pathog. 2005;1(1):e1. doi: 10.1371/journal.ppat.0010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Murawski IJ, et al. The C3H/HeJ inbred mouse is a model of vesico-ureteric reflux with a susceptibility locus on chromosome 12. Kidney Int. 2010;78(3):269–278. doi: 10.1038/ki.2010.110. [DOI] [PubMed] [Google Scholar]
- 84.Murawski IJ, Watt CL, Gupta IR. Vesico-ureteric reflux: using mouse models to understand a common congenital urinary tract defect. Pediatr Nephrol. 2011;26(9):1513–1522. doi: 10.1007/s00467-011-1821-1. [DOI] [PubMed] [Google Scholar]
- 85.van Adelsberg J, Edwards JC, Takito J, Kiss B, al-Awqati Q. An induced extracellular matrix protein reverses the polarity of band 3 in intercalated epithelial cells. Cell. 1994;76(6):1053–1061. doi: 10.1016/0092-8674(94)90382-4. [DOI] [PubMed] [Google Scholar]
- 86.Xie Y, et al. Identification of N-(quinolin-8-yl)benzenesulfonamides as agents capable of down-regulating NFκB activity within two separate high-throughput screens of NFκB activation. Bioorg Med Chem Lett. 2008;18(1):329–335. doi: 10.1016/j.bmcl.2007.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schmidt-Ott KM, et al. β-Catenin/TCF/Lef controls a differentiation-associated transcriptional program in renal epithelial progenitors. Development. 2007;134(17):3177–3190. doi: 10.1242/dev.006544. [DOI] [PubMed] [Google Scholar]
- 88.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 89.Chassin C, et al. Renal collecting duct epithelial cells react to pyelonephritis-associated Escherichia coli by activating distinct TLR4-dependent and -independent inflammatory pathways. J Immunol. 2006;177(7):4773–4784. doi: 10.4049/jimmunol.177.7.4773. [DOI] [PubMed] [Google Scholar]
- 90.Schwartz GJ, Barasch J, Alawqati Q. Plasticity of functional epithelial polarity. Nature. 1985;318(6044):368–371. doi: 10.1038/318368a0. [DOI] [PubMed] [Google Scholar]
- 91.Wagner CA, et al. Regulation of the expression of the Cl–/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int. 2002;62(6):2109–2117. doi: 10.1046/j.1523-1755.2002.00671.x. [DOI] [PubMed] [Google Scholar]
- 92.Shohl AT, Janney JH. The growth of Bacillus coli in urine at varying hydrogen ion concentrations. J Urol. 1917;1(2):211–229. [Google Scholar]
- 93.Martinez JJ, Mulvey MA, Schilling JD, Pinkner JS, Hultgren SJ. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000;19(12):2803–2812. doi: 10.1093/emboj/19.12.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nielubowicz GR, Mobley HLT. Host-pathogen interactions in urinary tract infection. Nat Rev Urol. 2010;7(8):430–441. doi: 10.1038/nrurol.2010.101. [DOI] [PubMed] [Google Scholar]
- 95.Ramanathan B, Davis EG, Ross CR, Blecha F. Cathelicidins: microbicidal activity, mechanisms of action, and roles in innate immunity. Microbes Infect. 2002;4(3):361–372. doi: 10.1016/S1286-4579(02)01549-6. [DOI] [PubMed] [Google Scholar]
- 96.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3(9):710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 97.Gauer S, et al. IL-18 is expressed in the intercalated cell of human kidney. Kidney Int. 2007;72(9):1081–1087. doi: 10.1038/sj.ki.5002473. [DOI] [PubMed] [Google Scholar]
- 98.Barasch J, et al. Mesenchymal to epithelial conversion in rat metanephros is induced by LIF. Cell. 1999;99(4):377–386. doi: 10.1016/S0092-8674(00)81524-X. [DOI] [PubMed] [Google Scholar]
- 99.Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol. 2002;13(7):1837–1846. doi: 10.1097/01.ASN.0000016444.90348.50. [DOI] [PubMed] [Google Scholar]
- 100.Yamaguchi Y, Yonemura S, Takada S. Grainyhead-related transcription factor is required for duct maturation in the salivary gland and the kidney of the mouse. Development. 2006;133(23):4737–4748. doi: 10.1242/dev.02658. [DOI] [PubMed] [Google Scholar]
- 101.Watts RE, et al. Contribution of siderophore systems to growth and urinary tract colonization of asymptomatic bacteriuria Escherichia coli. Infect Immun. 2012;80(1):333–344. doi: 10.1128/IAI.05594-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bachman MA, Miller VL, Weiser JN. Mucosal lipocalin 2 has pro-inflammatory and iron-sequestering effects in response to bacterial enterobactin. PLoS Pathog. 2009;5(10):e1000622. doi: 10.1371/journal.ppat.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bao G, et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol. 2010;6(8):602–609. doi: 10.1038/nchembio.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123(7):1293–1305. doi: 10.1016/j.cell.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 105.Subcommittee on Urinary Tract Infection, Steering Committee on Quality Improvement Management, Roberts KB. Urinary tract infection: clinical practice guideline for the diagnosis and management of the initial UTI in febrile infants and children 2 to 24 months. Pediatrics. 2011;128(3):595–610. doi: 10.1542/peds.2011-1330. [DOI] [PubMed] [Google Scholar]
- 106.Hagberg L, Hull R, Hull S, McGhee JR, Michalek SM, Svanborg Eden C. Difference in susceptibility to gram-negative urinary tract infection between C3H/HeJ and C3H/HeN mice. Infect Immun. 1984;46(3):839–844. doi: 10.1128/iai.46.3.839-844.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schilling JD, Martin SM, Hung CS, Lorenz RG, Hultgren SJ. Toll-like receptor 4 on stromal hematopoietic cells mediates innate resistance to uropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2003;100(7):4203–4208. doi: 10.1073/pnas.0736473100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Karoly E, et al. Heat shock protein 72 (HSPA1B) gene polymorphism Toll-like receptor (TLR) 4 mutation are associated with increased risk of urinary tract infection in children. Pediatr Res. 2007;61(3):371–374. doi: 10.1203/pdr.0b013e318030d1f4. [DOI] [PubMed] [Google Scholar]
- 109.Hopkins WJ, Gendron-Fitzpatrick A, Balish E, Uehling DT. Time course and host responses to Escherichia coli urinary tract infection in genetically distinct mouse strains. Infect Immun. 1998;66(6):2798–2802. doi: 10.1128/iai.66.6.2798-2802.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schilling JD, Mulvey MA, Vincent CD, Lorenz RG, Hultgren SJ. Bacterial invasion augments epithelial cytokine responses to Escherichia coli through a lipopolysaccharide-dependent mechanism. J Immunol. 2001;166(2):1148–1155. doi: 10.4049/jimmunol.166.2.1148. [DOI] [PubMed] [Google Scholar]
- 111.Nelson AL, Ratner AJ, Barasch J, Weiser JN. Interleukin-8 secretion in response to aferric enterobactin is potentiated by siderocalin. Infect Immun. 2007;75(6):3160–3168. doi: 10.1128/IAI.01719-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Agace W, Hedges S, Andersson U, Andersson J, Ceska M, Svanborg C. Selective cytokine production by epithelial cells following exposure to Escherichia coli. Infect Immun. 1993;61(2):602–609. doi: 10.1128/iai.61.2.602-609.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ko YC, et al. Elevated interleukin-8 levels in the urine of patients with urinary tract infections. Infect Immun. 1993;61(4):1307–1314. doi: 10.1128/iai.61.4.1307-1314.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Heyman SN, Rosenberger C, Rosen S. Experimental ischemia-reperfusion: biases and myths-the proximal vs. distal hypoxic tubular injury debate revisited. Kidney Int. 2010;77(1):9–16. doi: 10.1038/ki.2009.347. [DOI] [PubMed] [Google Scholar]
- 115.di Mari JF, Davis R, Safirstein RL. MAPK activation determines renal epithelial cell survival during oxidative injury. Am J Physiol. 1999;277(2 pt 2):F195–F203. doi: 10.1152/ajprenal.1999.277.2.F195. [DOI] [PubMed] [Google Scholar]
- 116.Arany I, Faisal A, Nagamine Y, Safirstein RL. p66shc inhibits pro-survival epidermal growth factor receptor/ERK signaling during severe oxidative stress in mouse renal proximal tubule cells. J Biol Chem. 2008;283(10):6110–6117. doi: 10.1074/jbc.M708799200. [DOI] [PubMed] [Google Scholar]
- 117.Rosenberger C, et al. Adaptation to hypoxia in the diabetic rat kidney. Kidney Int. 2008;73(1):34–42. doi: 10.1038/sj.ki.5002567. [DOI] [PubMed] [Google Scholar]
- 118.Brezis M, Rosen S. Hypoxia of the renal medulla — its implications for disease. N Engl J Med. 1995;332(10):647–655. doi: 10.1056/NEJM199503093321006. [DOI] [PubMed] [Google Scholar]
- 119. DuBose TD Jr, Alpern R. Renal Tubular Acidosis. 8th ed. New York, New York, USA: McGraw-Hill; 2001. [Google Scholar]
- 120.Brown D, Breton S. Mitochondria-rich, proton-secreting epithelial cells. J Exp Biol. 1996;199(pt 11):2345–2358. doi: 10.1242/jeb.199.11.2345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.