

Abstract
The accumulation of somatic mutations, to which the cellular genome is permanently exposed, often leads to cancer. Analysis of any tumour shows that, besides the malignant cells, one finds other ‘supporting’ cells such as fibroblasts, immune cells of various types and even blood vessels. Together, these cells generate the microenvironment that enables the malignant cell population to grow and ultimately lead to disease. Therefore, understanding the dynamics of tumour growth and response to therapy is incomplete unless the interactions between the malignant cells and normal cells are investigated in the environment in which they take place. The complex interactions between cells in such an ecosystem result from the exchange of information in the form of cytokines- and adhesion-dependent interactions. Such processes impose costs and benefits to the participating cells that may be conveniently recast in the form of a game pay-off matrix. As a result, tumour progression and dynamics can be described in terms of evolutionary game theory (EGT), which provides a convenient framework in which to capture the frequency-dependent nature of ecosystem dynamics. Here, we provide a tutorial review of the central aspects of EGT, establishing a relation with the problem of cancer. Along the way, we also digress on fitness and of ways to compute it. Subsequently, we show how EGT can be applied to the study of the various manifestations and dynamics of multiple myeloma bone disease and its preceding condition known as monoclonal gammopathy of undetermined significance. We translate the complex biochemical signals into costs and benefits of different cell types, thus defining a game pay-off matrix. Then we use the well-known properties of the EGT equations to reduce the number of core parameters that characterize disease evolution. Finally, we provide an interpretation of these core parameters in terms of what their function is in the ecosystem we are describing and generate predictions on the type and timing of interventions that can alter the natural history of these two conditions.
Keywords: cancer ecology, evolutionary game theory, somatic evolution of cancer, cell population dynamics, fitness driven dynamics, frequency-dependent selection
1. Introduction
Cancer is the result of the accumulation of somatic mutations, to which the cellular genome is continuously exposed [1,2]. Serial accumulation of mutations can produce a cell that ignores the mechanisms which regulate growth control and in addition may acquire an ability to invade other tissues—i.e. the full cancer phenotype [2,3]. The human haploid genome is ca 3 × 109 bp in length, and an adult human body has approximately 1014 cells. DNA polymerases have an error rate of around 1 × 10−9/base/replication [4] or approximately 1 × 10−7/gene/replication [5,6]. These observations alone make it likely that there is at least one cell harbouring any one possible mutation in our body [7]. Moreover, the presence of free radicals generated by metabolism and environmental genotoxic agents such as radiation, chemicals including therapeutic agents (e.g. alkylating agents and benzene) and viruses (e.g. integrating retroviruses), can also damage the genome. Such diverse insults to the genome also in part explain the great diversity in the mutation landscape observed in tumours [8]. However, the transformed cells alone do not constitute the tumour. Indeed, analysis of any tumour shows that apart from the malignant cells, one finds many ‘supporting’ cells such as fibroblasts, immune cells of various types and blood vessels due to activation of angiogenesis. Together, these cells create a local microenvironment that enables the malignant cell population to grow and ultimately lead to disease [9]. Therefore, understanding the dynamics of tumour growth and response to therapy is incomplete unless the interactions between the malignant cells and their environment are taken into consideration. The complex interplay between the malignant cells and their environment is due to the exchange of information in the form of cytokines and adhesion-dependent interactions. Such a complex signalling process imposes costs and benefits to the participant cells that can be conveniently recast in the form of a game pay-off matrix, the ensuing dynamics being well described in terms of evolutionary game theory [10]. In the following, we provide some basic aspects of evolutionary game theory (EGT) followed by a concrete example from a well-defined disease process to illustrate the utility of the application of EGT to cancer.
2. A quick motivation for evolutionary game theory
The appearance of mutated cells that may undergo unregulated replication may be conveniently described in terms of a new species that attempts to invade a resident species (wild-type) of normal cells. We shall implement such an ecological approach resorting to the tools of EGT that, in the settings we shall adopt here, provides a description equivalent to that of the traditional equations of ecology [10].
The central equation of EGT is the so-called replicator equation (RE), which makes use of the ubiquitous—yet hard to define (see below)—concept of fitness. Let us suppose that we have a population A of normal cells of initial size NA(0), in which all cells replicate each at a rate α, which we assume to be constant. Assuming infinite resources, the equation governing the time evolution of the population is simply
Similarly, denoting by β (also assumed constant) the rate of replication of mutated cells, the mutant population B evolves in time according to the equation
In this unrealistic scenario, in which the overall population size increases exponentially, all that matters is the ratio α/β: if α/β > 1, the normal cell population will outnumber the mutant population, the opposite happening whenever α/β < 1.
A more realistic scenario is to assume that the available resources impose that the total population stays (approximately) constant at a fixed value (carrying capacity). We can include this constraint by modifying the equations above in the following way [11]:
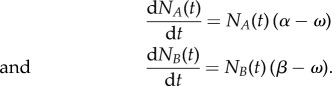 |
Imposing the conservation of total population size (NT ≡ const. = NA(t) + NB(t)) leads to NTω = NA(t)α + NB(t)β. In other words, ω stands as the average reproductive rate of the population, and now all that matters is how α and β compare with ω: the population in which cells replicate at a higher than average rate will outcompete the other. Conservation of the total size of the population means that we need only one equation to describe the two-population dynamics. Choosing the resident population, we may write
Assuming population size is large enough to convert absolute cell numbers NA into cell frequencies x (our continuous description implicitly requires this assumption), we may write
| 2.1 |
This nonlinear ordinary differential equation is a particular case of the RE. It describes the evolutionary dynamics of a sub-population of cells which (all) replicate at the same constant rate α in the presence of another sub-population of cells, all of which replicate at a constant rate β, provided the total population size is constant.
The rate of cell replication provides a convenient (and also conventional) definition of cell fitness. Indeed, in such a simple model, the fact that those cells that replicate faster outcompete the other cells, makes it natural to state that such cells have a higher fitness than the others, implicitly assuming an equivalence between these two concepts. This, however, may prove insufficient to identify cell fitness, as we demonstrate in §3.
Furthermore, in the previous example, the fitness of cells of each sub-population remains constant in time, regardless of the total number of cells of a given species. In other words, it does not matter if there is 1 cell or 1000 cells of the same type, their replication rate is the same. This is clearly a crude assumption. There are many examples (and practitioners in the field know it very well) in which (sub-population) size matters, and this feature is in no way included in the equation above. Importantly, this feature also severely constrains the type of disease progression which one can describe, leaving out several simple scenarios, such as the one we shall use to describe the homoeostatic state of disease-free individuals.
EGT provides one possible means to overcome such type of shortcomings. If size matters, then fitness should be frequency-dependent. If we replace α in equation (2.1) by φA(x) and β by φB(x), we may now write the general form of the RE (replacing, as usual, the time derivative of x by 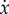 ),
),
| 2.2 |
In its simplest form, the frequency dependence of the fitness results, in EGT, from assuming that cells engage in a game in which every cell interacts with any other cell with the same likelihood (well-mixed approximation). The well-mixed approximation, also known as mean-field approximation, is well suited to discuss cell populations with high mobility, as one usually finds in haematopoiesis (and, in multiple myeloma (MM) bone disease that we will address in detail below). The approximation is less justifiable in, e.g. solid tumours, where space and limited (if any) cell motility may play a very important role. With this proviso in mind, one may appreciate the beauty and simplicity of the formalism that unfolds under this approximation. When two cells interact, the result can be computed by inspection of the so-called pay-off matrix
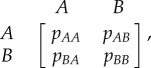 |
where pik stands for what a cell of type i gets from interacting with a cell of type k. Interaction here is used in a very general sense, to include competition for space and nutrients as well as exchange of information through cytokines and growth factors. In fact, one may assume that this is how the fitness of a cell is (directly or indirectly) modified, when this cell is in the presence of another cell (which can be of the same type or of another type). In the well-mixed approximation, it is trivial to compute the average fitness of a cell of each type. We may write
thus explicitly specifying the form of frequency dependence that results from treating cell competition in a population of constant size by means of EGT. Naturally, it is trivial to recover the frequency independent scenario that we started with: whenever pAA = pAB = α and pBA = pBB = β, we recover equation (2.1) and the population dynamics no longer exhibits a frequency-dependent behaviour. This, in turn, makes it clear the meaning of frequency independence in this framework: it does not matter with which cell type a focal cell interacts, the result is always the same.
The generalization of these results for the case of three cell types—that we shall address in the following—is trivial: we now have to deal with two REs instead of one, as now the number of independent frequencies is 2, given that x + y + z = 1 (x, y and z are the frequencies of species 1, 2 and 3, respectively) and the pay-off matrix will be a 3 × 3 matrix. This generalization is carried out in the appendix A, where some properties that derive from the general behaviour of EGT are also provided, and which are relevant for the discussion that follows, in which we apply EGT to MM bone disease. But before, we digress on the concept of fitness.
3. Hierarchical tissue organization and cell fitness
Given the inescapable appearance of mutations, one would expect that evolution has selected for a tissue architecture that minimizes the risk of retention of mutant populations. Indeed, the probability that a particular mutation occurs in a given tissue is proportional to the population of cells at risk, the mutation rate and the average lifetime of cells in that population [12]. Most tissues (including haematopoiesis and epithelia) have a tree-like architecture in which the vast majority of cells have a relatively short lifetime. At the root of this tree lie (tissue specific) stem cells that are operationally defined by their ability to self-renew and give rise to progeny cells that can differentiate and repopulate the entire variety of lineages and are hence able to generate and maintain a specific tissue. In general, the stem cell population is but a tiny fraction of the cells making every tissue, but the definition of a stem cell does not require this characteristic. Stem cell division gives rise to more differentiated cells that often replicate at faster rates but which contribute to tissue maintenance for shorter periods of time. Mature cells in the tissue are eliminated, on average, at a constant rate by, for example, apoptosis in haematopoiesis or by shedding from the surface of epithelia.
Linking haematopoietic stem cells and mature blood cells is often a hierarchically organized process where cells divide and become increasingly differentiated (figure 1). Such a hierarchical tissue architecture has been shown to minimize the impact of most mutations that occur in its cells [13], given the fact that most of them occur in short-lived cell lineages [7]. To capture the dynamics and architecture of haematopoiesis, one needs to consider at least two fundamental processes: cellular reproduction and differentiation [14] (figure 1), processes which are stochastic and coupled [15,16]. Needless to say, this is a minimal model of cell division. There is ample experimental evidence that cells are able to undergo asymmetric cell division, a process that is not explicitly considered in such a minimalistic treatment. The explicit inclusion of asymmetric cell division [17–19] requires additional parameters, although the process remains stochastic in nature.
Figure 1.
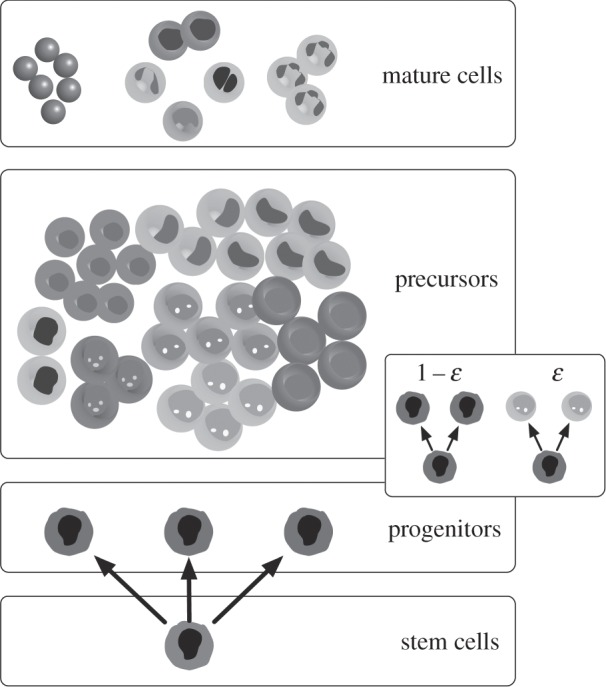
The tree-like structure of haematopoiesis. Haematopoiesis exhibits a hierarchical architecture so characteristic of general body tissues. At the root of this tree-like structure, one finds the tissue-specific stem cells, which can self-renew (reproduction) and differentiate into all other types of haematopoietic cells. The figure illustrates specifically the most common haematopoietic cell types, from progenitors to mature cells. Along this path of differentiation, we also find the so-called precursor cells. In the mathematical model of haematopoiesis referred to in the main text, cell reproduction and differentiation constitutes a coupled stochastic event, occurring with probability 1 − ɛ and ɛ, respectively, as illustrated on the right.
A hierarchical tissue organization provides an excellent example to demonstrate that fitness advantage, at the cell level, may not result exclusively from the rate of division. In haematopoiesis, for instance, the fact that besides reproduction, differentiation is also required to account for the tissue architecture, changes the relation between cell fitness and cell reproductive rate. As a result, fitness differences between cell types may arise from mutations that do not change cell reproductive rates: such is the case in chronic myeloid leukaemia (CML), where the fitness of cancer cells relative to that of normal cells can be written in terms of the differentiation rate ɛk of each cell type (figure 1) [7]
As shown in figure 1, ɛk gives the probability that, upon cell division, symmetric cell differentiation occurs. When combined with a hierarchical, tree-like structure, and the staggering number of haematopoietic cells in the bone marrow, any changes in the value of this probability drastically change the competitive odds of cancer cell populations. In haematopoiesis, ɛnormal = 0.84; fits to disease progression curves, in turn [20–23], show that for CML cells we have ɛcancer = 0.72 which means that φcancer ≈ 2φnormal without any changes in the rate of cell division, which remains unchanged according to the stochastic analysis of ref. [20]. Given that this tissue architecture is so ubiquitous, we should not only relate fitness simply to the reproductive cell rate, but also to the pattern of reproduction (symmetric versus asymmetric) [17,22], and the structural and spatial organization of cells within the specific tissue.
How does this translate into the equations of EGT? Given the form of the REs, all one has to do is to include, on their right-hand-side, the appropriate fitness of each species, together with the average fitness of the population. In other words, one may generally use the central equations of EGT, even if fitness does not result exclusively from the reproductive rates of whatever species. In fact, EGT has been widely applied, recently, to study the dynamics of social systems, in which case fitness relates to social success [24,25], and not to reproduction.
4. Application of evolutionary game theory to the ecology of multiple myeloma bone disease
We will now illustrate the use of EGT to model and understand a well-known form of cancer. MM is a malignant plasma cell neoplasm that is characterized by a variable constellation of symptoms, the cardinal of which are bone loss, anaemia, hypercalcaemia and renal failure (CRAB criteria). Bone disease is observed in up to 80% of patients with MM and is an important cause of morbidity due to pain, the risk of pathological fractures and neurological deficits (given the risk of spinal cord compression). Bone loss in MM follows two broad patterns: focal lytic disease [26,27] or diffuse loss leading to osteoporosis [28]. The generation of the first MM cell is a multistep process requiring a series of mutations that transform a normal plasma cell into an MM cell [29,30]. We shall assume that this (complex) process has already occurred, i.e. the first MM has already appeared, and we shall concentrate on investigating its possible clonal expansion. It appears that malignant plasma cells require the bone marrow microenvironment for their growth and survival. In most patients, myeloma is preceded by an asymptomatic premalignant state called monoclonal gammopathy of undetermined significance (MGUS) in which the clonal plasma cell burden in the bone marrow is lower than in myeloma and by definition the CRAB criteria are absent. Progression to myeloma is generally associated with an increase in the clonal plasma cell population and the appearance of symptoms as discussed above. This transition from MGUS to symptomatic myeloma may be relatively abrupt (often associated with the acquisition of additional genetic events such as loss of TP53, amplification of a segment of the short arm of chromosome 1, etc.) or the consequence of a slow but relentless expansion of the clonal plasma cell population.
Normal bone remodelling is a consequence of the dynamic balance between osteoclast-mediated (OC) bone resorption and bone formation due to osteoblast (OB) activity (see top of figure 3). The interplay between these two populations is complex but partly dependent on the receptor activator of nuclear factor-κB (RANK), RANK ligand (RANKL) and osteoprotegerin (OPG) axis as well as MIP-1α and interleukin (IL)-1β [31–33]. This dynamic balance can be captured trivially in the framework of EGT, but it requires an explicit frequency dependence to be in place. Indeed, frequency-independent population dynamics cannot capture such a simple process. Let us then assume that we have two cell species which exhibit a stable balance between them. This can be obtained by means of what is known in game theory as a coexistence game, which can be realized by a pay-off matrix satisfying pBA > pAA and pAB > pBB, e.g. (e and a are both positive reals),
 |
4.1 |
The designation coexistence game is intuitive, given the ensuing dynamics associated with the pay-off matrix above. Indeed, we may write
where x stands for the fraction of OC cells and (1 − x) for the fraction of OB cells. The RE, equation (2.2) now reads
with fixed points (that is, the values of x at which 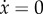 ) at x* = 0, x* = 1 and x* = a/(a + e), this last one associated with the condition φOC(x*) = φOB(x*). A typical plot of
) at x* = 0, x* = 1 and x* = a/(a + e), this last one associated with the condition φOC(x*) = φOB(x*). A typical plot of 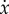 —the so-called gradient of selection—as a function of x has the behaviour depicted in figure 2. The horizontal arrows in figure 2 show the direction of selection associated with the sign of
—the so-called gradient of selection—as a function of x has the behaviour depicted in figure 2. The horizontal arrows in figure 2 show the direction of selection associated with the sign of 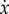 and show that both x* = 0 and x* = 1 are unstable fixed points, in the sense that any perturbation deviating the population from such a state will induce it to move away from the fixed point. The opposite happens for x* = a/(a + e), in which case any deviation from its position will induce a dynamics that restore it to that fixed point. Hence, the interior fixed point is stable, thus reflecting the dynamical balance we wanted to reproduce.
and show that both x* = 0 and x* = 1 are unstable fixed points, in the sense that any perturbation deviating the population from such a state will induce it to move away from the fixed point. The opposite happens for x* = a/(a + e), in which case any deviation from its position will induce a dynamics that restore it to that fixed point. Hence, the interior fixed point is stable, thus reflecting the dynamical balance we wanted to reproduce.
Figure 2.
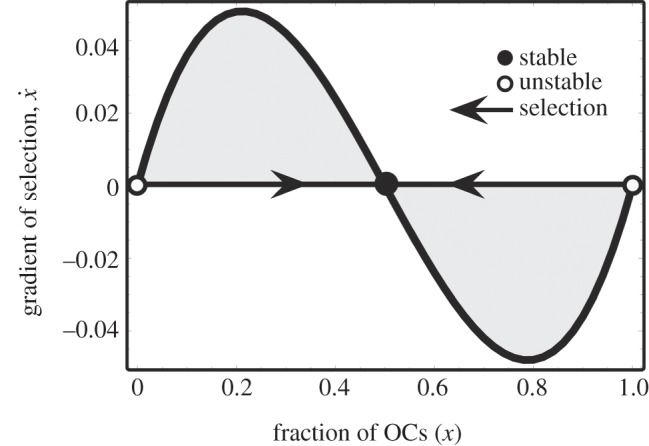
Gradient of selection for the coexistence dynamics among two cell species: OCs and OBs (a = e = 0.5, see equation (4.1)). In the absence of MM cells (see below), OB and OC engage in a coexistence game, the internal equilibrium of which reflects normal physiology. Any disturbance from this equilibrium will result in an evolutionary dynamics that will restore the original state, enabling normal tissue to adapt to changing demands and repair after injury.
The appearance and expansion of MM cells disrupts this dynamic equilibrium between OB and OC in favour of OC [27,31–33], with a disease progression time scale of the order of a few years [26]. The process can be subdivided into two components: (i) MM and stromal cells produce a variety of cytokines including IL-1β [34], RANKL [35] and MIP-1α [36] (summarized as ‘osteoclast-activating factors’, OAFs [31,33,37]) that recruit OC precursors and stimulate growth of the OC population; and (ii) secretion of Dickkopf-1 (Dkk-1) by myeloma cells directly inhibits Wnt3a-regulated differentiation of OBs, reduces OPG expression and alters the OPG-RANKL axis against OB activity [32,37–40]. The biochemical interactions between MM, OC and OB cells highlight the dependence of MM cells on the bone marrow microenvironment, at least early in the course of the disease [30,41]. Here, the microenvironment refers to the different cell populations within the bone marrow, including OC and OB cells together with blood vessels that provide the conditions necessary for myeloma cell growth and survival.
As illustrated in figure 3, IL-6 [42] and osteopontin [43], produced by OC cells stimulate growth of the MM cell population, hence conferring a net benefit to MM cells. On the other hand, production of OAF by MM cells (i) confers a net benefit to OC cells. Mechanism (ii) also leads to a potential disadvantage for OB cells in the presence of MM cells, while MM cells are unaffected by the presence of OB cells. Thus, figure 3 provides a convenient means to translate the complex exchange of chemical signals between different cell types into net benefits and costs of each cell type. We can therefore recast the frequency-dependent balance between these cell populations in terms of an evolutionary game encompassing three unconditional strategies, or cell types—OC, OB and MM, with the following (extended with respect to equation (4.1)) pay-off matrix:
 |
4.2 |
In the matrix above, all parameters a, b, c, d and e are non-negative, which means that the only pay-off entry that is negative is that associated with –d. The present formulation is the mathematical equivalent of an ecosystem where the interactions between species (MM, OC and OB) are determined by matrix (4.2). Without loss of generality (see appendix A), we assume that interactions between cells of the same type are neutral (with null entries). In the absence of MM, we recover the 2 × 2 matrix (4.1) written before.
Figure 3.
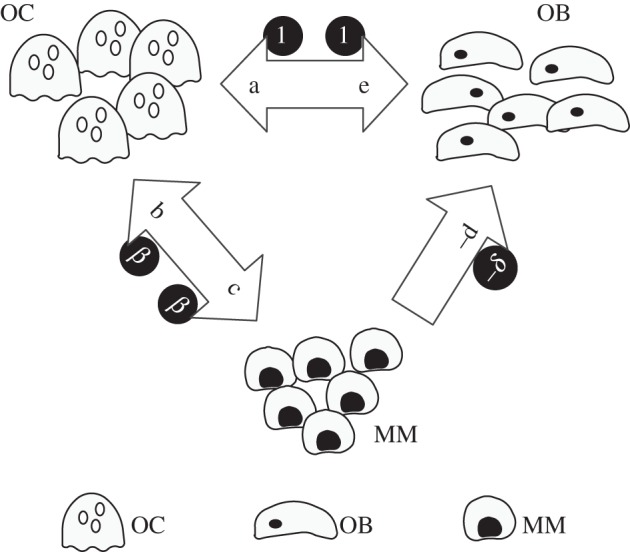
Pathological bone turnover. The presence of MM cells alters bone homoeostasis by cytokine production (e.g. IL-1β, RANKL, MIP-1α) that recruit and activate OC, increasing bone resorption. OC may also produce growth factors (IL-6) for MM cells, which may also secrete cytokines that suppress OB activity. The synergy between MM and OC confers them an advantage with respect to OB. The arrows indicate the net effects associated with pay-off matrix entries in equations (4.2) and (4.3). The invariance of the evolutionary dynamics under projective transformations (see main text for details) leads to the definition of the minimal pay-off matrix in equation (4.3), with parameters depicted inside circles, that accounts for all dynamical scenarios encompassing the coevolution of these three cell types.
What happens now when we embed the OB–OC dynamics above in the more general framework that includes MM cells? As shown in appendix A, we can make use of some very general properties that accrue to EGT combined with the RE, to further reduce the number of parameters in matrix (4.2) from 5 to 2 (β and δ), without changing the nature of the dynamics entwining MM, OB and OC cells. This is illustrated in figure 3 by placing the new variables β and δ inside circles. The minimal pay-off matrix, that is, the pay-off matrix with the smallest number of parameters compatible with the dynamics involving MM, OB and OC cells now reads
 |
4.3 |
5. Results and discussion
Figure 4 provides an overview of the possible scenarios that emerge from the evolutionary dynamics associated with pay-off matrix (4.3), which provides a minimal description of the disease and identifies the core elements that determine tumour behaviour and dynamics. Unlike the OB–OC dynamics, which could be analysed along the segment 0 ≤ x ≤ 1, the OB–OC–MM dynamics proceeds in a two-dimensional space (called simplex) that can be represented by the equilateral triangles in figure 4. Each vertex of the simplex represents a monotypic population, that is, with populations in which 100% of the cells are of only one type. Edges of the simplex represent population configurations in which at least one of the cell types is missing. The interior of the simplex, in turn, corresponds to population configurations in which all cell types coexist, albeit with different fractions in different points. The unique feature of such a simplex representation is that, at every point, the sum of the fractions of the three cell types is 100%.
Figure 4.
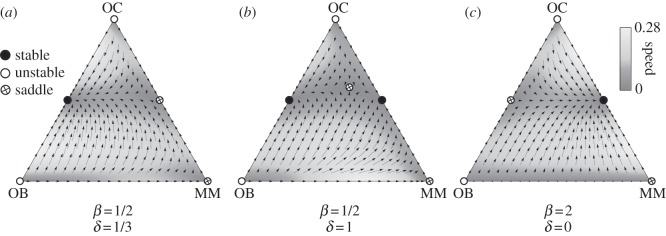
Evolutionary dynamics of OC, OB and MM cell types. Vertices mean that cell population is monotypic. Bone homoeostasis occurs in the absence of MM (OC–OB line), remaining stable in the presence of MM if β < 1 (a) and unstable otherwise (b,c). Whenever β < 1 but β + δ > 1 (b), a saddle point appears in the interior of the simplex, which maintains normal homoeostasis as a possible outcome of the coevolution of the three cells types. Unfortunately, most of the existing scenarios suggest that β > 1 is the rule, which means that the scenario depicted in panel (c) applies, even in the limit when δ = 0 (see main text for details).
It is important to stress that the non-trivial identification of the key parameters β and δ could not be anticipated from the rationale underlying the model set-up, as illustrated in figure 3 and out of which matrix (4.2) was derived. Instead, it results from the properties of EGT and the RE, which dictate that the evolutionary outcome of the disease can be equivalently analysed in terms of matrix (4.3). Thus, under the present model assumptions, β and δ are sufficient to characterize myeloma bone disease.
The three strategy game leads to unstable equilibria in the sense described before whenever the population is monotypic: the presence of a single cell of any of the other types leads to the coexistence of the two strategies, as the pairwise games OB–OC and OC–MM turn out to be coexistence games, with the same structure as already discussed in connection with equation (4.1) [44]. The number and nature of the fixed points in the simplex will naturally depend on the relative balance between β and δ in the pay-off matrix. If the net benefit that OC cells obtain from MM cells is smaller than what they get from OB cells (β < 1), the population of MM cells can go extinct, and OB and OC may again re-establish the stable dynamic equilibrium (figure 4a). Also, whenever β < 1 but β + δ > 1 (figure 4b), there appears a typical saddle point structure in the interior of the simplex (because the simplex now spans a two-dimensional space), which still ensures that normal homoeostasis is a possible ‘end-game’ of the coevolutionary process. In this case (see below), therapies that change β may provide important contributions to overall disease eradication. Unfortunately, various studies suggest that in general, β > 1 [41,45]. In this case, the only stable equilibrium is the coexistence of MM and OC cells (figure 4c). In this extreme situation, a part of bone is completely devoid of OB and as the bone approaches this state is at increasing risk of fracture, a common feature of MM [26,46]. Such a coexistence between MM and OC (figure 5c) will prevail even if δ = 0 (MM and OB are neutral with respect to each other). However, changes in δ may have a significant impact on the life history of the disease and associated progression time [47].
Figure 5.

Type and timing of MM treatment. (a) Typical scenario of MM disease progression portrayed in the simplex, where at a certain moment, BMT is carried out, here represented by the straight dashed arrow. This means that the ratio OC/OB remains unaffected while the MM disease burden is significantly reduced (though not eliminated). This ultimately leads to relapse, as shown in (b) where the explicit time dependence of the different cell types is shown. In (c,d), we depict what one would expect via administration of a drug that successfully alters β from the conventional regime (β = 3.0 > 1 and δ = 1, panels (a,b)) to the more favourable regime β < 1 (β = 1/2 and δ = 1, panels (c,b)), leading to the appearance of a saddle point structure. If therapy is administered early on during disease progression, then therapy alone can be curative, as intervention occurs in the non-shaded area of the simplex. However, if therapy is provided later (shaded area of the simplex, see panel (d)), then it cannot, alone, lead to cure, as disease progression will still proceed towards the MM–OC dire equilibrium. In this case, however, BMT may bring the patient into the non-shaded area, in which the stable equilibrium will correspond to normal homoeostasis. In other words, when combined with therapy, BMT may become curative and, in many cases, crucial. Other parameters: fraction of MM cells before transplant equal to 0.2 (panels (a–c)) and 0.4 (panel (d)); MM burden reduction upon BMT is 10−4 in all panels; in panel (b), t0 stands for the total progression time for the given parameters.
Increasing β will lead to more bone destruction, higher tumour burden and faster tumour progression. This behaviour is observed clinically: patients with higher MIP-1α levels (increasing β) tend to have more bone resorption and lytic bone lesions [48,49] and shorter survival due to a higher tumour burden [48]. Consequently, therapies which suppress or reduce β (inhibiting, e.g. MIP-1α secretion or IL-1β [50,51]) should decrease the number of lytic lesions and the speed of disease progression, prolonging survival [52]. Similarly, any therapy that decreases δ (e.g. Dkk-1) should reduce the myeloma burden, slow the progression of the disease and improve bone mass [53]. On the contrary, increasing δ for fixed β—that is, increasing the disadvantage of OB cells in the presence of MM cells—leads to disease dynamics in which considerable bone loss occurs without a significant increase in the MM population. This can explain at least two clinical scenarios: (i) instances of myeloma-induced osteoporosis without a massive MM cell burden and (ii) loss of cortical bone thickness and alteration of its microarchitecture in patients with the premalignant state known as MGUS [54,55]. In the latter scenario, patients with MGUS have been found to have higher levels of the Wnt pathway inhibitor Dkk-1 that inhibits OB growth [54]. Similarly, the model would suggest that any therapy designed to suppress OC growth should indirectly improve outcomes in patients with myeloma due to its effects on MM cells. Indeed, this is the case as shown by the UK MRC Myeloma IX trial where zoledronic acid therapy, directed at OC cells, improved survival in a large cohort of patients [56].
As stated before, under the present model assumptions, β and δ are sufficient to characterize myeloma bone disease. However, it is well known that the natural history of the disease is variable, presumably due to genetic and epigenetic differences in myeloma cells from different patients [57,58]. Normal physiology also varies from individual to individual. Such variability is actually taken into consideration in pay-off matrix (4.2). Any pay-off matrix of the form of matrix (4.2) will exhibit the core dynamics just described, resulting from the analysis of matrix (4.3). However, the location of the stable, saddle and unstable fixed points of the dynamics will change for different set of values of the pay-off entries {a,b,c,d,e}, which are expected to be patient specific, reflecting tumour–host interactions and variability of disease due to differences in the host rather than the malignant cell genotype. Thus, while matrix (4.3) describes the core dynamics that accrues to MM bone disease, its manifestation may change from patient to patient as a result of a different pay-off matrix (4.2).
The model reviewed here shows that, under normal conditions (when β > 1) the evolutionary dynamics of MM leads always to a very negative prognosis, a feature which is very well known to clinicians. In fact, and whenever possible, autologous bone marrow transplantation (BMT) is chosen to ameliorate the patient's condition by reducing the disease burden further. However, it is trivial to show that, whenever β > 1, BMT cannot be curative in MM. This is shown in figure 5a, where the impact of BMT is depicted in the simplex. Assuming that the relative proportion of OB and OC cells does not change under BMT, then BMT corresponds to drawing a straight line joining the MM vertex and the population configuration at the time of BMT (squares in figure 5, see 5a). BMT will lead to a significant reduction of the disease burden (by reducing by several logs the number of MM cells) but, as shown both in figure 5a and in the time evolution depicted in figure 5b, the dynamics will inevitably resume to the standard case already discussed. As a result, BMT will postpone the ‘end-game’, but relapse is inevitable.
On the other hand, development of a drug treatment that changes β to values lower than 1 will lead to the appearance of the saddle point already shown in figure 4b. Then, it all depends when the drug is provided to the patient, as shown in figure 5c,d. If the patient is treated well in the beginning of the disease, there is hope that the drug treatment alone will be curative, as β < 1 will fundamentally change the dynamics of disease progression (figure 5c). However, in those cases where patients are diagnosed too late (that is, in configurations falling in the shaded area, see figure 5d), BMT can now be curative, when combined with such a putative drug treatment. Indeed, BMT is now able to bring the patient into the area of the simplex where treatment alone will be curative, as opposed to the initial case, where treatment alone would be helpless.
We also note that the model can well explain the slow but inexorable progression of plasma cell expansion and further bone loss seen in some patients with MGUS as they evolve into MM. In addition, one can envision a scenario where the small plasma cell population can acquire additional mutations that result in changes in β and/or δ that can lead to a rapid change in the dynamics as is sometimes observed in this disease. Our model would hypothesize that patients with MGUS who have high MIP-1α or Dkk-1 levels would be at high risk of progression to myeloma and perhaps these biomarkers can be used to estimate the risk of progression of MGUS to myeloma and identify a cohort of patients that may require more careful observation. It is possible that therapeutic intervention in these ‘higher risk’ MGUS patients may not only prevent symptomatic bone loss but also slow or prevent progression to active myeloma and perhaps cure some of these individuals.
The approach developed here is general and readily applicable to other diseases. Furthermore, it provides a novel paradigm for dealing with cancer eradication: to harness the power of evolutionary forces in the multi-species cell dynamics to eradicate the cancer cells. In this sense, therapies should aim at changing the rules of engagement between different cell types: in our case, therapies should act to change parameters β and δ. To the extent that such therapies change the dynamics, enabling normal cells to outcompete the malignant clone, one gets total remission as a result. Last but not least, the game at stake need not be a simple two-species game as the one employed here. Situations where the game involves many cells at once in each interaction are easily conceivable. Work along these lines is in progress.
Funding statement
This work was supported by Mayo Foundation (D.D.) and FCT-Portugal (J.M.P. and F.C.S.) through grant nos. PTDC/MAT/122897/2010 and EXPL/EEI-SII/2556/2013, and by multi-annual funding of CBMA and INESC-ID (under the projects PEst-OE/BIA/UI4050/2014 and PEst-OE/EEI/LA0021/2013) also provided by FCT-Portugal.
Appendix A
We employ the RE to describe the frequency-dependent evolutionary dynamics of a well-mixed populations involving three cell types [10]. Consequently, we assume that (i) no new mutations occur during tumour dynamics except those which initiated the tumour and (ii) deterministic cell population dynamics. Tumour dynamics is conveniently represented in the simplex (an equilateral triangle for three strategies) at every point of which we have the relative frequencies of OB, OC and MM populations that sum up to 1. Let us denote by xi(t), the relative frequencies of the cell types: x1(t) (OC cells), x2(t) (OB cells) and x3(t) (MM cells). The REs read
where the fitness of each cell type is given in terms of a game pay-off matrix Aij by
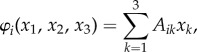 |
whereas the average fitness of the population reads
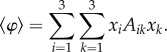 |
The benefits and costs resulting from the interacting cell populations are captured in the initial pay-off matrix (1.2), here associated with matrix Aij. We may reduce this matrix to the minimal pay-off matrix (4.3) by taking into account that the nature of the fixed points of the evolutionary dynamics (though not their location) remains unaffected under a projective transformation of the relative cells frequencies [10], leading to [47] β = c/e and δ = dc/be. Note further, that the REs and associated dynamics remain unaffected if we add an arbitrary constant to each column of the pay-off matrix. In other words, it is always possible to zero all the diagonal elements of the game pay-off matrix.
The fixed points (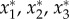 ) of the evolutionary dynamics under matrix Bij are readily found. Two vertices of the simplex, (0, 1, 0) and (1, 0, 0) are unstable fixed points, whereas the third is a saddle point. The fixed point (1/2, 1/2, 0) associated with normal physiology is unstable whenever β > 1, being stable otherwise; the fixed point (1/2, 0, 1/2) is a stable fixed point whenever β > 1 or whenever β < 1 but β + δ > 1; in this last situation, a saddle point arises in the interior of the simplex [47], located at (η−1 = (1 − β)1 + δ+ β(δ + β − 2)) q* = (ηδ, ηβ(δ + β − 1), η(1 − β)).
) of the evolutionary dynamics under matrix Bij are readily found. Two vertices of the simplex, (0, 1, 0) and (1, 0, 0) are unstable fixed points, whereas the third is a saddle point. The fixed point (1/2, 1/2, 0) associated with normal physiology is unstable whenever β > 1, being stable otherwise; the fixed point (1/2, 0, 1/2) is a stable fixed point whenever β > 1 or whenever β < 1 but β + δ > 1; in this last situation, a saddle point arises in the interior of the simplex [47], located at (η−1 = (1 − β)1 + δ+ β(δ + β − 2)) q* = (ηδ, ηβ(δ + β − 1), η(1 − β)).
References
- 1.Cairns J. 1975. Mutation selection and the natural history of cancer. Nature 255, 197–200. ( 10.1038/255197a0) [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144, 646–674. ( 10.1016/j.cell.2011.02.013) [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Kinzler KW. 2004. Cancer genes and the pathways they control. Nat. Med. 10, 789–799. ( 10.1038/nm1087) [DOI] [PubMed] [Google Scholar]
- 4.Kunkel TA, Bebenek K. 2000. DNA replication fidelity 1. Annu. Rev. Biochem. 69, 497–529. ( 10.1146/annurev.biochem.69.1.497) [DOI] [PubMed] [Google Scholar]
- 5.Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R, Luzzatto L. 2005. A quantitative measurement of the human somatic mutation rate. Cancer Res. 65, 8111–8117. ( 10.1158/0008-5472.CAN-04-1198) [DOI] [PubMed] [Google Scholar]
- 6.Peruzzi B, Araten DJ, Notaro R, Luzzatto L. 2010. The use of PIG-A as a sentinel gene for the study of the somatic mutation rate and of mutagenic agents in vivo. Mutat. Res. Rev. Mutat. Res. 705, 3–10. ( 10.1016/j.mrrev.2009.12.004) [DOI] [PubMed] [Google Scholar]
- 7.Traulsen A, Pacheco JM, Luzzatto L, Dingli D. 2010. Somatic mutations and the hierarchy of hematopoiesis. Bioessays 32, 1003–1008. ( 10.1002/bies.201000025) [DOI] [PubMed] [Google Scholar]
- 8.Kandoth C, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. ( 10.1038/nature12634) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Junttila MR, de Sauvage FJ. 2013. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354. ( 10.1038/nature12626) [DOI] [PubMed] [Google Scholar]
- 10.Hofbauer J, Sigmund K. 1998. Evolutionary games and population dynamics. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 11.Nowak MA. 2006. Evolutionary dynamics. Cambridge, MA: Belknap/Harvard. [Google Scholar]
- 12.Traulsen A, Pacheco JM, Imhof LA. 2006. Stochasticity and evolutionary stability. Phys. Rev. E Stat. Nonlin. Soft Matter. Phys. 74, 021905 ( 10.1103/PhysRevE.74.021905) [DOI] [PubMed] [Google Scholar]
- 13.Lopes JV, Pacheco JM, Dingli D. 2007. Acquired hematopoietic stem-cell disorders and mammalian size. Blood 110, 4120–4122. ( 10.1182/blood-2007-05-089805) [DOI] [PubMed] [Google Scholar]
- 14.Dingli D, Traulsen A, Pacheco JM. 2007. Compartmental architecture and dynamics of hematopoiesis. PLoS ONE 2, e345 ( 10.1371/journal.pone.0000345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon M, Blackett N. 1994. Routes to repopulation: a unification of the stochastic model and separation of stem-cell subpopulations. Leukemia 8, 1068–1072; discussion 1072–3. [PubMed] [Google Scholar]
- 16.Abkowitz JL, Catlin SN, Guttorp P. 1996. Evidence that hematopoiesis may be a stochastic process in vivo. Nat. Med. 2, 190–197. ( 10.1038/nm0296-190) [DOI] [PubMed] [Google Scholar]
- 17.Dingli D, Traulsen A, Michor F. 2007. (A) symmetric stem cell replication and cancer. PLoS Comput. Biol. 3, e53 ( 10.1371/journal.pcbi.0030053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shahriyari L, Komarova NL. 2013. Symmetric vs. asymmetric stem cell divisions: an adaptation against cancer? PLoS ONE 8, e76195 ( 10.1371/journal.pone.0076195) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wodarz D, Komarova NL. 2005. Computational biology of cancer: lecture notes and mathematical modeling. Singapore: World Scientific. [Google Scholar]
- 20.Lenaerts T, Pacheco JM, Traulsen A, Dingli D. 2010. Tyrosine kinase inhibitor therapy can cure chronic myeloid leukemia without hitting leukemic stem cells. Haematologica 95, 900–907. ( 10.3324/haematol.2009.015271) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lenaerts T, Castagnetti F, Traulsen A, Pacheco JM, Rosti G, Dingli D. 2011. Explaining the in vitro and in vivo differences in leukemia therapy. Cell cycle 10, 1540–1544. ( 10.4161/cc.10.10.15518) [DOI] [PubMed] [Google Scholar]
- 22.Traulsen A, Pacheco JM, Dingli D. 2010. Reproductive fitness advantage of BCR-ABL expressing leukemia cells. Cancer Lett. 294, 43–48. ( 10.1016/j.canlet.2010.01.020) [DOI] [PubMed] [Google Scholar]
- 23.Dingli D, Traulsen A, Pacheco JM. 2008. Chronic myeloid leukemia: origin, development, response to therapy, and relapse. Clin. Leuk. 2, 133–139. ( 10.3816/CLK.2008.n.017) [DOI] [Google Scholar]
- 24.Santos FC, Pacheco JM. 2011. Risk of collective failure provides an escape from the tragedy of the commons. Proc. Natl Acad. Sci. USA 108, 10 421–10 425. ( 10.1073/pnas.1015648108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vasconcelos VV, Santos FC, Pacheco JM. 2013. A bottom-up institutional approach to cooperative governance of risky commons. Nat. Clim. Change 3, 797–801. ( 10.1038/nclimate1927) [DOI] [Google Scholar]
- 26.Kyle RA, Rajkumar SV. 2004. Multiple myeloma. N. Engl. J. Med. 351, 1860–1873. ( 10.1056/NEJMra041875) [DOI] [PubMed] [Google Scholar]
- 27.Berenson JR. 2001. Bone disease in myeloma. Curr. Treat. Options Oncol. 2, 271–283. ( 10.1007/s11864-001-0041-5) [DOI] [PubMed] [Google Scholar]
- 28.Harper KD, Weber TJ. 1998. Secondary osteoporosis. Diagnostic considerations. Endocrinol. Metab. Clin. North Am. 27, 325–348. ( 10.1016/S0889-8529(05)70008-6) [DOI] [PubMed] [Google Scholar]
- 29.Bergsagel PL, Kuehl WM. 2005. Molecular pathogenesis and a consequent classification of multiple myeloma. J. Clin. Oncol. 23, 6333–6338. ( 10.1200/JCO.2005.05.021) [DOI] [PubMed] [Google Scholar]
- 30.Kuehl WM, Bergsagel PL. 2002. Multiple myeloma: evolving genetic events and host interactions. Nat. Rev. Cancer 2, 175–187. ( 10.1038/nrc746) [DOI] [PubMed] [Google Scholar]
- 31.Roodman GD. 2004. Pathogenesis of myeloma bone disease. Blood Cells Mol. Dis. 32, 290–292. ( 10.1016/j.bcmd.2004.01.001) [DOI] [PubMed] [Google Scholar]
- 32.Terpos E, Sezer O, Croucher P, Dimopoulos MA. 2007. Myeloma bone disease and proteasome inhibition therapies. Blood 110, 1098–1104. ( 10.1182/blood-2007-03-067710) [DOI] [PubMed] [Google Scholar]
- 33.Raje N, Roodman GD. 2011. Advances in the biology and treatment of bone disease in multiple myeloma. Clin. Cancer Res. 17, 1278–1286. ( 10.1158/1078-0432.CCR-10-1804) [DOI] [PubMed] [Google Scholar]
- 34.Dinarello CA. 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550. ( 10.1146/annurev.immunol.021908.132612) [DOI] [PubMed] [Google Scholar]
- 35.Croucher PI, et al. 2001. Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood 98, 3534–3540. ( 10.1182/blood.V98.13.3534) [DOI] [PubMed] [Google Scholar]
- 36.Choi SJ, Cruz JC, Craig F, Chung H, Devlin RD, Roodman GD, Alsina M. 2000. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood 96, 671–675. [PubMed] [Google Scholar]
- 37.Roux S, Mariette X. 2004. The high rate of bone resorption in multiple myeloma is due to RANK (receptor activator of nuclear factor-κB) and RANK ligand expression. Leuk. Lymphoma 45, 1111–1118. ( 10.1080/10428194310001593193) [DOI] [PubMed] [Google Scholar]
- 38.Qiang YW, Barlogie B, Rudikoff S, Shaughnessy JD., Jr 2008. Dkk1-induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone 42, 669–680. ( 10.1016/j.bone.2007.12.006) [DOI] [PubMed] [Google Scholar]
- 39.Qiang YW, Chen Y, Stephens O, Brown N, Chen B, Epstein J, Barlogie B, Shaughnessy JD., Jr 2008. Myeloma-derived Dickkopf-1 disrupts Wnt-regulated osteoprotegerin and RANKL production by osteoblasts: a potential mechanism underlying osteolytic bone lesions in multiple myeloma. Blood 112, 196–207. ( 10.1182/blood-2008-01-132134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiang YW, Shaughnessy JD, Jr, Yaccoby S. 2008. Wnt3a signaling within bone inhibits multiple myeloma bone disease and tumor growth. Blood 112, 374–382. ( 10.1182/blood-2007-10-120253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Epstein J, Yaccoby S. 2003. Consequences of interactions between the bone marrow stroma and myeloma. Hematol. J. 4, 310–314. ( 10.1038/sj.thj.6200313) [DOI] [PubMed] [Google Scholar]
- 42.Jelinek DF. 1999. Mechanisms of myeloma cell growth control. Hematol. Oncol. Clin. North Am. 13, 1145–1157. ( 10.1016/S0889-8588(05)70117-9) [DOI] [PubMed] [Google Scholar]
- 43.Abe M, et al. 2004. Osteoclasts enhance myeloma cell growth and survival via cell–cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood 104, 2484–2491. ( 10.1182/blood-2003-11-3839) [DOI] [PubMed] [Google Scholar]
- 44.Maynard-Smith J. 1982. Evolution and the theory of games. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 45.Roodman GD. 2002. Role of the bone marrow microenvironment in multiple myeloma. J. Bone Miner. Res. 17, 1921–1925. ( 10.1359/jbmr.2002.17.11.1921) [DOI] [PubMed] [Google Scholar]
- 46.Bataille R, Harousseau JL. 1997. Multiple myeloma. N. Engl. J. Med. 336, 1657–1664. ( 10.1056/NEJM199706053362307) [DOI] [PubMed] [Google Scholar]
- 47.Dingli D, Chalub FACC, Santos FC, Van Segbroeck S, Pacheco JM. 2009. Cancer phenotype as the outcome of an evolutionary game between normal and malignant cells. Br. J. Cancer 101, 1130–1136. ( 10.1038/sj.bjc.6605288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terpos E, Politou M, Rahemtulla A. 2003. New insights into the pathophysiology and management of bone disease in multiple myeloma. Br. J. Haematol. 123, 758–769. ( 10.1046/j.1365-2141.2003.04712.x) [DOI] [PubMed] [Google Scholar]
- 49.Hashimoto T, Abe M, Oshima T, Shibata H, Ozaki S, Inoue D, Matsumoto T. 2004. Ability of myeloma cells to secrete macrophage inflammatory protein (MIP)-1alpha and MIP-1beta correlates with lytic bone lesions in patients with multiple myeloma. Br. J. Haematol. 125, 38–41. ( 10.1111/j.1365-2141.2004.04864.x) [DOI] [PubMed] [Google Scholar]
- 50.Dinarello CA. 2009. Targeting the pathogenic role of interleukin 1β in the progression of smoldering/indolent myeloma to active disease. Mayo Clin. Proc. 84, 105–107. ( 10.4065/84.2.105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lust JA, et al. 2009. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1β-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 84, 114–122. ( 10.4065/84.2.114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi SJ, Oba Y, Gazitt Y, Alsina M, Cruz J, Anderson J, Roodman GD. 2001. Antisense inhibition of macrophage inflammatory protein 1-alpha blocks bone destruction in a model of myeloma bone disease. J. Clin. Invest. 108, 1833–1841. ( 10.1172/JCI200113116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD., Jr 2007. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood 109, 2106–2111. ( 10.1182/blood-2006-09-047712) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ng AC, et al. 2011. Bone microstructural changes revealed by high-resolution peripheral quantitative computed tomography imaging and elevated DKK1 and MIP-1α levels in patients with MGUS. Blood 118, 6529–6534. ( 10.1182/blood-2011-04-351437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Farr JN, Zhang W, Kumar SK, Jacques RM, Ng AC, McCready LK, Rajkumar SV, Drake MT. 2014. Altered cortical microarchitecture in patients with monoclonal gammopathy of undetermined significance. Blood 123, 647–649. ( 10.1182/blood-2013-05-505776) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morgan GJ, et al. 2013. Long-term follow-up of MRC Myeloma IX Trial: survival outcomes with bisphosphonate and thalidomide treatment. Clin. Cancer Res. 19, 6030–6038. ( 10.1158/1078-0432.CCR-12-3211) [DOI] [PubMed] [Google Scholar]
- 57.Chapman MA, et al. 2011. Initial genome sequencing and analysis of multiple myeloma. Nature 471, 467–472. ( 10.1038/nature09837) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar SK, et al. 2012. Lenalidomide, cyclophosphamide, and dexamethasone (CRd) for light-chain amyloidosis: long-term results from a phase 2 trial. Blood 119, 4860–4867. ( 10.1182/blood-2012-01-407791) [DOI] [PMC free article] [PubMed] [Google Scholar]