

Abstract
Hybridization is thought to play an important role in plant evolution by introducing novel genetic combinations and promoting genome restructuring. However, surprisingly little is known about the impact of hybridization on transposable element (TE) proliferation and the genomic response to TE activity. In this paper, we first review the mechanisms by which homoploid hybrid species may arise in nature. We then present hybrid sunflowers as a case study to examine transcriptional activity of long terminal repeat retrotransposons in the annual sunflowers Helianthus annuus, Helianthus petiolaris and their homoploid hybrid derivatives (H. paradoxus, H. anomalus and H. deserticola) using high-throughput transcriptome sequencing technologies (RNAseq). Sampling homoploid hybrid sunflower taxa revealed abundant variation in TE transcript accumulation. In addition, genetic diversity for several candidate genes hypothesized to regulate TE activity was characterized. Specifically, we highlight one candidate chromatin remodelling factor gene with a direct role in repressing TE activity in a hybrid species. This paper shows that TE amplification in hybrid lineages is more idiosyncratic than previously believed and provides a first step towards identifying the mechanisms responsible for regulating and repressing TE expansions.
Keywords: transposable elements, genome evolution, hybridization, RNAseq, Helianthus
1. Introduction
Botanists have long recognized that new species may arise as a consequence of hybridization between genetically differentiated lineages [1–5]. Hybrid speciation occurs most commonly via duplication of a hybrid genome or allopolyploidy [3,6,7]. Genome duplication solves the two main challenges associated with hybrid speciation: hybrid sterility and the preservation of fit gene combinations [2,8,9]. Early-generation plant hybrids often exhibit not only reduced fertility due to abnormal meiotic pairing [10–12], but also increased vigour or heterosis [13–15]. The doubling of a hybrid's chromosomal complement restores normal pairing and fertility [13,16] and reduces recombination between homeologous chromosomes, thereby fixing heterotic gene combinations [17]. In addition, changes in ploidy confer partial reproductive isolation between the new hybrid and parental populations [3,18], contributing to their divergent evolutionary trajectories.
Hybrid speciation can occur without a change in ploidy (homoploid hybrid speciation), but the conditions are much more restrictive than for allopolyploidy [19–21]. In homoploid hybrid speciation, ecological and fertility selection are expected to lead to the establishment of fit hybrid segregants [3,22]. However, long-term stabilization of these hybrid segregants requires reproductive isolation, as fit gene combinations will be disrupted by gene flow with parental genotypes or with other hybrids [23]. Reproductive isolation may occur through behavioural [24], habitat [25–28], pollinator [29], karyotypic [30,31], geographical [32] and/or mating system [33] mechanisms. However, these mechanisms do not provide the immediate and strong reproductive barriers associated with ploidy differences. In addition, unlike allopolyploidy, which is marked by a change in chromosome number, homoploid hybrid speciation lacks an easily diagnosable feature [34]. As a consequence, prior to the development of molecular approaches, there were no unambiguous examples of homoploid hybrid speciation identified in nature.
Gottlieb's classic paper on the homoploid hybrid origin of Stephanomeria diegensis [34] revolutionized the study of hybrid speciation by (i) providing the first rigorously documented example of homoploid hybrid speciation in plants, and (ii) showing how molecular markers (in this case, allozymes) allow critical evaluation of the evolutionary outcomes of hybridization. Building on the Stephanomeria example (recently confirmed by Sherman & Burke [35]), numerous examples of plant homoploid hybrid species were convincingly demonstrated in the 1990s (reviewed in [36]), followed by a deluge of animal examples in the 2000s (reviewed in [8,37–39]).
The publication of unambiguous examples of homoploid hybrid speciation has stimulated interest in the genomic changes that accompany and possibly facilitate this speciation mode [9,30,31,40–47]. Homoploid hybrid species often differ from their parental species in karyotype [47,48], gene expression patterns [41–43] and transposable element (TE) copy number and expression level [49,50]. However, surprisingly little is known about the impact of these genomic changes on the origin and evolution of homoploid hybrid species. Karyotypic changes have the most obvious role in hybrid speciation, as they can contribute directly to reproductive isolation of the new hybrid lineage [31]. The roles of gene expression alteration and TE proliferation in homoploid hybrid speciation are less clear, although both processes are known to generate novel phenotypic variation that can be moulded by selection. In addition, there appears to be a connection between TE evolution and hybrid inviability and sterility [51], but this has not been explored in homoploid hybrid species.
Studies of wild sunflowers in the genus Helianthus have considerably advanced our understanding of homoploid hybrid speciation. Three different hybrid species (Helianthus anomalus, Helianthus paradoxus and Helianthus deserticola) have originated independently from the same two parents, Helianthus annuus and Helianthus petiolaris. These hybrid species have different geographical and temporal origins, and two species appear to have multiple origins [52–54]. All five species are diploid (2n = 34), self-incompatible and native to central and western North America (figure 1). The parental species have widespread overlapping distributions across the central and western USA [55,56]. Helianthus annuus is found in mesic, clay-based soils, whereas H. petiolaris occurs in drier, sandier soils [10]. By contrast, the three hybrid species are extremophiles, occurring in sand dune (H. anomalus), desert floor (H. deserticola) and salt marsh (H. paradoxus) habitats [57]. Genetic and ecological studies indicate that transgressive phenotypic variation (i.e. variation outside the range of their parental species) and the hybrid gene combinations underlying this variation allowed the hybrid species to colonize these extreme habitats [25,26,58,59]. In addition to habitat isolation, the hybrid lineages are reproductively isolated by large-scale karyotypic changes [31,48,60]. Analyses of the sizes and distribution of parental chromosomal segments in the three hybrid species further indicate that reproductive isolation probably arose quickly during speciation [61,62] and that the process is surprisingly repeatable [40,58].
Figure 1.

Phylogenetic network based on a random subset of 11 522 high-quality SNPs genotyped for all individuals.
Intriguingly, the sunflower homoploid hybrid species feature large genome sizes relative to parental species. While the genomes of H. petiolaris and H. annuus are approximately 3.3 and 3.5 Gb, respectively, the genomes of the hybrid species range from approximately 5.3 Gb in H. deserticola and H. paradoxus to 5.6 Gb in H. anomalus [63], with the difference in size largely accounted for by proliferation of TEs in each of the separate hybrid lineages [49,50]. Numerous classes of TEs exist in plant genomes, and long terminal repeat (LTR) retrotransposons are the most abundant and variable. Related to infectious retroviruses, these elements transpose through an RNA intermediate and thus individual elements can give rise to numerous daughter copies capable of inserting elsewhere in the genome [64]. Regulation of TEs in host genomes is mainly controlled by epigenetic mechanisms [65], and under most circumstances the vast majority of them are suppressed and rendered inactive. Only under specific conditions, such as during hybridization or stress, can a breakdown in gene silencing mechanisms reactivate these elements [66,67]. In many plant species, including the three sunflower homoploid hybrid species, the high replicative capacity of these elements has been associated with genome expansion [49,68].
The origins of H. deserticola, H. paradoxus and H. anomalus via hybridization between the same two parental species allow for unique comparative analysis of LTR retrotransposon activity and proliferation, because elements in the hybrid species are necessarily derived from the parental species genomes. While both major classes of LTR retrotransposons (i.e. Gypsy and Copia) have undergone proliferation events, the dynamics differ among species. For example, Gypsy sequences exhibit clear patterns of large-scale proliferation in all three sunflower hybrid species [49], whereas Copia sequences show differential patterns of proliferation, with H. paradoxus having experienced larger-scale proliferation of these sequences compared with H. deserticola and H. anomalus [68]. Interestingly, proliferation of LTR retrotransposons, while of massive scale in the sunflower hybrid species, does not appear to be a common feature of contemporary H. annuus × H. petiolaris natural hybrid populations [69]. Both Gypsy and Copia sequences remain transcriptionally active, however, in the parental species (H. annuus and H. petiolaris), in early-generation H. annuus × H. petiolaris hybrid genotypes generated through controlled crosses and found naturally, and in the homoploid hybrid species themselves [69,70].
The objectives of this study are to examine transcriptional activity of LTR retrotransposons (hereafter referred as TEs) within and between the annual sunflowers H. annuus, H. petiolaris and their homoploid hybrid derivatives using high-throughput transcriptome sequencing technologies (RNAseq). In addition, we characterized genetic diversity in these species for candidate genes hypothesized to regulate TE activity. RNAseq advances previous studies of TE activity in hybrid sunflowers by providing precise identification of TE variants and their estimated transcriptional activity. Expanded sampling of homoploid hybrid taxa reveals that abundant variation in TE transcript accumulation occurs within species. Comparison of hybrid species' TE transcript levels to those of parental species indicates that relatively few of the TEs examined (less than 5%) differ in expression between hybrids and parents, and that only a few TEs are overexpressed in multiple hybrid species relative to parent species. Analyses of sequence and expression diversity of candidate TE regulatory loci suggest the influence of divergent selection on these loci in hybrid species lineages, possibly contributing to differences in observed patterns of TE activity. Finally, we highlight one candidate gene displaying multiple hallmarks consistent with a direct role in repressing TE activity in one of the hybrid species.
2. Material and methods
(a). Plant collection and transcriptome sequencing
Achenes (single seeded fruits) representing 14 Helianthus annuus, 14 H. petiolaris, eight H. petiolaris × H. annuus F1 hybrids, three H. anomalus, seven H. deserticola and eight H. paradoxus spanning the range of each species were acquired either from USDA collections, previous sampling efforts or laboratory crosses for F1 hybrids (figure 1 and the electronic supplementary material, table S1). For each individual, we extracted RNA from young leaves and stems using a modified TRIzol reagent protocol (Invitrogen, Carlsbad, CA). All reads were sequenced on an Illumina (San Diego, CA) GAII or HiSeq next-generation sequencing platform (paired end reads, 2 × 100 bp, non-normalized libraries). Note that H. petiolaris and H. annuus individuals were sequenced as part of a larger study on genomic islands of divergence in wild sunflowers and are reported in detail in Renaut et al. [71]. Raw sequences from the three hybrid species (H. anomalus, H. deserticola and H. paradoxus) have been described and made publicly available previously [72]. Finally, the F1 hybrid sequences were previously described in Rowe & Rieseberg [73].
(b). Reference datasets and alignments
We used a reference dataset of 914 TEs (table 1) derived from the sequences of 96 randomly chosen BACs and described in details in Gill et al. [74]. This set of candidate TE nucleotide sequences was annotated (tblastx) using Uniprot Protein NR database (release-2013_01), and queried against GO databases (blast2GO, [75], electronic supplementary material, table S3). Additionally, candidate TEs were aligned to a published [76] Helianthus TE reference set (blastn, e-value < 10−10, only best hit retained). The combined results were used to classify sequences as LTR retrotransposon Copia, LTR retrotransposon Gypsy or Other element (table 1).
Table 1.
Summary of the TEs identified by Gill et al. [74], 914 contigs. The group ‘Other elements’ is further broken into subgroups based on BLAST results.
| transposable element family | no. elements | mean length of elements (base pairs) |
|---|---|---|
| Gypsy | 100 | 6603 |
| Copia | 37 | 6018 |
| Other elements | 777 | 3592 |
| non-LTR retro | 2 | 2961 |
| unclass retro | 35 | 3971 |
| uncharacterized | 17 | 9121 |
| DNA (pingpong) | 1 | 1341 |
| top hit non-TE | 44 | 6047 |
| no hits | 678 | 3280 |
Reads were aligned against a reference H. annuus transcriptome using the Burrows–Wheeler aligner (bwa, aln and sampe commands [77]). The transcriptome reference consisting of 51 468 contigs (51.3 M base pairs) is available on dryad (www.datadryad.org) and described in Renaut et al. [71]. Reads were aligned to the reference set of 914 TEs using the same approach. Aligned files (.bam format) were sorted using samtools sort utility.
(c). Gene and transposable element expression
Raw estimates of transcript accumulation were obtained using bedtools (coverageBed) to determine the number of sequence reads mapping to each contig [78]. Comparisons of the accumulation of individual transcripts between sample groups per species were conducted within the R [79] package DESeq [80]. This program normalizes raw read counts based on aligned library size but not sequence length, as all comparisons are performed within a given transcript using a modified Fisher's exact test of data fit to a binomial distribution. Adjusted p-values (q-values < 0.05 [81]) were used to determine statistical significance of comparisons. We then normalized TE transcript estimates first by the total number of reads aligned for each sample and then by the length of the reference gene or TE (fragments per kilobase per million fragments mapped (FPKM) value [82]). TE expression estimates (normalized read counts per sample) were summed across element classes Copia, Gypsy and Other elements to create TE expression phenotypes. Species differences in these aggregate TE expression phenotypes were assessed via linear modelling in R.
A previously identified set of 107 LTR retrotransposons with insertion age estimates for the H. annuus genome [76] were matched to our own reference set of TEs by blast search. We then calculated whether element age was correlated with levels of transcript accumulation in any of the five sunflower species.
(d). Evaluation of candidate transposable element regulatory genes
We constructed a reference dataset of candidate genes presumed to be involved in repressing TEs in Arabidopsis thaliana. First, DNA sequences of A. thaliana genes assigned to the Gene Ontology term GO:0016441 (post-transcriptional gene silencing, 131 genes) were obtained from The Arabidopsis Information Resource (www.arabidopsis.org). Literature searches identified 14 additional genes with empirical evidence of involvement in TE repression [65,83–85]. These genes were then compared (tblastx, e-value < 1 × 10−10) with all 51 468 genes in the H. annuus reference transcriptome. Two hundred and forty-five genes in the reference transcriptome matched these criteria and were subjected to analysis as candidate TE regulatory genes.
We used the weighted gene co-expression network analysis (WGCNA) package in R to cluster these candidate regulatory genes by principal component analysis [86]. Genes showing no transcript variance were excluded from the analysis. First, we evaluated the strength of module membership for individual genes (correlation of individual gene transcript accumulation estimate with module eigenvector). We then evaluated the correlation of individual candidate regulatory genes and co-expressed gene modules with TE expression phenotypes (Pearson correlation coefficient with Bonferroni-corrected p-values). Genes significantly correlated with TE expression phenotypes and with Pearson correlation coefficients greater than |0.4| were retained for sequence diversity analyses and hereafter referred to as TE regulator genes.
(e). Variant calling, population genetics and selection
Because relationships among populations may not conform to a tree-like bifurcating pattern owing to introgression and shared ancestral polymorphisms, we performed a phylogenetic network analysis using the neighbour-net method implemented in splitstree4 [87]. We used samtools (mpileup and bcftools [88]) to call single nucleotide polymorphisms (SNPs) using information from all samples for a random set of 1000 genes. From this, we compiled an artificial nucleotide sequence comprising 11 522 high-quality (overall missing data less than 10%) SNPs. We then used these markers to generate a phylogenetic network in splitstree4 using default parameters (figure 1).
We used samtools (mpileup and bcftools, [88]) to identify SNPs between each pair of samples (table 2). SNPs with more than 20% missing data were removed. We also filtered out SNPs as described previously in Renaut et al. [71]. Briefly, SNPs with low expected heterozygosity (He < 0.2) were removed given that they probably represent either sequencing errors or rare alleles with little information content for interspecific comparisons. We also filtered out SNPs with very high observed heterozygosity (Ho > 0.6) because they probably represent paralogous sequence variants. From this curated dataset, FST values [89] were calculated for each marker and each species pair, using the R package hierfstat [90]. We also calculated genetic diversity (π) using sites [91].
Table 2.
Summary statistics of sequence alignments for the six species analysed here.
| species | transcriptome reference (51 468 contigs) |
TE reference (914 contigs) |
||||
|---|---|---|---|---|---|---|
| no. reads aligned in millions (% total reads) | mean (95% CI) no. reads aligned per reference contig | no. contigs with > 2 reads aligned | no. reads aligned in thousands (% total reads) | mean (95% CI) no. reads aligned per TE | no. contigs with > 2 reads aligned | |
| H. annuus | 17.0 (62%) | 331 (284–378) | 26 139 | 34 (0.11%) | 37.6 (18–57) | 460 |
| H. petiolaris | 24.8 (66%) | 464 (376–552) | 24 782 | 38 (0.08%) | 34 (7–62) | 421 |
| F1 hybrids | 15.8 (55%) | 306 (297–317) | 32 850 | 34 (0.12%) | 42 (13–71) | 445 |
| H. anomalus | 28.8 (66%) | 598 (553–643) | 24 345 | 71 (0.15%) | 77 (34–121) | 497 |
| H. deserticola | 23.5 (64%) | 442 (393–490) | 24 560 | 78 (0.19%) | 85 (52–118) | 517 |
| H. paradoxus | 21.0 (64%) | 428 (364–492) | 24 351 | 73 (0.19%) | 77 (50–104) | 471 |
We predicted open reading frames in our reference transcriptome and tested whether the ratio of non-synonymous to synonymous fixed differences was greater than the ratio of non-synonymous to synonymous polymorphisms using a g-test [92]. As an extension of this approach, we estimated the average proportion of amino acid substitutions driven by positive selection (alpha [93]). Based on empirical distributions of FST values that showed a bimodal distribution (see electronic supplementary material, figure S1), SNPs with an FST value greater or equal to 0.8 were considered as substitutions (D). We calculated alpha per species pair, first for all polymorphic genes, then for the subset of TE regulator genes. Significance values were calculated by resampling (with replacement, number of resamples equal to number of TE regulator genes) for each species pair.
3. Results
(a). Alignments and summary statistics
Between 17 and 29 million reads (approx. 65% of all reads) aligned to the reference transcriptome of 51 468 contigs (table 2). A much smaller proportion of reads aligned to the TE reference of 914 contigs (0.1–0.2%). Nevertheless, this represents approximately 55 000 reads aligned per individual to the TE reference (table 3), which is sufficient to produce quantitative estimates of TE expression variation.
Table 3.
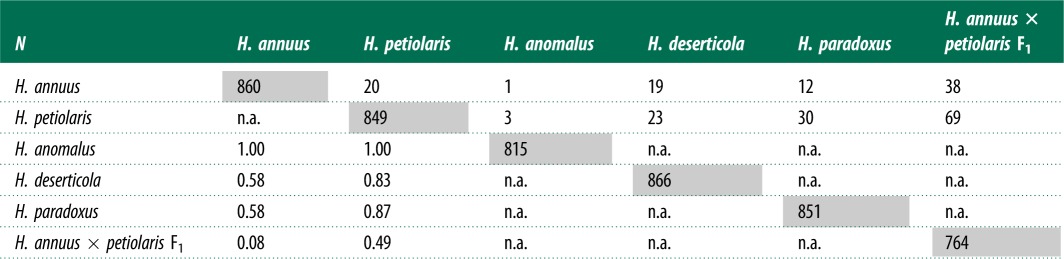
Interspecific comparisons of transcriptional activity for 914 putative TEs identified in H. annuus genomic DNA sequence. Comparisons were performed between parental and hybrid species only. Diagonal (grey box): number of putative TEs showing evidence of transcription (>2 read pairs aligned); above the diagonal: number of putative TEs showing significant (q-value < 0.05) differences in pairwise comparison of transcriptional activity; below the diagonal: proportion of significant comparisons where the hybrid shows higher transcription levels. Note that because only significant comparisons are shown here, the proportions reported for H. anomalus in the lower diagonal are based on very few comparisons (1 and 3), which accounts for apparent differences relative to figure 2.
 |
Based on a concatenated sequence of 11 522 SNPs, we plotted a phylogenetic network of all individuals (figure 1). While most individuals cluster as expected, H. anomalus did not form a distinct cluster. Limited sampling of this species or multiple hybrid origins [52] may contribute to the poor clustering (figure 1).
(b). Gene and transposable element expression
Of 914 candidate TE sequences extracted from H. annuus genomic sequence, only 14 showed no evidence of transcription across all 54 samples (electronic supplementary material, table S3). However, for another 104 putative TEs, no sample contained more than four reads aligning to the reference sequence. Fewer than 10% of putative TEs showed mean expression across all samples greater than one FPKM, with sample groups ranging from 7.1% of putative TEs transcribed at greater than one FPKM in H. petiolaris to 13.6% in H. deserticola.
TE expression phenotypes (aggregate transcript levels for elements assigned to Gypsy, Copia, or Other elements) show positive transgression in hybrid species, with both H. paradoxus and H. deserticola displaying transcript levels significantly higher than either parental species for all three TE categories (figure 2). The third hybrid species, H. anomalus, exhibited positive transgression for Copia, but not for Gypsy or Other elements. By contrast, F1 hybrids express intermediate (or additive) transcript levels with respect to the parental species (figure 2). Intraspecific variation in TE transcript accumulation varied among species (Levene's test of equality of variances, F5,48 = 3.0, 4.0 and 2.8, p-value = 0.02, 0.004 and 0.03 for the Gypsy, Copia and Other elements categories, respectively). In addition, variance appeared higher in H. paradoxus and H. deserticola (but not H. anomalus) than in the parental species or F1 hybrids (figure 2).
Figure 2.
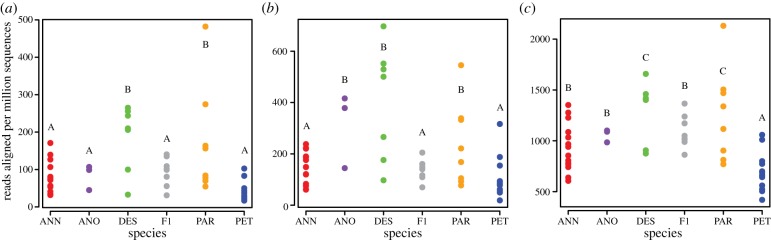
Abundance of transcript reads from individual samples aligned to putative transposable element sequences classified as (a) Gypsy-like, (b) Copia-like or (c) Other elements. Sample groups (‘species’) are arrayed along the x-axes, with each point representing an individual sample. y-Axes indicate transcript estimates normalized by library size. ANOVA revealed significant differences among species per sample groups (Gypsy: F = 6.2, p-value = 1.7 × 10−4; Copia: F = 7.8, p-value = 2.0 × 10−5, Other elements: F = 7.7, p-value = 2.2 × 10−5). Within a given element class, species per sample groups labelled with the same letter (A, B, C) do not significantly differ (pairwise t-test, p-value > 0.05).
Pairwise comparisons of transcript accumulation per individual TEs among all groups revealed that 782 (85%) showed no significant difference in inferred transcript accumulation (number of aligned reads) for any comparison (table 3). The number of TEs showing significant differences between the parental species and their hybrid species derivatives ranged from as low as one between H. annuus and H. anomalus (presumably owing to low sample size for H. anomalus) to as high as 30 between H. petiolaris and H. paradoxus (table 3). As expected, the majority of significant expression changes were due to increased expression in the hybrid species. While comparisons between F1s and parental species revealed a greater number of significant differences in TE expression, less than half of these TEs showed increased transcript in the F1 hybrids relative to the parental species.
In addition, comparisons were performed within H. annuus (between H. annuus and subspecies H. annuus texanus), and within H. petiolaris (between subspecies H. petiolaris petiolaris and H. petiolaris fallax), with no significant subspecies differences in transcription of putative TEs identified (Fisher's exact test, q-value > 0.05 for all comparisons, data not shown).
Element age from a previously identified set of 107 LTR retrotransposons [76] does not appear to correlate with levels of transcript accumulation in any of the five species examined here (data for H. annuus in the electronic supplementary material, figure S2, all other comparisons similar and n.s.).
(c). Evaluation of candidate transposable element regulatory genes
Analysis of transcript patterns for 236 candidate genes within WGCNA indicated that these formed two clusters of 167 and 28 genes, with 41 genes remaining unclustered (electronic supplementary material, table S2). The larger cluster (labelled turquoise; figure 3) showed strong correlation with TE expression phenotypes (Pearson's r (p-value): Gypsy 0.84 (2 × 10−15), Copia 0.81 (8 × 10−14), Other elements 0.74 (2 × 10−10)). Cluster membership (correlation of individual locus transcript level with the cluster eigenvalue) was strongly and positively correlated with TE expression phenotypes (Pearson's r (p-value): Gypsy 0.91 (5.5 × 10−65), Copia 0.71 (6.4 × 10−27), Other elements 0.83 (1.1 × 10−43)).
Figure 3.
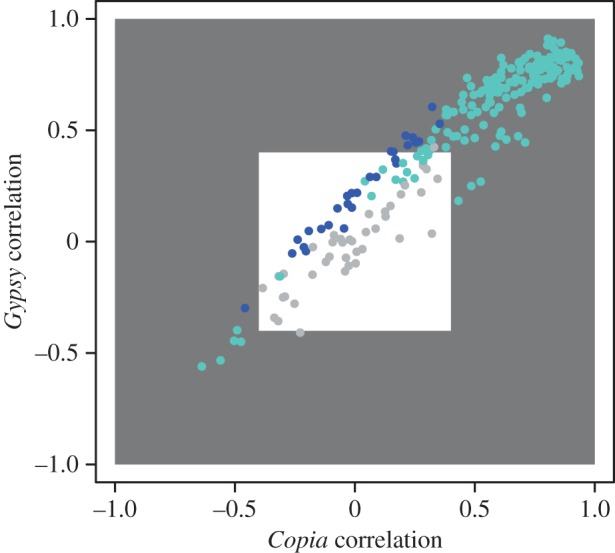
Correlation between aggregate Gypsy and Copia transcript abundance. Dots are coloured according to whether they belong to the turquoise, blue or grey (unassigned) modules according to WGCNA. Grey area represents genes that had a correlation coefficient above |0.4| and were labelled as TE regulator genes in subsequent analyses.
A set of 170 genes significantly correlated with TE expression phenotypes and with Pearson correlation coefficients greater than |0.4| were retained for sequence diversity analyses (grey area in figure 3). These are hereafter referred to TE regulator genes.
(d). Variant calling, population genetics and selection
We compared population genetic aspects of these TE regulator genes identified through WGCNA with a larger set of expressed genes. The total number of SNPs identified for each of the seven species pairs was between 162 and 224 thousand (mean = 191 thousand per species pair, approx. three SNPs per kb of reference sequence). By contrast, few SNPs were identified in the putative TE sequences (mean = 63 per species pair, approximately 0.02 SNPs per kb of reference sequence).
Next, we calculated global FST for each of seven species pairs (figure 4 and the electronic supplementary material, figure S1). While FST varied among species pairs (Kruskal–Wallis rank-sum test, χ2 (6, N = 3 139 052) = 252 035, p-value < 2 × 10−16 for the species pair effect), estimates remained similar whether based on all genes or the subset TE regulator genes (Kruskal–Wallis rank-sum test, χ2 (1, N = 3 139 052) = 0.05, p-value = 0.83 for the gene category effect).
Figure 4.

Boxplot of FST values for the subset of TE regulator genes against all other genes for the parental species pairs (H. annuus versus H. petiolaris) and their hybrids (H. anomalus, H. deserticola, H. paradoxus).
Similarly, we calculated genetic diversity (π) for each of the five species (figure 5). Genetic diversity (log(π)) varied among species (one-way ANOVA: F1,72 752 = 572.3, p-value < 2 × 10−16 for the species effect). In addition, intraspecific genetic diversity was lower for the TE regulator genes versus all others (one-way ANOVA: F1,72 752 = 36.2, p-value = 1.8 × 10−9 for the gene category effect).
Figure 5.

Boxplot of genetic diversity (π) values for the subset of TE regulator genes against all other genes for the parental species (H. annuus and H. petiolaris) and their hybrids (H. anomalus, H. deserticola, H. paradoxus).
Finally, we calculated the estimated proportion of amino acid substitutions driven to fixation by positive selection (alpha) for each of the seven species pairs individually (figure 6). Alpha varied among species pairs (one-way ANOVA on resampled data: F6,8044 = 837, p-value < 2.2 × 10−16) and between gene categories (F1,8044 = 24.2, p-value = 8.9 × 10−7). In addition, alpha for TE regulatory genes compared with all other genes was substantially higher in comparisons involving the hybrid species H. deserticola and H. paradoxus (Tukey's HSD test, all p-values < 0.0001, except for the H. petiolaris—H. paradoxus comparison where p-value > 0.05). However, alpha does not appear to be higher for the TE regulatory genes in the parental species comparison and the two comparisons involving H. anomalus (figure 6).
Figure 6.

Barplot of the estimated proportion of amino acid substitutions driven to fixation by positive selection (alpha) for the subset of TE regulator genes against all other genes for the parental species pairs (H. annuus versus H. petiolaris) and their hybrids (H. anomalus, H. deserticola, H. paradoxus).
(e). Identification of a candidate transposable element repressor gene
Here, we present one promising gene as an example of how combining different lines of evidence can suggest candidates for TE regulation. First, a blast search revealed that this candidate gene is similar to a chromatin remodelling factor of the CHD3 group in Arabidopsis thaliana, a member of a conserved group of negative transcriptional regulators (figure 7a) [94,95]. Additionally, this transcribed sequence is overexpressed in hybrid species compared with parental species (figure 7b), shows strong correlation with aggregate Gypsy transcript levels (figure 7b), and possesses four non-synonymous fixed mutations that differentiate H. paradoxus from both parental species, in addition to one synonymous mutation that differentiates H. paradoxus from H. petiolaris (figure 7c).
Figure 7.

Example of a candidate gene. (a) BLAST and Gene Ontology annotation; (b) correlation between gene and TE Expression (aggregate Gypsy elements) and (c) genetic divergence (fixed or polymorphic synonymous and non-synonymous sites).
4. Discussion
Hybridization in plants can reactivate dormant TEs, contributing to genome expansion and restructuring [96]. Yet, for viable hybrid populations to persist, mosaic genomes exposed to novel TEs must prevent selfish elements from proliferating uncontrollably [97]. The fact that the hybrid species show unique cases of transgressive TE expression suggests that these lineages have potential redundancies for preventing the expansion of some elements, while perhaps having lost the capacity to suppress others. Fundamentally, it implies an evolutionary arms race between the host and its genetic parasites where both players must adapt to one another in order to reproduce and thrive.
Here, sampling homoploid hybrid sunflower taxa revealed abundant variation in TE transcript accumulation. High-throughput transcriptome sequencing (RNAseq) allowed simultaneous quantification of gene expression for a large number of genes, identification of polymorphic sites and measurement of genetic divergence in loci potentially involved in TE repression. Patterns of transgressive TE expression in these hybrids suggest that relatively few elements are highly transcribed, and that few elements show consistent differences in expression across species. Combining TE transcript patterns with model species annotation identified TE regulatory candidate genes that show patterns of sequence diversity consistent with evolution under divergent natural selection.
(a). Expression divergence
The majority of sequences in our reference set of 914 putative TEs, extracted from H. annuus genomic sequence, showed some level of transcriptional activity within our set of 54 transcriptome samples from five sunflower species and one set of first generation interspecific hybrids. As observed for systems such as Arabidopsis spp. (thaliana and lyrata), maize and rice, a relatively small proportion of elements appear to account for the bulk of transcription [98,99]. The hybrid species showed an overall trend towards higher accumulation of putative TE transcripts, both in summed levels of TE transcript observed and the number of individual elements contributing transcripts. F1 hybrids showed aggregate TE transcript levels similar to those of parental species, and for individual TEs showing significant differences in transcript levels between F1 and parental species, F1 transcript estimates were often lower (figure 2 and table 3). Once possible explanation for this pattern is the concerted action of dominant alleles from both parental species to reduce TE transcription in F1 hybrids, and if this were the case, homologous recombination should break up co-adapted allele combinations in the next generation. Additionally, both parental species contain substantial levels of genetic variation that may combine to produce effects on TE expression that were not observed in this or prior studies. While it will be extremely insightful to expand our studies to include later-generation hybrids and a broader sampling of existing variation, at this point, and in combination with prior observations of early-generation H. annuus × H. petiolaris hybrids [70], our results argue that increased TE activity in hybrid species is not an immediate or necessary consequence of hybridization [69,70].
These results are in general accord with Ungerer & Kawakami [70] who found non-additive expression of Gypsy elements for H. deserticola and H. paradoxus, although here this pattern extends to additional classes of elements, probably owing to the increased sensitivity of RNAseq. For the group of putative elements not specified as Gypsy or Copia (i.e. Other elements), H. petiolaris actually showed significantly lower TE-associated transcript levels than any other sample group (figure 2). Building a genomic reference specific for H. petiolaris may help to clarify whether this difference reflects more strict control of TE transcription in H. petiolaris or the presence of divergent TEs not present in the H. annuus genome.
While mean values for aggregate TE expression phenotypes demonstrate that the hybrid species accumulate higher levels of TE-associated transcripts than parental species or F1 hybrid genotypes, the sunflower hybrid species were quite variable both in overall TE transcript levels and the identity of elements contributing to these aggregate TE expression phenotypes. This suggests that the mechanism(s) regulating transcription of TEs in these hybrid species are not highly specific to particular elements and that intraspecific variation in TE regulation is present. It is currently unclear whether elevated transcriptional activity of TEs in the hybrid species underlies differences in genomic copy number of these sequences in the hybrid versus parental sunflower taxa and whether amplification of these elements and genome expansion in the hybrid species is an ongoing phenomenon. Additional experiments in the five sunflower species designed to assay insertional activity of these sequences will be required to address this issue. For example, copy number variation could be assessed through genome resequencing efforts [100,101], which are currently underway for several species of sunflowers.
Moreover, while the initial set of candidate genes gathered from the literature represents a good starting point for further study, the specific genetic mechanisms that lead to TE expression differences remain nebulous. Do elevated expression levels for particular variants in the hybrid species simply reflect higher copy numbers for these sequences, or might expression differences result from epigenetic TE silencing mechanisms [65,102] that are differentially effective among these species? It is also unknown what triggered the amplification of TEs and what maintains elevated transcriptional activity of these sequences in contemporary populations of these hybrid species. Detailed analyses of genomic sequence from these hybrid species may provide insights into how observed variation in transcription contributes to variation in genome size and structure.
(b). Genetic divergence
Following quantification of expression divergence, we calculated several genetic parameters to compare the complete reference transcriptome with a subset of 170 candidate TE regulator genes associated with TE transcript phenotypes through WGCNA. While there was no difference in FST between all genes and the subset of TE regulator genes, these candidate genes nevertheless appear to be less polymorphic than the transcriptome average. One plausible explanation is that these genes undergo stronger or more frequent selective sweeps, which reduce variation. The estimated proportion of mutations fixed by natural selection (alpha) is consistent with a greater effect of selection for the TE regulator genes in the hybrid H. deserticola and H. paradoxus (low sample size in H. anomalus precludes any strong conclusion for this species). It therefore suggests that divergent natural selection could have greater impact on the evolution of TE regulator genes in hybrid species. This might be expected given that the response of regulator genes to the proliferation of TEs probably plays an important role in genome stabilization during hybrid speciation [97].
(c). Combining evidence towards identifying candidate regulatory genes
Combining experimental evidence targeting different biological levels (e.g. variation at the DNA, gene expression and phenotypic levels) represents the best strategy towards deciphering the genetic bases of evolutionary change [103]. Here, we showed one example of a chromatin remodelling factor gene which is involved in post-transcriptional gene silencing, one of the main pathways used by cells to repress TE activity [97]. Several lines of evidence combining functional (figure 7a), expression (figure 7b) and molecular evolution (figure 7c) components suggest that it evolved in at least one of the hybrid species to regulate and restrain the expansion of TEs, presumably owing to an enhanced ‘threat’ to genomic stability imposed by elevated expression of TEs. More detailed analyses, including analysing sequence variation of the candidate gene regulatory regions and sampling a larger number of individuals, would help to confirm these results.
5. Conclusions and future directions
The outcomes of hybridization are difficult to predict owing to simultaneous beneficial (heterosis, increased resilience due to increased genetic variation) and negative (hybrid incompatibilities, maladaptive gene combinations) evolutionary impacts. The role of TE proliferation in hybrid evolution poses a similar conundrum, in which the potentially negative impact of TE proliferation on fitness may be counterbalanced by the generation of genetic and phenotypic novelty. This paper shows that TE amplification in hybrid lineages is more idiosyncratic than previously documented and provides a first step towards identifying the gene(s) and evolutionary mechanisms responsible for regulating and repressing TE expansions.
In the future, it would be useful to expand the study of TE evolution both horizontally—to assess whether our findings in Helianthus hybrid lineages can be extended to other organismal groups such as the Stephanomeria system studied by Gottlieb—and vertically to functionally validate the candidate transcription regulator genes found in this study. Both kinds of studies offer the opportunity to assess the repeatability of genomic changes in hybrid evolution and may provide clues regarding potential abiotic or epigenetic factors that trigger TE amplification in the first place.
Several other puzzles about TE evolution in hybrid lineages remain to be solved. Most importantly, we need to clarify the importance of hybridization (or the interaction of divergent parental genomes) versus other evolutionary processes in the TE expansions. Why, for example, do we see little evidence of TE amplification in contemporary F1 sunflower hybrids? A similar result from studies of interspecific Arabidopsis F1 crosses [104] suggests that upregulation of TEs is not a general phenomenon. Empirical evidence is also required to assess causality between the TE expansion reported here and the large-scale karyotypic and phenotypic changes observed in the homoploid hybrid species.
Thirty years ago, Gallez & Gotlieb [34] argued that the availability of electrophoretic techniques to identify large numbers of genetic loci would allow evolutionary biologists to estimate the likelihood of homoploid hybrid speciation. They were right, although it took hundreds (or thousands) of markers made available by DNA-sequence-based marker technologies to finally fulfil their prophecy. They also predicted that the most interesting examples of homoploid hybrid speciation would ‘be those in which the diploid parents appear to be strongly distinct in a genetic sense’. The many fascinating genomic changes that have accompanied homoploid hybrid speciation in sunflowers, which involve highly divergent parental species, supports this claim, although further studies of the genomic consequences of homoploid hybrid speciation across a continuum of parental divergence will be required to fully validate this second prediction.
Supplementary Material
Supplementary Material
Acknowledgements
We thank Navdeep Gill for providing us an advance access to the H. annuus TE reference.
Funding statement
This work was supported by an NIH Postdoctoral Fellowship to H.C.R., an NSERC Postdoctoral Fellowship to S.R., an NSERC grant no. (327475) to L.H.R., and NSF grant no. DEB-0742993 to M.C.U.
References
- 1.Winge O. 1917. The chromosomes. Their numbers and general importance. Comp. Rend. Travaux Lab. Carlsberg Copenhague 13, 131–275. [Google Scholar]
- 2.Stebbins GL. 1950. Variation and evolution in plants. New York, NY: Columbia University Press. [Google Scholar]
- 3.Grant V. 1981. Plant speciation. New York, NY: Columbia University Press. [Google Scholar]
- 4.Arnold ML. 1997. Natural hybridization and evolution. Oxford, UK: Oxford University Press. [Google Scholar]
- 5.Soltis DE, Buggs RJA, Doyle JJ, Soltis PS. 2010. What we still don't know about polyploidy. Taxon 59, 1387–1403. [Google Scholar]
- 6.Brochmann C, Brysting AK, Alsos IG, Borgen L, Grundt HH, Scheen AC, Elven R. 2004. Polyploidy in arctic plants. Biol. J. Linn. Soc. 82, 521–536. ( 10.1111/j.1095-8312.2004.00337.x) [DOI] [Google Scholar]
- 7.Doyle JJ, Flagel LE, Paterson AH, Rapp RA, Soltis DE, Soltis PS, Wendel JF. 2008. Evolutionary genetics of genome merger and doubling in plants. Annu. Rev. Genet. 42, 443–461. ( 10.1146/annurev.genet.42.110807.091524) [DOI] [PubMed] [Google Scholar]
- 8.Mallet J. 2007. Hybrid speciation. Nature 446, 279–283. ( 10.1038/nature05706) [DOI] [PubMed] [Google Scholar]
- 9.Abbott R, et al. 2013. Hybridization and speciation. J. Evol. Biol. 26, 229–246. ( 10.1111/j.1420-9101.2012.02599.x) [DOI] [PubMed] [Google Scholar]
- 10.Heiser CB., Jr 1947. Hybridization between the sunflower species Helianthus annuus and H. petiolaris. Evolution 1, 249–262. ( 10.2307/2405326) [DOI] [Google Scholar]
- 11.Morgan DT. 1950. A cytogenetic study of inversion in Zea mays. Genetics 35, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levin DA. 2002. Role of chromosomal change in plant evolution. New York, NY: Oxford University Press. [Google Scholar]
- 13.Stebbins GL. 1958. The inviability, weakness, and sterility of interspecific hybrids. Adv. Genet. Incorporating Mol. Genet. Med. 9, 147–215. ( 10.1016/s0065-2660(08)60162-5) [DOI] [PubMed] [Google Scholar]
- 14.Lippman ZB, Zamir D. 2007. Heterosis: revisiting the magic. Trends Genet. 23, 60–66. ( 10.1016/j.tig.2006.12.006) [DOI] [PubMed] [Google Scholar]
- 15.Birchler JA, Yao H, Chudalayandi S, Vaiman D, Veitia RA. 2010. Heterosis. Plant Cell 22, 2105–2112. ( 10.1105/tpc.110.076133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobzhansky T. 1933. On the sterility of the interracial hybrids in Drosophila pseudoobscura. Proc. Natl Acad. Sci. USA 19, 397–403. ( 10.1073/pnas.19.4.397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roose ML, Gottlieb LD. 1976. Genetic and biochemical consequences of polyploid in Tragopogon. Evolution 30, 818–830. ( 10.2307/2407821) [DOI] [PubMed] [Google Scholar]
- 18.Ramsey J, Schemske DW. 2002. Neopolyploidy in flowering plants. Annu. Rev. Ecol. Syst. 33, 589–639. ( 10.1146/annurev.ecolysis.33.010802.150437) [DOI] [Google Scholar]
- 19.Buerkle CA, Morris RJ, Asmussen MA, Rieseberg LH. 2000. The likelihood of homoploid hybrid speciation. Heredity 84, 441–451. ( 10.1046/j.1365-2540.2000.00680.x) [DOI] [PubMed] [Google Scholar]
- 20.Turelli M, Barton NH, Coyne JA. 2001. Theory and speciation. Trends Ecol. Evol. 16, 330–343. ( 10.1016/s0169-5347(01)02177-2) [DOI] [PubMed] [Google Scholar]
- 21.Coyne JA, Orr HA. 2004. Speciation. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 22.Templeton AR. 1981. Mechanisms of speciation: a population genetic approach. Annu. Rev. Ecol. Syst. 12, 23–48. ( 10.1146/annurev.es.12.110181.000323) [DOI] [Google Scholar]
- 23.Rieseberg LH. 1997. Hybrid origins of plant species. Annu. Rev. Ecol. Syst. 28, 359–389. ( 10.1146/annurev.ecolsys.28.1.359) [DOI] [Google Scholar]
- 24.Mavarez J, Salazar CA, Bermingham E, Salcedo C, Jiggins CD, Linares M. 2006. Speciation by hybridization in Heliconius butterflies. Nature 441, 868–871. ( 10.1038/nature04738) [DOI] [PubMed] [Google Scholar]
- 25.Lexer C, Welch ME, Raymond O, Rieseberg LH. 2003. The origin of ecological divergence in Helianthus paradoxus (Asteraceae): selection on transgressive characters in a novel hybrid habitat. Evolution 57, 1989–2000. ( 10.1111/j.0014-3820.2003.tb00379.x) [DOI] [PubMed] [Google Scholar]
- 26.Ludwig F, Rosenthal DM, Johnston JA, Kane N, Gross BL, Lexer C, Dudley SA, Rieseberg LH, Donovan LA. 2004. Selection on leaf ecophysiological traits in a desert hybrid Helianthus species and early-generation hybrids. Evolution 58, 2682–2692. ( 10.1111/j.0014-3820.2004.tb01621.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor SJ, Willard RW, Shaw JP, Dobson MC, Martin NH. 2011. Differential response of the homoploid hybrid species Iris nelsonii (Iridaceae) and its progenitors to abiotic habitat conditions. Am. J. Bot. 98, 1309–1316. ( 10.3732/ajb.1100012) [DOI] [PubMed] [Google Scholar]
- 28.Ma F, Zhao CM, Milne R, Ji MF, Chen LT, Liu JQ. 2010. Enhanced drought-tolerance in the homoploid hybrid species Pinus densata: implication for its habitat divergence from two progenitors. New Phytol. 185, 204–216. ( 10.1111/j.1469-8137.2009.03037.x) [DOI] [PubMed] [Google Scholar]
- 29.Taylor SJ, Rojas LD, Ho SW, Martin NH. 2013. Genomic collinearity and the genetic architecture of floral differences between the homoploid hybrid species Iris nelsonii and one of its progenitors, Iris hexagona. Heredity 110, 63–70. ( 10.1038/hdy.2012.62) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rieseberg LH, Vanfossen C, Desrochers AM. 1995. Hybrid speciation accompanied by genomic reorganization in wild sunflowers. Nature 375, 313–316. ( 10.1038/375313a0) [DOI] [Google Scholar]
- 31.Lai Z. 2005. Extensive chromosomal repatterning and the evolution of sterility barriers in hybrid sunflower species. Genetics 171, 291–303. ( 10.1534/genetics.105.042242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James JK, Abbott RJ. 2005. Recent, allopatric, homoploid hybrid speciation: the origin of Senecio squalidus (Asteraceae) in the British Isles from a hybrid zone on Mount Etna, Sicily. Evolution 59, 2533–2547. ( 10.1111/j.0014-3820.2005.tb00967.x) [DOI] [PubMed] [Google Scholar]
- 33.Stebbins GL. 1957. The hybrid origin of microspecies in the Elymus glaucuscomplex. Cytol. Suppl. 36, 336–340. [Google Scholar]
- 34.Gallez GP, Gottlieb LD. 1982. Genetic evidence for the hybrid origin of the diploid plant Stephanomeria diegensis. Evolution 36, 1158–1167. ( 10.2307/2408150) [DOI] [PubMed] [Google Scholar]
- 35.Sherman NA, Burke JM. 2009. Population genetic analysis reveals a homoploid hybrid origin of Stephanomeria diegensis (Asteraceae). Mol. Ecol. 18, 4049–4060. ( 10.1111/j.1365-294X.2009.04349.x) [DOI] [PubMed] [Google Scholar]
- 36.Gross BL, Rieseberg LH. 2005. The ecological genetics of homoploid hybrid speciation. J. Hered. 96, 241–252. ( 10.1093/jhered/esi026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mavarez J, Linares M. 2007. Homoploid hybrid speciation in animals. Mol. Ecol. 17, 4181–4185. ( 10.1111/j.1365-294X.2008.03898.x) [DOI] [PubMed] [Google Scholar]
- 38.Brelsford A, Mila B, Irwin DE. 2011. Hybrid origin of Audubon's warbler. Mol. Ecol. 20, 2380–2389. ( 10.1111/j.1365-294X.2011.05055.x) [DOI] [PubMed] [Google Scholar]
- 39.Hermansen JS, Saether SA, Elgvin TO, Borge T, Hjelle E, Saetre G-P. 2011. Hybrid speciation in sparrows I: phenotypic intermediacy, genetic admixture and barriers to gene flow. Mol. Ecol. 20, 3812–3822. ( 10.1111/j.1365-294X.2011.05183.x) [DOI] [PubMed] [Google Scholar]
- 40.Rieseberg LH, Sinervo B, Linder CR, Ungerer MC, Arias DM. 1996. Role of gene interactions in hybrid speciation: evidence from ancient and experimental hybrids. Science 272, 741–745. ( 10.1126/science.272.5262.741) [DOI] [PubMed] [Google Scholar]
- 41.Lai Z, Gross BL, Zou YI, Andrews J, Rieseberg LH. 2006. Microarray analysis reveals differential gene expression in hybrid sunflower species. Mol. Ecol. 15, 1213–1227. ( 10.1111/j.1365-294X.2006.02775.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hegarty MJ, Hiscock SJ. 2005. Hybrid speciation in plants: new insights from molecular studies. New Phytol. 165, 411–423. ( 10.1111/j.1469-8137.2004.01253.x) [DOI] [PubMed] [Google Scholar]
- 43.Hegarty MJ, Barker GL, Brennan AC, Edwards KJ, Abbott RJ, Hiscock SJ. 2009. Extreme changes to gene expression associated with homoploid hybrid speciation. Mol. Ecol. 18, 877–889. ( 10.1111/j.1365-294X.2008.04054.x) [DOI] [PubMed] [Google Scholar]
- 44.Staton SE, Ungerer MC, Moore RC. 2009. The genomic organization of TY3/gypsy-like retrotransposons in Helianthus (Asteraceae) homoploid hybrid species. Am. J. Bot. 96, 1646–1655. ( 10.3732/ajb.0800337) [DOI] [PubMed] [Google Scholar]
- 45.Soltis PS, Soltis DE. 2009. The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60, 561–588. ( 10.1146/annurev.arplant.043008.092039) [DOI] [PubMed] [Google Scholar]
- 46.Abbott RJ, Hegarty MJ, Hiscock SJ, Brennan AC. 2010. Homoploid hybrid speciation in action. Taxon 59, 1375–1386. [Google Scholar]
- 47.Dunn B, et al. 2013. Recurrent rearrangement during adaptive evolution in an interspecific yeast hybrid suggests a model for rapid introgression. PLoS Genet. 9, e1003366 ( 10.1371/journal.pgen.1003366) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rieseberg LH, Linder C, Seiler GJ. 1995. Chromosomal and genic barriers to introgression in Helianthus. Genetics 141, 1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ungerer MC, Strakosh SC, Zhen Y. 2006. Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr. Biol. 16, R872–R873. ( 10.1016/j.cub.2006.09.020) [DOI] [PubMed] [Google Scholar]
- 50.Ungerer MC, Strakosh SC, Stimpson KM. 2009. Proliferation of Ty3/gypsy-like retrotransposons in hybrid sunflower taxa inferred from phylogenetic data. BMC Biol. 7, 40 ( 10.1186/1741-7007-7-40) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khurana JS, et al. 2011. Adaptation to P element transposon invasion in Drosophila melanogaster. Cell 147, 1551–1563. ( 10.1016/j.cell.2011.11.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwarzbach AE, Rieseberg LH. 2002. Likely multiple origins of a diploid hybrid sunflower species. Mol. Ecol. 11, 1703–1715. ( 10.1046/j.1365-294X.2002.01557.x) [DOI] [PubMed] [Google Scholar]
- 53.Welch ME, Rieseberg LH. 2002. Habitat divergence between a homoploid hybrid sunflower species, Helianthus paradoxus (Asteraceae), and its progenitors. Am. J. Bot. 89, 472–478. ( 10.3732/ajb.89.3.472) [DOI] [PubMed] [Google Scholar]
- 54.Gross BL, Schwarzbach AE, Rieseberg LH. 2003. Origin(s) of the diploid hybrid species Helianthus deserticola (Asteraceae). Am. J. Bot. 90, 1708–1719. ( 10.3732/ajb.90.12.1708) [DOI] [PubMed] [Google Scholar]
- 55.Rieseberg LH, Carter R, Zona S. 1990. Molecular tests of the hypothesized hybrid origin of 2 diploid Helianthus species (Asteraceae). Evolution 44, 1498–1511. ( 10.2307/2409332) [DOI] [PubMed] [Google Scholar]
- 56.Rieseberg LH. 1991. Homoploid reticulate evolution in Helianthus (Asteraceae): evidence from ribosomal genes. Am. J. Bot. 78, 1218–1237. ( 10.2307/2444926) [DOI] [Google Scholar]
- 57.Heiser CB, Smith DM, Clevenger SB, Martin WC. 1969. The North American sunflowers (Helianthus). Memoirs Torrey Botanical Club 22, 218. [Google Scholar]
- 58.Rieseberg LH, et al. 2003. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301, 1211–1216. ( 10.1126/science.1086949) [DOI] [PubMed] [Google Scholar]
- 59.Gross BL, Kane NC, Lexer C, Ludwig F, Rosenthal DM, Donovan LA, Rieseberg LH. 2004. Reconstructing the origin of Helianthus deserticola: survival and selection on the desert floor. Am. Nat. 164, 145–156. ( 10.1086/422223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rieseberg LH. 2000. Crossing relationships among ancient and experimental sunflower hybrid lineages. Evolution 54, 859–865. ( 10.1111/j.0014-3820.2000.tb00086.x) [DOI] [PubMed] [Google Scholar]
- 61.Ungerer MC, Baird SJE, Pan J, Rieseberg LH. 1998. Rapid hybrid speciation in wild sunflowers. Proc. Natl Acad. Sci. USA 95, 11 757–11 762. ( 10.1073/pnas.95.20.11757) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buerkle CA, Rieseberg LH. 2008. The rate of genome stabilization in homoploid hybrid species. Evolution 62, 266–275. ( 10.1111/j.1558-5646.2007.00267.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baack EJ, Whitney KD, Rieseberg LH. 2005. Hybridization and genome size evolution: timing and magnitude of nuclear DNA content increases in Helianthus homoploid hybrid species. New Phytol. 167, 623–630. ( 10.1111/j.1469-8137.2005.01433.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sabot F, Schulman AH. 2006. Parasitism and the retrotransposon life cycle in plants: a hitchhiker's guide to the genome. Heredity 97, 381–388. ( 10.1038/sj.hdy.6800903) [DOI] [PubMed] [Google Scholar]
- 65.Lisch D. 2009. Epigenetic regulation of transposable elements in plants. Annu. Rev. Plant Biol. 60, 43–66. ( 10.1146/annurev.arplant.59.032607.092744) [DOI] [PubMed] [Google Scholar]
- 66.Wessler SR. 1996. Plant retrotransposons: turned on by stress. Curr. Biol. 6, 3 ( 10.1016/S0960-9822(02)00638-3) [DOI] [PubMed] [Google Scholar]
- 67.Grandbastien MA. 1998. Activation of plant retrotransposons under stress conditions. Trends Plant Sci. 3, 181–187. ( 10.1016/S1360-1385(98)01232-1) [DOI] [Google Scholar]
- 68.Kawakami T, Strakosh SC, Zhen Y, Ungerer MC. 2010. Different scales of Ty1/copia-like retrotransposon proliferation in the genomes of three diploid hybrid sunflower species. Heredity 104, 341–350. ( 10.1038/hdy.2009.182) [DOI] [PubMed] [Google Scholar]
- 69.Kawakami T, Dhakal P, Katterhenry AN, Heatherington CA, Ungerer MC. 2011. Transposable element proliferation and genome expansion are rare in contemporary sunflower hybrid populations despite widespread transcriptional activity of LTR retrotransposons. Genome Biol. Evol. 3, 156–167. ( 10.1093/gbe/evr005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ungerer MC, Kawakami T. 2013. Transcriptional dynamics of LTR retrotransposons in early generation and ancient sunflower hybrids. Genome Biol. Evol. 5, 329–337. ( 10.1093/gbe/evt006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Renaut S, Grassa CJ, Yeaman S, Moyers BT, Lai Z, Kane NC, Bowers JE, Burke JM, Rieseberg LH. 2013. Genomic islands of divergence are not affected by geography of speciation in sunflowers. Nat. Commun. 4, 1827 ( 10.1038/ncomms2833) [DOI] [PubMed] [Google Scholar]
- 72.Genomic Resources Development Consortium, King MG, Renaut S, Rieseberg LH, Rowe HC. 2013. Genomic resources notes accepted 1 February 2013–31 March 2013. Mol. Ecol. Resour. 13, 759 ( 10.1111/1755-0998.12123) [DOI] [PubMed] [Google Scholar]
- 73.Rowe HC, Rieseberg LH. 2013. Genome-scale transcriptional analyses of first-generation interspecific sunflower hybrids reveals broad regulatory compatibility. BMC Genomics 14, 342 ( 10.1096/fj.10-170639) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gill N, Buti M, Kane N, Bellec A, Helmstetter N, Berges H, Rieseberg LH. 2014. Sequence-based analysis of structural organization and composition of the cultivated sunflower (Helianthus annuus L.) genome. Biology 3, 295–319. ( 10.3390/biology3020295) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gotz S, et al. 2008. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. ( 10.1093/nar/gkn176) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Staton SE, et al. 2012. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 72, 142–153. ( 10.1111/j.1365-313X.2012.05072.x) [DOI] [PubMed] [Google Scholar]
- 77.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. ( 10.1093/bioinformatics/btp324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. ( 10.1093/bioinformatics/btq033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.R Core Team. 2012. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 80.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol. 11, R106 ( 10.1186/gb-2010-11-10-r106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Storey JD. 2002. A direct approach to false discovery rates. J. Royal Stat. Soc. B (Stat. Methodol.) 64, 479–498. ( 10.1111/1467-9868.00346) [DOI] [Google Scholar]
- 82.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. ( 10.1038/nmeth.1226) [DOI] [PubMed] [Google Scholar]
- 83.Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, Jacobsen SE, Carrington JC. 2004. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2, e104 ( 10.1371/journal.pbio.0020104.sg002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mirouze M, et al. 2009. Selective epigenetic control of retrotransposition in Arabidopsis. Nature 461, 427–430. ( 10.1038/nature08328) [DOI] [PubMed] [Google Scholar]
- 85.Tsukahara S, Kobayashi A, Kawabe A, Mathieu O, Miura A, Kakutani T. 2009. Bursts of retrotransposition reproduced in Arabidopsis. Nature 461, 423–426. ( 10.1038/nature08351) [DOI] [PubMed] [Google Scholar]
- 86.Langfelder P, Horvath S. 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 ( 10.1186/1471-2105-9-559.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huson DH. 2005. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. ( 10.1093/molbev/msj030) [DOI] [PubMed] [Google Scholar]
- 88.Li H, et al. 2009. The sequence alignment/map format and SAM tools. Bioinformatics 25, 2078–2079. ( 10.1093/bioinformatics/btp352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weir B, Cockerham CC. 1984. Estimating F-statistics for the analysis of population-structure. Evolution 38, 1358–1370. ( 10.2307/2408641) [DOI] [PubMed] [Google Scholar]
- 90.Goudet J. 2005. HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. 5, 184–186. ( 10.1111/j.1471-8278.2004.00828.x) [DOI] [Google Scholar]
- 91.SITES (Hey Lab Distributed Software). See http://genfaculty.rutgers.edu/hey/%E2%80%A8software#SITES/ (accessed on 08 October 2012).
- 92.McDonald JH, Kreitman M. 1991. Adaptive protein evolution at the ADH locus in Drosophila. Nature 351, 652–654. ( 10.1038/351652a0) [DOI] [PubMed] [Google Scholar]
- 93.Smith NGC, Eyre-Walker A. 2002. Adaptive protein evolution in Drosophila. Nature 415, 1022–1024. ( 10.1038/4151022a) [DOI] [PubMed] [Google Scholar]
- 94.Woodage T, Basrai MA, Baxevanis AD, Hieter P, Collins FS. 1997. Characterization of the CHD family of proteins. Proc. Natl Acad. Sci. USA 94, 11 472–11 477. ( 10.1073/pnas.94.21.11472) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ogas J, Kaufmann S, Henderson J, Somerville C. 1999. PICKLE is a CHD3 chromatin-remodeling factor that regulates the transition from embryonic to vegetative development in Arabidopsis. Proc. Natl Acad. Sci. USA 96, 13 839–13 844. ( 10.1073/pnas.96.24.13839) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McClintock B. 1984. The significance of responses of the genome to challenge. Science 226, 792–810. ( 10.1126/science.15739260) [DOI] [PubMed] [Google Scholar]
- 97.Michalak P. 2009. Epigenetic, transposon and small RNA determinants of hybrid dysfunctions. Heredity 102, 45–50. ( 10.1038/hdy.2008.48) [DOI] [PubMed] [Google Scholar]
- 98.de Meaux J, Pecinka A. 2012. The Arabidopsis genus: an emerging model to elucidate the molecular basis of interspecific differences in transposable element activity. Mobile Genet. Elem. 2, 142–144. ( 10.4161/mge.21111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vicient C. 2010. Transcriptional activity of transposable elements in maize. BMC Genomics 11, 601 ( 10.1186/1471-2164-11-601) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Magi A, et al. 2013. EXCAVATOR: detecting copy number variants from whole-exome sequencing data. Genome Biol. 14, R120 ( 10.1186/gb-2013-14-10-r120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bonchev G, Parisod C. 2013. Transposable elements and microevolutionary changes in natural populations. Mol. Ecol. Resour. 13, 765–775. ( 10.1111/1755-0998.12133) [DOI] [PubMed] [Google Scholar]
- 102.Slotkin RK, Martienssen R. 2007. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285. ( 10.1038/nrg2072) [DOI] [PubMed] [Google Scholar]
- 103.Stinchcombe JR, Hoekstra HE. 2007. Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity 100, 158–170. ( 10.1038/sj.hdy.6800937) [DOI] [PubMed] [Google Scholar]
- 104.Josefsson C, Dilkes B, Comai L. 2006. Parent-dependent loss of gene silencing during interspecies hybridization. Curr. Biol. 16, 1322–1328. ( 10.1016/j.cub.2006.05.045) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.