

Abstract
The first total synthesis of marine-derived penicimonoterpene (±)-1 has been achieved in four steps from 6-methylhept-5-en-2-one using a Reformatsky reaction as the key step to construct the basic carbon skeleton. A total of 24 new derivatives of 1 have also been designed and synthesized. Their structures were characterized by analysis of their 1H NMR, 13C NMR and HRESIMS data. Some of them showed significant antibacterial activity against Aeromonas hydrophila, Escherichia coli, Micrococcus luteus, Staphylococcus aureus, Vibrio anguillarum, V. harveyi and/or V. parahaemolyticus, and some showed activity against plant-pathogenic fungi (Alternaria brassicae, Colletotrichum gloeosporioides and/or Fusarium graminearum). Some of the derivatives exhibited antimicrobial MIC values ranging from 0.25 to 4 μg/mL, which were stronger than those of the positive control. Notably, Compounds 3b and 10 showed extremely high selectively against plant-pathogenic fungus F. graminearum (MIC 0.25 μg/mL) and pathogenic bacteria E. coli (MIC 1 μg/mL), implying their potential as antimicrobial agents. SAR analysis of 1 and its derivatives indicated that modification of the carbon-carbon double bond at C-6/7, of groups on the allylic methylene unit and of the carbonyl group at C-1, effectively enhanced the antimicrobial activity.
Keywords: (±)-penicimonoterpene, total synthesis, antimicrobial activity, structure-activity relationship
1. Introduction
In recent years, there has been increasing interest in research on marine natural products, since an enormous range of chemically diverse biologically active metabolites have been discovered from marine organisms [1,2,3,4]. Recently, marine-derived fungi have triggered our interest, due to their ability to produce structurally interesting bioactive secondary metabolites [5,6,7,8,9].
Terpenoids comprise an important class of bioactive agents. Despite their relatively simple structures, some of these compounds exhibit interesting biological activities [10,11,12,13,14]. We have recently reported the isolation and identification of a new monoterpenoid, penicimonoterpene (+)-1 (Figure 1), from a marine-derived endophytic isolate of the fungus, Penicillium chrysogenum QEN-24S [15]. This compound exhibited potent activity against the plant pathogen, Alternaria brassicae, comparable to that of a positive control [15].
Figure 1.
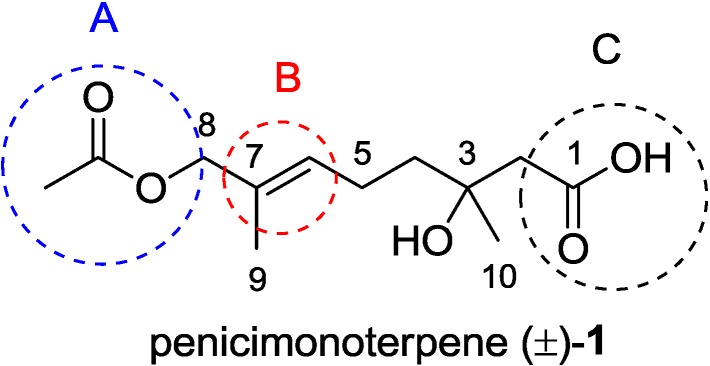
Structure of penicimonoterpene (±)-1.
Herein, we report a short synthesis of (±)-1 featuring a Reformatsky reaction as a key step. This approach has the merits of low cost, mild reaction conditions and easy access to diversity-oriented derivatives for potential structure-activity relationship (SAR) investigation. In antimicrobial assays, penicimonoterpene (±)-1 not only displayed antifungal activity against A. brassicae, but also showed potent antibacterial activity against marine bacteria, including Aeromonas hydrophila, Vibrio harveyi, and V. parahaemolyticus. Therefore, we became interested in designing and synthesizing diverse antibacterial inhibitors using (±)-1 as a model compound. Modifications were focused on variation of the substituents at the eight-position (Section A in Figure 1), the carbon-carbon double bond at C-6/7 (Section B) and carboxyl substituents at C-1 (Section C).
2. Results and Discussion
2.1. Chemistry
2.1.1. Synthesis of Penicimonoterpene (±)-1
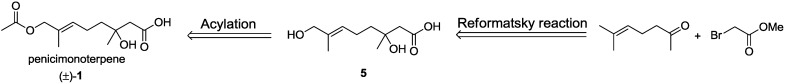
We report herein a practical and efficient racemic synthesis of 1 from 6-methylhept-5-en-2-one using a Reformatsky reaction as the key step. Retrosynthetic analysis (Figure 2) led to 5 as the key target intermediate, which could be accessible via three routes, as listed in Scheme I.
Figure 2.
Retrosynthetic analysis for penicimonoterpene (±)-1.
Scheme I.

Three routes for the total synthesis of penicimonoterpene (±)-1. Reagents and conditions: (a) SeO2 (0.1 eq), t-BuOOH (3.0 eq), CH2Cl2, room temperature (rt.), 19 h, 38%; (b) BrCH2COOCH3 (2.0 eq), Zn (2.0 eq), THF, reflux, 2 h, 4%; (c) KOH (2.2 eq), MeOH: H2O (v/v, 3:1), reflux 5 h, 85%; (d) BrCH2COOCH3 (2.0 eq), Zn (2.0 eq), THF, reflux, 2 h, 85%; (e) KOH (2.2 eq), MeOH: H2O (v/v, 3:1), reflux 5 h, 94%; (f) SeO2 (0.1 eq), t-BuOOH (3.0 eq), CH2Cl2, rt., 19 h, 11%; (g) SeO2 (0.1 eq), t-BuOOH (3.0 eq), CH2Cl2, rt., 19 h, 25% for 4a, 29% for 3a; (h) NaBH4 (1.0 eq), MeOH, rt., 1 h, 69%; (i) Ac2O (1.5 eq), 4-dimethylaminopyridine (DMAP) catalyst (cat.), Et3N (1.5 eq), CH2Cl2, rt., overnight, 55%.
In Route I, selective oxidation of 6-methylhept-5-en-2-one with SeO2 in CH2Cl2 [16,17,18] yielded allylic alcohol 7, which was converted to methyl ester 3a by a Reformatsky reaction using 2.0 eq of BrCH2COOCH3 in the presence of 2.0 eq of Zn powder in refluxing THF. Methyl ester 3a was then hydrolyzed with KOH to afford key intermediate 5. However, this route was not considered satisfactory, because the yield of the Reformatsky reaction step was only 4%.
In Route II, the Reformatsky reaction of 6-methylhept-5-en-2-one with BrCH2COOCH3 yielded 2a in a good yield (85%). After refluxing with 2.2 eq of KOH in a mixture of MeOHH2O (v/v, 3/1), Compound 2a was hydrolyzed in excellent yield (94%) to produce Compound 6, which was subsequently oxidized to give 5 with 11% yield. Unfortunately, Compound 5 proved to be difficult to purify from by-products formed in the reaction, leading to the poor yield obtained for this step. In Route III, treatment of Compound 2a with 3.0 eq of t-BuOOH in the presence of 0.1 eq of SeO2 afforded a chromatographically separable mixture of aldehyde 4a (25%) and alcohol 3a (29%). The former could be converted into the latter (69%) by reduction with NaBH4 in MeOH. In an effort to improve the efficiency of this process, we studied the conditions for the selective oxidation of the allylic methyl groups of 2a. A microwave irradiation method using SeO2 and t-BuOOH adsorbed on SiO2 [19] also afforded 4a, but in low yield (13%). After optimization by extensive variation of the number of equivalents of SeO2 and the reaction time, it was found that the best results were obtained with 0.1 eq of SeO2, and 3.0 eq t-BuOOH in CH2Cl2, reacting for 19 h at room temperature, yielded Compound 3a in 29% yield and Compound 4a in 25% yield. The key intermediate, 5, was obtained in 85% yield by saponification of 3a with no protection of the tertiary hydroxyl group. Treatment of intermediate 5 with 1.5 eq of Ac2O, a catalytic amount of 4-dimethylaminopyridine (DMAP) and 1.5 eq of Et3N at room temperature for 12 h gave 1 in 55% yield.
As expected, based on the selectivity rules and site for the SeO2-mediated oxidation reaction [20], as well as the mechanism of this process [21], the Δ6,7-double bond in alcohol 3a was found to have the E-configuration on the basis of a NOESY correlation between H-6 (δ 5.39) and H2-8 (δ 3.98). The 1H NMR, 13C NMR, MS and HRMS data for synthetic (±)-1 were identical to those of reported natural penicimonoterpene [15].
2.1.2. Synthesis of Diverse Derivatives of (±)-1
In order to investigate the influence of structural changes on the antimicrobial activities of 1, a series of derivatives of 1 were synthesized by modification of three main structural features of 1, including the 8-acetoxy group, the C-6/7 double bond and the carboxyl group at C-1. The syntheses of these derivatives are summarized in Scheme II and Table 1.
Scheme II.

Synthetic routes for diverse derivatives of penicimonoterpene (±)-1 (2–10, 12, 16 and 18). Reagents and conditions: (a) BrCH2COOCH3 or BrCH2COOCH2CH3 (2.0 eq), Zn (2.0 eq), THF, reflux, 2 h, 85% for 2a, 95% for 2b; (b) SeO2 (0.1 eq), t-BuOOH (3.0 eq), CH2Cl2, rt., 19 h, 81% for 3b and 16% for 4b; (c) KOH (2.2 eq), MeOH: H2O (v/v, 3:1), reflux 5 h, 91%; (d) Ac2O (1.5 eq), DMAP (cat.), Et3N (1.5 eq), CH2Cl2, rt., 12 h, 55% for 1, 18% for 10; (e) Ac2O (2.0 eq), DMAP (cat.), Et3N (2.0 eq), CH2Cl2, rt., 24 h, 85%; (f) Ac2O (1.5 eq), DMAP (cat.), Et3N (1.5 eq), CH2Cl2, rt., 12 to 18 h, 91%–95% for 8a and 8b; R-(−)-O-acyl mandelic acid (1.1 eq), N,N′-dicyclohexylcarbodiimide (DCC, 1.1 eq), DMAP (0.1 eq) CH2Cl2, rt., 16 h, 81% for 8c; (g) PPh3 (1.5 eq), NCS (N-chlorosuccinimide) (1.5 eq), dry CH2Cl2, 0 °C, overnight, 69%–77% for 9a and 9b; (h) KOH (2.2 eq), MeOH: H2O (v/v, 3:1), reflux 5 h, 94%; (i) LiAlH4 (3.0 eq), THF, 0 °C for 2 h, then 65 °C for 12 h, 97% from 2a; (j) Pd/C, H2, EtOAc, rt., 12 h, 89%.
Table 1.
Hydrogenation of diverse derivatives of penicimonoterpene (±)-1 to afford 11, 13–15, 17, 19 and 20.
| Entry | Substrate | Product | Yield (%) |
|---|---|---|---|
| 1 | 2a | 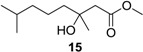 |
93 |
| 2 | 8a | 76 | |
| 3 | 9a | 25 | |
| 4 | 2b | 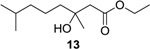 |
90 |
| 5 | 8b | 88 | |
| 6 | 8c | 96 | |
| 7 | 9b | 15 | |
| 8 | 10 |  |
93 |
| 9 | 18 | 89 | |
| 10 | 1 | 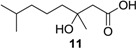 |
92 |
| 11 | 6 | 67 | |
| 12 | 5 | 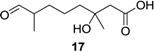 |
45 |
| 13 | 3a | 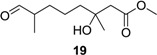 |
27 |
| 14 | 3b |  |
25 |
Reagents and conditions: Pd/C, H2, EtOAc, rt., 12–36 h, 15%–96%.
For the synthesis of these derivatives, BrCH2COOCH2CH3 was used to react with 6-methylhept-5-en-2-one in refluxing THF in the presence of Zn powder to generate 2b, which was then oxidized with SeO2 and t-BuOOH to yield Compounds 3b (81% yield) and 4b (16% yield) (Scheme II). Saponification of 3a or 3b in the presence of KOH, then adjusting the pH to 2~3, yielded Compound 5 in 85%–91% yield with no detectable formation of a cyclic by-product [22]. The use of LiOH for this process gave a very poor yield (11%). Acetylation of 5 was attempted by treatment with 1.5 eq of Ac2O in the presence of a catalytic amount of 4-dimethylaminopyridine (DMAP) and 1.5 eq of Et3N in CH2Cl2 at room temperature for 12 h. The major product was rac-penicimonoterpene 1 (55% yield), with anhydride product 10 forming in 18% yield. Using the same acetylation method, anhydride 18 was obtained in 85% yield from 6. Upon treatment of 2a with 3.0 eq of LiAlH4 in anhydrous THF at 0 °C for 2 h, then raising the temperature to 65 °C for 12 h, reduction product 12 was obtained in 97% yield. Treatment of 12 with H2 in the presence of 10% Pd/C gave 16 in 89% yield.
In an attempt to enhance the antibacterial activities, we envisaged modification at C-8 by introducing electron-withdrawing groups, such as Cl and Br (Scheme II). Chlorine-substituted products 9a and 9b were successfully prepared in 69%–77% yield by using 1.5 eq of NCS (N-chlorosuccinimide) as the chlorinating reagent in the presence of PPh3 at 0 °C overnight. Unfortunately, NBS (N-bromosuccinimide) treatment failed to give the targeted bromination products, possibly because NBS had a tendency to be more reactive than NCS, leading to the bromine-substituted products being less stable, or because other products were formed.
Hydrogenation of 1, 2a–b, 3a–b, 5, 6, 8a–c, 9a–b, 10 and 18 proceeded with H2 using 10% Pd/C as the catalyst to give corresponding products, as shown in Table 1. The Pd-catalyzed isomerization of primary allylic alcohols [23], 3a, 3b, and 5, into the corresponding saturated aldehydes have been achieved at room temperature. Another interesting result was that allylic ester groups (e.g., in 8b and 8c) were easily removed by catalytic hydrogenation in addition to the reduction of the olefin unit at room temperature with excellent yields. It is worth noting that ester substrates 1, 8a–c and 10, prepared by acylation of the corresponding alcohols, as well as chlorine-substituted products 9a and 9b were transformed into related methyl products 11, 13, 14 and 15 by hydrogenolysis using 10% Pd/C as the catalyst with good yields (76%–96%), while subjecting the parent allylic alcohols, 3a, 3b and 5, to the same conditions did not lead to the corresponding methyl products. These results indicated that chloro, as well as ester groups, might be as valuable as protecting groups, since they could be easily removed by catalytic hydrogenation in high yields.
In summary, we have efficiently synthesized penicimonoterpene (±)-1 for the first time, as well as 24 racemic derivatives via simple routes and at low cost. Of 24 derivatives, Compounds 3a–b, 4a–b, 8a–c, 9a–b, 10, 14 and 17–20 were new, and these products were fully characterized by NMR and HRESIMS (see Supplementary Information).
2.2. Antibacterial Activity and SAR Analysis
Synthetic Compounds 1–20 were tested in vitro for antibacterial activity against two Gram-positive bacteria (Micrococcus luteus and Staphylococcus aureus) and five Gram-negative bacteria, including Escherichia coli and the marine bacteria, Aeromonas hydrophila, Vibrio anguillarum, V. harveyi and V. parahaemolyticus. Many of these derivatives exhibited antibacterial effects. Compounds 13–17 showed significant activity, with MIC values ranging from 0.25 to 64 μg/mL, as listed in Table 2. Several observations were made regarding structure-activity relationships among these compounds. Reduction of the carbon-carbon double bond at C-6/7, significantly increasing the antibacterial activity (e.g., 13 vs. 2b, 14 vs. 18, 15 vs. 2a, 16 vs. 12, except for Compounds 19 and 20, which contain an aldehyde group). For compounds containing the double bond at C-6/7, the general order of antibacterial potency for substituents at C-1 was COOH > COOCH2CH3 > COOCH3 > COOCOCH3 (except in the case of E. coli, for which Compound 10 showed better selective activity). Hydroxylation at C-8 (as in 3a and 3b) resulted in loss of activity.
Table 2.
Minimum inhibitory concentration (MIC, μg/mL) of 1 and its derivatives against seven bacterial strains a,b.
| Compounds | A. h. | V. a. | V. h. | V. p. | E. c. | M. l. | S. a. |
|---|---|---|---|---|---|---|---|
| 1 | 64 | >64 | 16 | 64 | 16 | 64 | 8 |
| 2a | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 2b | 32 | 64 | >64 | >64 | >64 | 64 | >64 |
| 3a | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 3b | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 4a | >64 | 32 | 64 | >64 | 64 | >64 | >64 |
| 4b | 64 | 32 | 64 | 32 | 32 | 16 | 32 |
| 5 | >64 | >64 | 32 | >64 | >64 | >64 | 64 |
| 6 | >64 | >64 | 32 | >64 | >64 | >64 | 2 |
| 8a | 64 | >64 | 32 | >64 | >64 | >64 | 64 |
| 8b | 32 | 64 | >64 | >64 | 64 | >64 | 32 |
| 8c | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 9a | 64 | >64 | 8 | >64 | >64 | >64 | >64 |
| 9b | 32 | >64 | 8 | 0.5 | >64 | >64 | 64 |
| 10 | 64 | >64 | >64 | >64 | 1 | >64 | >64 |
| 11 | 16 | >64 | >64 | >64 | 8 | 32 | 4 |
| 12 | >64 | >64 | 32 | 0.5 | >64 | >64 | >64 |
| 13 | 8 | 0.5 | 32 | 32 | 32 | 64 | 2 |
| 14 | 1 | 0.25 | 2 | 0.5 | 1 | 4 | 0.25 |
| 15 | 0.5 | 0.25 | 0.25 | 0.25 | 0.5 | 64 | 0.25 |
| 16 | 0.5 | 0.25 | 0.25 | 0.25 | 0.5 | 16 | 0.5 |
| 17 | 0.5 | 0.25 | 0.5 | 0.25 | 0.5 | 16 | 0.25 |
| 18 | 32 | >64 | 16 | 16 | 8 | 16 | 64 |
| 19 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 20 | 32 | >64 | 64 | 32 | 64 | >64 | 64 |
| Ch c | 0.5 | 4 | 0.5 | 0.5 | 2 | 2 | 8 |
a A. h., A. hydrophila; E. c., E. coli; M. l., M. luteus; S. a., S. aureus; V. a., V. anguillarum; V. h., V. harveyi; V. p., V. parahaemolyticus; Ch, Chloramphenicol; b Average of three replicates; c Positive control.
Notably, hydrogenated Compounds 13–17 and aldehyde 4b showed inhibitory activity to all tested bacterial strains, and in many cases, the activity was stronger than or comparable to the corresponding positive controls (Table 2). For V. parahaemolyticus, Compounds 9b and 12 showed activity similar to that of the positive control (the common antibacterial agent, chloramphenicol). Compound 10 exhibited better selective activity against E. coli with a MIC value of 1 μg/mL, which is 16-fold more potent than that of 1 and two-fold more active than chloramphenicol. Compound 11 exhibited an MIC value of 4 μg/mL against S. aureus, making it more potent than chloramphenicol in the assay (MIC = 8 μg/mL).
2.3. Antifungal Activity and SAR Analysis
The inhibitory effects of Compounds 1–20 against three plant-pathogenic fungi are summarized in Table 3. Compound 1 exhibited activity against A. brassicae with an MIC value of 64 μg/mL. Compounds 4b, 11, 13–15 and 19 showed activity against C. gloeosporioides, and the activity of Compound 14 (MIC = 8 μg/mL) in this assay was twice as potent as that of the positive control (amphotericin B, MIC = 16 μg/mL). In assays against F. graminearum, eight compounds, including 1, 3b, and 11–16, displayed activity, with MIC values ranging from 0.25 to 64 μg/mL. Among them, Compound 3b showed particularly noteworthy activity (MIC = 0.25 μg/mL), as it was 128-fold more potent than amphotericin B (MIC = 32 μg/mL) in this assay. Compound 3b showed extremely high selectively against F. graminearum and might have potential as an antifungal agent. Four other compounds (11–15) were also more potent (two- to eight-fold) than the positive control.
Table 3.
Minimum inhibition concentration (MIC) of 1 and its derivatives (μg/mL) against three plant-pathogenic fungi a.
| Compounds | A. brassicae | C. gloeosporioides | F. graminearum |
|---|---|---|---|
| 1 | 64 | >64 | 32 |
| 2a | >64 | >64 | >64 |
| 2b | >64 | >64 | >64 |
| 3a | >64 | >64 | >64 |
| 3b | >64 | >64 | 0.25 |
| 4a | >64 | >64 | >64 |
| 4b | >64 | 32 | >64 |
| 5 | >64 | >64 | >64 |
| 6 | >64 | >64 | >64 |
| 8a | >64 | >64 | >64 |
| 8b | >64 | >64 | >64 |
| 8c | >64 | >64 | >64 |
| 9a | >64 | >64 | >64 |
| 9b | >64 | >64 | >64 |
| 10 | >64 | >64 | >64 |
| 11 | >64 | 32 | 4 |
| 12 | >64 | >64 | 16 |
| 13 | >64 | 32 | 16 |
| 14 | >64 | 8 | 8 |
| 15 | >64 | 16 | 16 |
| 16 | >64 | >64 | 64 |
| 17 | >64 | >64 | >64 |
| 18 | >64 | >64 | >64 |
| 19 | >64 | 64 | >64 |
| 20 | >64 | >64 | >64 |
| Amphotericin B b | 16 | 16 | 32 |
a Average of three replicates; b positive control.
Based on the above data, newly synthesized Compounds 3b, 11, 14 and 15 were found to be the most effective, especially against F. graminearum. From an SAR standpoint, it is noteworthy that the oxidation of the methyl to a hydroxymethyl group at C-8 (2b vs. 3b) or replacement of the methyl ester group at C-1 by an ethyl ester (3a vs. 3b) significantly increased the antifungal activity. Compounds with a reduced double bond at C-6/7 (11 and 13–15) also showed better inhibitory activities against C. gloeosporioides and F. graminearum (except for those containing an aldehyde group, such as 17, 19 and 20).
3. Experimental Section
3.1. General
Chemicals and instruments: 2-Methyl-2-hepten-6-one was purchased from TCI Company. t-Butyl hydroperoxide (70% in water), BrCH2COOCH3 and BrCH2COOCH2CH3 were purchased from J&K Company (Qingdao, China). THF was dried over LiAlH4 and distilled prior to use. CH2Cl2 was distilled over CaH2. The reagents were all analytically or chemically pure. All solvents and liquid reagents were dried by standard methods in advance and distilled before use according to Perrin and Armarego [24]. Thin-layer chromatography (TLC) was performed using silica gel GF-254 plates (Qing-Dao Chemical Company, Qingdao, China) with detection by UV (254 nm). NMR spectra were recorded on Bruker Advance 500 spectrometers with tetramethylsilane (TMS) as an internal standard. Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m) and broad (br). Mass spectra were recorded by electrospray ionization (ESI) using a VG Autospec 3000 mass spectrometer.
Plant-pathogenic fungi: All strains of fungi were provided by Qindao Agricultural University. The strains were retrieved from a storage tube and incubated in potato dextrose agar media (PDA) at 28 °C for a week to get new mycelia for the antifungal assay.
Bacterial strains: The strains were provided by the Institute of Oceanology, Chinese Academy of Sciences, and cultured at 28 or 37 °C in nutrient agar (NA).
3.2. Synthesis of Compounds 1–20
3.2.1. Synthesis of Methyl (±)-3-Hydroxy-3,7-dimethyloct-6-enoate (2a)
2-Methyl-2-hepten-6-one (3.16 g, 25.0 mol) and BrCH2COOCH3 (7.65 g, 50.0 mol) in 50 mL of anhydrous THF were added to a suspension of Zn (3.27 g, 50.0 mol) in anhydrous THF (50 mL) at reflux. After the reaction was initiated, the other portion of the solution was added dropwise to the reaction mixture. The mixture was allowed to stir at reflux until the color of the Zn changed from gray to brownish. After 2 h, the reaction mixture was cooled to room temperature, and the reaction was quenched with 100 mL of 10% AcOH. The reaction mixture was extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine and dried with anhydrous Na2SO4. The solvent was evaporated in vacuo, and the crude product was purified by column chromatography (EtOAc–petroleum ether, 1:7) to yield 2a (4.25 g, 85%) as a pale yellow oil; Rf 0.32 (EtOAc–petroleum ether, 1:7); 1H NMR (500 MHz, CDCl3) δ 5.07 (t, J = 6.3 Hz, 1H, CH2CH=), 3.69 (s, 3H, OCH3), 3.08 (s, 1H, OH), 2.51 (d, J = 15.5 Hz, 1H, CH2COOCH3), 2.43 (d, J = 15.5 Hz, 1H, CH2COOCH3), 2.02 (m, 2H, =CHCH2), 1.66 (s, 3H, CH3), 1.59 (s, 3H, CH3), 1.52 (m, 2H, =CHCH2CH2), 1.22 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.4, 131.9, 124.2, 71.0, 51.7, 44.9, 41.9, 26.8, 25.7, 22.8, 17.7; HRESIMS m/z 201.1486 [M + H]+ (calcd. for C11H21O3, 201.1485).
3.2.2. Synthesis of Ethyl (±)-3-Hydroxy-3,7-dimethyloct-6-enoate (2b)
This compound was obtained from BrCH2COOCH2CH3 by a method analogous to that used for 2a. 2b (3.05 g, 95%); Rf 0.35 (EtOAc–petroleum ether, 1:7); 1H NMR (500 MHz, CDCl3) δ 5.06 (t, J = 6.3 Hz, 1H, CH2CH=), 4.14 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.39 (br s, 1H, OH), 2.48 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.40 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.01 (m, 2H, =CHCH2), 1.64 (s, 3H, CH3), 1.57 (s, 3H, CH3), 1.50 (td, J = 7.3, 3.5 Hz, 2H, =CHCH2CH2), 1.24 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.21 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.1, 131.8, 124.2, 71.0, 60.7, 45.0, 41.9, 26.7, 25.7, 22.7, 17.6, 14.2; HRESIMS m/z 215.1640 [M + H]+ (calcd. for C12H23O3, 215.1642).
3.2.3. Syntheses of Methyl (E)-(±)-3,8-Dihydroxy-3,7-dimethyloct-6-enoate (3a) and Methyl (E)-(±)-3-Hydroxy-3,7-dimethyl-8-oxooct-6-enoate (4a)
Compound 2a (3.20 g, 15.9 mmol) was dissolved in CH2Cl2 (150 mL) and added dropwise to a solution of SeO2 (825 mg, 7.4 mmol) and t-BuOOH (70% in water, 3.2 mL, 22.3 mmol) in CH2Cl2 (50 mL) at room temperature. After stirring at room temperature for 19 h, the reaction mixture was quenched with aqueous 10% NaOH, H2O and brine; then, the organic layer was dried and concentrated. The resulting yellow oil was purified by flash column chromatography (EtOAc–petroleum ether, 1:7) on silica gel to yield 4a as a colorless oil (624 mg, 25%); Rf 0.33 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 9.38 (s, 1H, CHO), 6.48 (t, J = 6.9 Hz, 1H, CH2CH=), 3.72 (s, 3H, OCH3), 2.55 (d, J = 15.7 Hz, 1H, CH2COOCH3), 2.49 (d, J = 15.7 Hz, 1H, CH2COOCH3), 2.46 (m, 2H, CH2CH2CH=), 1.75 (s, 3H, CH3), 1.67 (m, 2H, CH2CH2CH=), 1.28 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 195.3, 173.3, 154.3, 139.6, 70.7, 51.9, 44.9, 40.2, 26.7, 23.7, 9.3; HRESIMS m/z 237.1100 [M + Na]+ (calcd. for C11H18O4Na, 237.1097). Compound 3a was obtained in an analogous fashion; colorless oil (732 mg, 29%); Rf 0.22 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.39 (t, J = 7.1 Hz, 1H, CH2CH=), 3.98 (s, 2H, CH2OH), 3.71 (s, 3H, OCH3), 2.53 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.46 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.12 (m, 2H, CH2CH=), 1.66 (s, 3H, CH3C=), 1.56 (m, 2H, CH2CH2CH=), 1.25 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.5, 135.3, 125.8, 71.0, 68.9, 51.8, 45.0, 41.6, 26.8, 22.4, 13.7; HRESIMS m/z 239.1252 [M + Na]+ (calcd. for C11H20O4Na, 239.1254). Additionally, 841 mg of material were recovered. The E-configuration of Compound 3a was verified by a NOESY correlation from CH2CH= (δ 5.39) to CH2OH (δ 3.98).
3.2.4. Reduction of Aldehyde 4a to Alcohol 3a
NaBH4 (117 mg, 3.1 mmol) was added to a stirred solution of aldehyde 4a (840 mg, 3.1 mmol) in MeOH (50 mL). The mixture was stirred for 1 h at room temperature, then concentrated under reduced pressure. The residue thus obtained was treated with EtOAc (20 mL) and saturated NH4Cl solution (50 mL); then, the separated aqueous phase was extracted with EtOAc (3 × 30 mL). The combined organic phases were dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography (EtOAc–petroleum ether, 1:5) on silica gel to yield a colorless oil, 3a (460 mg, 69%).
3.2.5. Syntheses of Ethyl (E)-(±)-3,8-Dihydroxy-3,7-dimethyloct-6-enoate (3b) and Ethyl (E)-(±)-3-Hydroxy-3,7-dimethyl-8-oxooct-6-enoate (4b)
These compounds were obtained from 2b in a manner similar to that described for the preparation of 3a and 4a. Colorless oil 4b (3.98 g, 16%); Rf 0.35 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 9.39 (s, 1H, CHO), 6.49 (t, J = 7.1 Hz, 1H, CH2CH=), 4.20 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.54 (d, J = 15.8 Hz, 1H, CH2COOCH2CH3), 2.48 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.45 (m, 2H, CH2CH=), 1.76 (s, 3H, CH3), 1.67 (m, 2H, CH2CH2CH=), 1.29 (s, 3H, COHCH3), 1.29 (t, J = 7.0 Hz, 3H, OCH2CH3); 13C NMR (125 MHz, CDCl3) δ 195.3, 173.0, 154.3, 139.6, 70.7, 61.0, 45.1, 40.2, 26.7, 23.8, 14.3, 9.3; HRESIMS m/z 251.1253 [M + Na]+ (calcd. for C12H20O4Na, 251.1254). Colorless oil 3b (20.91 g, 81%); Rf 0.25 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.38 (t, J = 6.7 Hz, 1H, CH2CH=), 4.16 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.97 (s, 2H, CH2OH), 2.50 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.11 (m, 2H, =CHCH2), 1.65 (s, 3H, CH3), 1.54 (m, 2H, =CHCH2CH2), 1.26 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.24 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.1, 135.2, 125.7, 71.0, 68.9, 60.8, 45.1, 41.6, 26.7, 22.3, 14.3, 13.7; HRESIMS m/z 253.1415 [M + Na]+ (calcd. for C12H22O4Na, 253.1410).
3.2.6. Synthesis of (E)-(±)-3,8-Dihydroxy-3,7-dimethyloct-6-enoic Acid (5)
To a solution of 3a (133 mg, 0.6 mmol) in MeOH (30 mL) and water (10 mL) was added KOH (76 mg, 1.4 mmol). The resulting reaction mixture was heated under reflux for 3 h, and the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was acidified with 1 N HCl to pH 2~3, followed by extraction with CHCl3 (3 × 20 mL). The combined organic layers were dried (MgSO4) and concentrated in vacuo. The residue was chromatographed over silica gel (EtOAc–petroleum ether, 1:1) to afford the acid, 5 (105 mg, 85%), as a colorless oil; Rf 0.25 (EtOAc:CHCl3, 1:1); 1H NMR (500 MHz, CDCl3) δ 5.41 (t, J = 5.4 Hz, 1H, CH2CH=), 4.00 (s, 2H, CH2OH), 2.58 (d, J = 15.7 Hz, 1H, CH2COOH), 2.51 (d, J = 15.7 Hz, 1H, CH2COOH), 2.13 (m, 2H, CH2CH=), 1.67 (s, 3H, CH3C=), 1.63 (m, 2H, CH2CH2CH=), 1.31 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 175.5, 135.4, 125.7, 71.5, 68.9, 44.9, 41.5, 26.8, 22.4, 13.8; HRESIMS m/z 225.1076 [M + Na]+ (calcd. for C10H18O4Na, 225.1103).
3.2.7. Syntheses of Acetic (E)-(±)-8-Acetoxy-3-hydroxy-3,7-dimethyloct-6-enoic Anhydride (10) and Penicimonoterpene (±)-1
Compound 5 (250 mg, 1.2 mmol) was dissolved in CH2Cl2 (20 mL) at room temperature. To this solution were added DMAP (3 mg, 0.03 mmol), Et3N (225 μL, 1.6 mmol) and, finally, Ac2O (152 μL, 1.6 mmol). The reaction was stirred overnight. After completion of the reaction as detected by TLC, the solvents were evaporated under reduced pressure, and the crude product was purified by column chromatography (EtOAc–petroleum ether, 1:1) to afford 10 and (±)-1.
10, colorless oil (65 mg, 18%); Rf 0.75 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.44 (t, J = 6.5 Hz, 1H, CH2CH=), 4.44 (s, 2H, CH2OAc), 3.05 (d, J = 14.5 Hz, 1H, CH2COOAc), 2.90 (d, J = 14.5 Hz, 1H, CH2COOAc), 2.10 (m, 2H, CH2CH=), 2.07 (s, 3H, COCH3), 2.03 (m, 1H, CH2CH2CH=), 2.00 (s, 3H, CH3COOCOCH2), 1.81 (m, 1H, CH2CH2CH=), 1.65 (s, 3H, CH3C=), 1.55 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 175.4, 171.2, 170.7, 130.9, 128.4, 81.2, 70.1, 42.5, 38.3, 24.1, 22.3, 22.1, 21.1, 14.0; HRESIMS m/z 309.1307 [M + Na]+ (calcd. for C14H22O6Na, 309.1309).
Penicimonoterpene (±)-1, colorless oil (165 mg, 55%); Rf 0.62 (EtOAc–petroleum ether, 2:1); 1H NMR (500 MHz, CDCl3) δ 5.39 (t, J = 6.7 Hz, 1H, CH2CH=), 4.38 (s, 2H, CH2O), 2.51 (d, J = 15.7 Hz, 1H, CH2COOH), 2.45 (d, J = 15.7 Hz, 1H, CH2COOH), 2.08 (m, 2H, CH2CH=), 2.01 (s, 3H, COCH3), 1.60 (s, 3H, CH3C=), 1.55 (m, 2H, CH2CH2CH=), 1.24 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 176.4, 171.5, 130.4, 129.0, 71.3, 70.3, 44.6, 41.0, 26.4, 22.3, 21.0, 13.8; HRESIMS m/z 267.1202 [M + Na]+ (calcd. for C12H20O5Na, 267.1203).
The spectral data (NMR, MS and HRMS) of (±)-1 were in agreement with those of the reported natural penicimonoterpene [15].
3.2.8. Synthesis of (±)-3-Hydroxy-3,7-dimethyloct-6-enoic Acid (6)
Compound 6 was obtained from 2a by a method similar to that described for the preparation of 5. Colorless oil 6 (443 mg, 94%); Rf 0.23 (EtOAc–CHCl3, 1:1); 1H NMR (500 MHz, CDCl3) δ 5.10 (t, J = 6.6 Hz, 1H, =CHCH2), 2.59 (d, J = 15.7 Hz, 1H, CH2COOH), 2.51 (d, J = 15.7 Hz, 1H, CH2COOH), 2.07 (m, 2H, =CHCH2), 1.68 (s, 3H, CH3), 1.61 (s, 3H, CH3), 1.58 (m, 2H, =CHCH2CH2), 1.30 (s, 3H, CH3COH); 13C NMR (125 MHz, CDCl3) δ 177.2, 132.3, 123.9, 71.5, 44.9, 41.8, 26.7, 25.8, 22.8, 17.8; HRESIMS m/z 187.1332 [M + H]+ (calcd. for C10H19O3, 187.1329).
3.2.9. Synthesis of (E)-7-Hydroxy-6-methylhept-5-en-2-one (7)
Compound 7 was obtained from 2-methyl-2-hepten-6-one in a manner similar to that described for the preparation of 3a. Yellow oil 7 (1.35 g, 38%); Rf 0.15 (EtOAc–petroleum ether, 1:5); 1H NMR (500 MHz, DMSO-d6) δ 5.26 (t, J = 7.2 Hz, 1H, =CHCH2), 4.64 (s, 1H, CH2OH), 3.75 (s, 2H, CH2OH), 2.46 (t, J = 7.4 Hz, 2H, CH2COCH3), 2.16 (q, J = 7.2 Hz, 2H, =CHCH2), 2.07 (s, 3H, COCH3), 1.54 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ 208.0, 135.9, 122.1, 66.2, 42.6, 29.6, 21.4, 13.4.
3.2.10. Synthesis of Methyl (E)-(±)-8-Acetoxy-3-hydroxy-3,7-dimethyloct-6-enoate (8a)
Compound 3a (130 mg, 0.6 mmol) was dissolved in CH2Cl2 (5 mL) at room temperature. To this solution were added DMAP (2 mg, 0.02 mmol), Et3N (125 μL, 0.9 mmol) and Ac2O (85 μL, 0.9 mmol). The reaction was stirred for 24 h. After completion of the reaction as detected by TLC, the solvents were evaporated under reduced pressure, and the crude products were purified by column chromatography (EtOAc–petroleum ether, 1:1) to afford 8a (141 mg, 91%) as a colorless oil; Rf 0.72 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.42 (t, J = 7.1 Hz, 1H, CH2CH=), 4.41 (s, 2H, CH2O), 3.69 (s, 3H, OCH3), 2.51 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.44 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.12 (m, 2H, CH2CH=), 2.04 (s, 3H, COCH3), 1.63 (s, 3H, CH3C=), 1.53 (m, 2H, CH2CH2CH=), 1.23 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 172.7, 170.4, 129.8, 128.6, 70.2, 69.5, 51.1, 44.2, 40.7, 26.1, 21.7, 20.4, 13.3; HRESIMS m/z 281.1359 [M + Na]+ (calcd. for C13H22O5Na, 281.1359).
3.2.11. Synthesis of Ethyl (E)-(±)-8-Acetoxy-3-hydroxy-3,7-dimethyloct-6-enoate (8b)
Compound 8b was obtained from 3b by a method similar to that described for the preparation of 8a. Colorless oil 8b (250 mg, 95%); Rf 0.75 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.43 (t, J = 7.0 Hz, 1H, CH2CH=), 4.43 (s, 2H, CH3COOCH2), 4.16 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.24 (br s, 1H, COHCH3), 2.50 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.13 (m, 2H, CH2CH=), 2.05 (s, 3H, CH3COOCH2), 1.64 (s, 3H, CH3C=), 1.54 (m, 2H, CH2CH2CH=), 1.26 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.23 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.0, 171.0, 130.5, 129.3, 70.9, 70.2, 60.8, 45.1, 41.4, 26.7, 22.4, 21.1, 14.3, 14.0; HRESIMS m/z 295.1518 [M + Na]+ (calcd. for C14H24O5Na, 295.1516).
3.2.12. Synthesis of Ethyl (E)-(±)-8-((R)-2-Acetoxy-2-phenylacetoxy)-3-hydroxy-3,7-dimethyloct-6-enoate (8c)
DCC (563 mg, 2.7 mmol) was dissolved in CH2Cl2 (20 mL), and R-(−)-O-acyl mandelic acid (530 mg, 2.7 mmol) and DMAP (30 mg, 0.3 mmol) were added at room temperature. After stirring for 16 h at room temperature, the solvent volume was reduced in vacuo, and the remaining mixture was purified by column chromatography (EtOAc–petroleum ether, 1:8) to afford 8c (819 mg, 81%) as a colorless oil; Rf 0.76 (EtOAc–petroleum ether, 1:2); 1H NMR (500 MHz, CDCl3) δ 7.47 (dd, J = 6.4, 2.8 Hz, 2H, ArH), 7.38 (m, 3H, ArH), 5.92 (s, 1H, CHArH), 5.33 (t, J = 6.9 Hz, 1H, CH2CH=), 4.50 (dd, J = 12.2 Hz, 2H, COOCH2CCH3=), 4.18 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.49 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.42 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.19 (s, 3H, CH3COO), 2.06 (m, 2H, CH2CH=), 1.50 (s, 3H, CH3C=), 1.48 (m, 2H, CH2CH2CH=), 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.22 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.1, 170.4, 168.8, 134.1, 129.9, 129.7, 129.3, 128.9 (two), 127.8 (two), 74.7, 71.1, 70.8, 60.8, 45.1, 41.3, 26.7, 22.4, 20.8, 14.3, 13.7; HRESIMS m/z 407.2065 [M + H]+ (calcd. for C22H31O7, 407.2064).
3.2.13. Synthesis of Methyl (E)-(±)-8-Chloro-3-hydroxy-3,7-dimethyloct-6-enoate (9a)
To a solution of 3a (76 mg, 0.3 mmol) and PPh3 (138 mg, 0.5 mmol) in 10 mL of dry CH2Cl2 was added NCS (70 mg, 0.5 mmol) at 0 °C under nitrogen. The reaction mixture was stirred at 0 °C for 1 h, then allowed to warm to room temperature and stirred for 2 h. The reaction was monitored by TLC. After completion of the reaction, the solvent volume was reduced in vacuo, and the remaining mixture was purified by column chromatography (EtOAc–petroleum ether, 1:5) to afford 9a (63 mg, 77%) as a colorless oil; Rf 0.63 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.49 (t, J = 7.0 Hz, 1H, CH2CH=), 3.97 (s, 2H, CH2Cl), 3.69 (s, 3H, OCH3), 3.20 (br s, 1H, COHCH3), 2.50 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.11 (m, 2H, CH2CH=), 1.71 (s, 3H, CH3C=), 1.55 (m, 2H, CH2CH2CH=), 1.22 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.3, 132.1, 130.4, 70.8, 52.3, 51.7, 44.9, 41.1, 26.7, 22.7, 14.1; HRESIMS m/z 257.0892 [M + Na]+ (calcd. for C11H19ClO3Na, 257.0920).
3.2.14. Synthesis of Ethyl (E)-(±)-8-Chloro-3-hydroxy-3,7-dimethyloct-6-enoate (9b)
This compound was obtained from 3b by a method similar to that used for 9a. Colorless oil 9b (157 mg, 69%); Rf 0.82 (EtOAc–petroleum ether, 1:2); 1H NMR (500 MHz, CDCl3) δ 5.51 (t, J = 7.0 Hz, 1H, CH2CH=), 4.17 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.99 (s, 2H, CH2Cl), 3.17 (br s, 1H, COHCH3), 2.50 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH2CH3), 2.13 (m, 2H, CH2CH=), 1.73 (s, 3H, CH3C=), 1.55 (m, 2H, CH2CH2CH=), 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.24 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 173.0, 132.1, 130.5, 70.8, 60.8, 52.4, 45.1, 41.2, 26.8, 22.8, 14.3, 14.2; HRESIMS m/z 249.1253 [M + H]+ (calcd. for C12H22ClO3, 249.1252).
3.2.15. Synthesis of (±)-3,7-Dimethyloct-6-ene-1,3-diol (12)
Compound 2a (100 mg, 0.5 mmol) in THF (10 mL) was added dropwise to a stirred slurry of LiAlH4 (57 mg, 1.5 mmol) in THF (10 mL) in an ice bath. After stirring in an ice bath for 2 h, the solution was heated to 65 °C about 10 h. The reaction was monitored by TLC. After completion of the reaction, the reaction mixture was successively treated with water (10 mL), 20% aqueous NaOH (10 mL) and then again with water (10 mL). The resulting white granular suspension was filtered, and the filter cake was washed with CHCl3 (3 × 10 mL). The organic layers were combined, dried (Na2SO4), and then, the solvent was evaporated under reduced pressure and the residue purified by chromatography (EtOAc–petroleum ether, 1:4) to afford 12 (83 mg, 97%) as a colorless oil; Rf 0.34 (EtOAc–petroleum ether, 1:2); 1H NMR (500 MHz, CDCl3) δ 5.09 (t, 1H, CH2CH=), 3.84 (m, 2H, CH2OH), 3.30 (s, 2H, 2 × OH), 2.01 (m, 2H, CH2CH=), 1.76 (m, 1H, CH2CH2OH), 1.66 (s, 3H, CH3),1.63 (m, 1H, CH2CH2OH), 1.59 (s, 3H, CH3), 1.51 (m, 2H, CH2CH2CH=), 1.21 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3) δ 131.9, 124.4, 73.9, 59.7, 42.5, 41.7, 26.7, 25.7, 22.8, 17.7; HRESIMS m/z 195.1335 [M + Na]+ (calcd. for C10H20O2Na, 195.1361).
3.2.16. Synthesis of Acetic (±)-3-Hydroxy-3,7-dimethyloct-6-enoic Anhydride (18)
Compound 6 (345 mg, 1.8 mmol) was dissolved in CH2Cl2 (20 mL) at room temperature. To this solution were added DMAP (23 mg, 0.18 mmol), Et3N (517 μL, 3.7 mmol) and, finally, Ac2O (350 μL, 3.7 mmol). The reaction was stirred overnight. After completion of the reaction as detected by TLC, the solvents were evaporated under reduced pressure, and the crude product was purified by column chromatography (EtOAc–petroleum ether, 1:10) to afford 18 as a colorless oil (360 mg, 85%); Rf 0.75 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 5.07 (t, J = 6.5 Hz, 1H, CH2CH=), 3.04 (d, J = 14.3 Hz, 1H, CH2COOCOCH3), 2.89 (d, J = 14.3 Hz, 1H, CH2COOCOCH3), 2.00 (s, 3H, COCH3), 1.99 (m, 2H, CH2CH=), 1.99 (m, 1H, CH2CH2CH=), 1.76 (m, 1H, CH2CH2CH=), 1.67 (s, 3H, CH3C=), 1.59 (s, 3H, CH3C=), 1.54 (s, 3H, COHCH3); 13C NMR (125 MHz, CDCl3)δ 176.1, 170.8, 132.2, 123.5, 81.4, 42.5, 38.9, 25.8, 24.1, 22.4, 22.3, 17.7; HRESIMS m/z 251.1260 [M + Na]+ (calcd. for C12H20O4Na, 251.1254).
3.2.17. Syntheses of Compounds 11, 13–17, 19 and 20
General procedure: A mixture of the starting material (i.e., a double-bond-containing monoterpene derivative) with 10% Pd/C in EtOAc was stirred at room temperature for an appropriate time (12 to 36 h). After the catalyst was removed by filtration, the filtrate was concentrated in vacuo to give a residue, which was purified by column chromatography on silica gel.
(±)-3-Hydroxy-3,7-dimethyloctanoic acid (11): Colorless oil (47 mg, 92%); Rf 0.52 (EtOAc–petroleum ether, 1:1); 1H NMR (500 MHz, CDCl3) δ 2.57 (d, J = 15.7 Hz, 1H, CH2COOH), 2.49 (d, J = 15.7 Hz, 1H, CH2COOH), 1.55 (m, 1H, CH2CH(CH3)2), 1.51 (m, 2H, CH2COH), 1.34 (m, 2H, CH2CH2CH2), 1.28 (s, 3H, COHCH3), 1.16 (m, 2H, CH2CH2CH2), 0.87 (s, 3H, CH(CH3)2), 0.86 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 177.2, 71.7, 44.8, 42.4, 39.4, 28.0, 26.7, 22.7, 22.6, 21.8; HRESIMS m/z 187.1353 [M − H]− (calcd. for C10H19O3, 187.1340).
Ethyl (±)-3-hydroxy-3,7-dimethyloctanoate (13): Colorless oil (61 mg, 96%); Rf 0.73 (EtOAc–petroleum ether, 1:5); 1H NMR (500 MHz, CDCl3) δ 4.17 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.50 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.42 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 1.53 (m, 1H, CH(CH3)2), 1.47 (m, 2H, CH2CH2CH2COHCH3), 1.33 (m, 2H, CH2CH2CH2COHCH3), 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.22 (s, 3H, COHCH3), 1.15 (m, 2H, CH2CH2CH2COHCH3), 0.87 (s, 3H, CH(CH3)2), 0.86 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 173.2, 71.2, 60.7, 45.0, 42.5, 39.5, 28.0, 26.8, 22.7, 22.6, 21.8, 14.3; HRESIMS m/z 239.1624 [M + Na]+ (calcd. for C12H24O3Na, 239.1618).
Acetic (±)-3-hydroxy-3,7-dimethyloctanoic anhydride (14): Colorless oil (45 mg, 93%); Rf 0.75 (EtOAc–petroleum ether, 1:1); 1H NMR (500 MHz, CDCl3) δ 3.02 (d, J = 14.4 Hz, 1H, CH2COOCOCH3), 2.88 (d, J = 14.4 Hz, 1H, CH2COOCOCH3), 1.99 (s, 3H, COCH3), 1.93 (m, 1H, CH2CH2CH2COHCH3), 1.73 (m, 1H, CH2CH2CH2COHCH3), 1.53 (m, 1H, CH(CH3)2), 1.52 (s, 3H, COHCH3), 1.31 (m, 2H, CH2CH2CH2COHCH3), 1.17 (m, 2H, CH2CH2CH2COHCH3), 0.87 (s, 3H, CH(CH3)2), 0.86 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 176.2, 170.8, 81.6, 42.6, 39.2, 39.1, 27.9, 24.2, 22.7, 22.4, 21.3; HRESIMS m/z 253.1409 [M + Na]+ (calcd. for C12H22O4Na, 253.1410).
Methyl (±)-3-hydroxy-3,7-dimethyloctanoate (15): Colorless oil (45 mg, 93%); Rf 0.72 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 3.71 (s, 3H, OCH3), 3.41 (br s, 1H, COHCH3), 2.52 (d, J = 15.1 Hz, 1H, CH2COOCH3), 2.45 (d, J = 15.6 Hz, 1H, CH2COOCH3), 1.54 (m, 1H, CH(CH3)2), 1.46 (m, 2H, CH2CH2CH2COHCH3), 1.33 (m, 2H, CH2CH2CH2COHCH3), 1.23 (s, 3H, COHCH3), 1.16 (m, 2H, CH2CH2CH2COHCH3), 0.87 (s, 3H, CH(CH3)2), 0.86 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 173.6, 71.2, 51.7, 44.9, 42.5, 39.5, 28.1, 26.9, 22.7, 22.6, 21.8; HRESIMS m/z 225.1461 [M + Na]+ (calcd. for C11H22O3Na, 225.1461).
(±)-3,7-Dimethyloctane-1,3-diol (16): Colorless oil (26 mg, 89%); Rf 0.52 (EtOAc–petroleum ether, 1:3); 1H NMR (500 MHz, CDCl3) δ 3.88 (m, 2H, CH2OH), 2.49 (s, 2H, 2 × OH), 1.79 (m, 1H, CH2CH2OH), 1.66 (m, 1H, CH2CH2OH), 1.55 (m, 1H, CH(CH3)2), 1.50 (m, 2H, CH2CH2CH2COHCH3), 1.31 (m, 2H, CH2CH2CH2COHCH3), 1.24 (s, 3H, COHCH3), 1.18 (m, 2H, CH2CH2CH2COHCH3), 0.88 (s, 3H, CH(CH3)2), 0.87 (s, 3H, CH(CH3)2); 13C NMR (125 MHz, CDCl3) δ 74.2, 60.1, 43.2, 41.7, 39.6, 28.1, 26.9, 22.7, 22.7, 21.9; HRESIMS m/z 197.1518 [M + Na]+ (calcd. for C10H22O2Na, 197.1512).
(±)-3-Hydroxy-3,7-dimethyl-8-oxooctanoic acid (17): Colorless oil (90 mg, 45%); Rf 0.33 (EtOAc–petroleum ether, 2:1); 1H NMR (500 MHz, CDCl3) δ 9.60 (s, 1H, CHO), 2.55 (d, J = 15.7 Hz, 1H, CH2COOH), 2.48 (d, J = 15.7 Hz, 1H, CH2COOH), 2.35 (m, 1H, CH3CHCHO), 1.70 (m, 1H, CH2CH2CH2COHCH3), 1.54 (m, 2H, CH2CH2CH2COHCH3), 1.41 (m, 2H, CH2CH2CH2COHCH3), 1.38 (m, 1H, CH2CH2CH2COHCH3), 1.26 (s, 3H, COHCH3), 1.09 (d, J = 7.0 Hz, 3H, CH3CHCHO); 13C NMR (125 MHz, CDCl3) δ 205.5, 177.0, 71.3, 46.3, 44.8, 41.9, 30.8, 26.7, 21.4, 13.5; HRESIMS m/z 225.1101 [M + Na]+ (calcd. for C10H18O4Na, 225.1097).
Methyl (±)-3-hydroxy-3,7-dimethyl-8-oxooctanoate (19): Colorless oil (94 mg, 27%); Rf 0.53 (EtOAc–petroleum ether, 1:1); 1H NMR (500 MHz, CDCl3) δ 9.61 (s, 1H, CHO), 3.71 (s, 3H, OCH3), 2.50 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.43 (d, J = 15.6 Hz, 1H, CH2COOCH3), 2.34 (m, 1H, CHCH3), 1.70 (m, 1H, CHCH2CH2CH2), 1.50 (m, 2H, CHCH2CH2CH2), 1.39 (m, 1H, CHCH2CH2CH2), 1.38 (m, 2H, CHCH2CH2CH2), 1.22 (s, 3H, COHCH3 ), 1.09 (d, J = 7.0 Hz, 3H, CHCH3); 13C NMR (125 MHz, CDCl3) δ 205.1, 173.5, 70.9, 51.8, 46.4, 44.8, 42.0, 31.0, 26.8, 21.4, 13.5; HRESIMS m/z 239.1260 [M + Na]+ (calcd. for C11H20O4Na, 239.1254).
Ethyl (±)-3-hydroxy-3,7-dimethyl-8-oxooctanoate (20): Colorless oil (15 mg, 25%); Rf 0.52 (EtOAc–petroleum ether, 1:2); 1H NMR (500 MHz, CDCl3) δ 9.56 (s, 1H, CHO), 4.12 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.44 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.37 (d, J = 15.5 Hz, 1H, CH2COOCH2CH3), 2.30 (m, 1H, CHCH3), 1.64 (m, 1H, CHCH2CH2CH2), 1.46 (m, 2H, CHCH2CH2CH2), 1.35 (m, 1H, CHCH2CH2CH2), 1.34 (m, 2H, CHCH2CH2CH2), 1.22 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.17 (s, 3H, COHCH3), 1.05 (d, J = 7.0 Hz, 3H, CHCH3); 13C NMR (125 MHz, CDCl3) δ 205.0, 172.9, 70.9, 60.7, 46.3, 45.0, 41.9, 30.9, 26.7, 21.4, 14.2, 13.4; HRESIMS m/z 231.1595 [M + H]+ (calcd. for C12H23O4, 231.1591).
3.3. Antibacterial Assay
Antibacterial assays against V. anguillarum, A. hydrophila, V. parahaemolyticus, V. harveyi, S. aureus, M. luteus and E. coli were carried out using the well diffusion method [25]. Chloramphenicol was used as a positive control.
3.4. Antifungal Assay
Antifungal assays against A. brassicae, C. gloeosporioides and F. graminearum were carried out using the well diffusion method [26]. Amphotericin B was used as a positive control.
4. Conclusions
We describe herein the first racemic synthesis of penicimonoterpene (±)-1 and 24 related derivatives. The structures of all of these products were confirmed on the basis of NMR and HRESIMS analysis. Compounds 3a–b, 4a–b, 8a–c, 9a–b, 10, 14 and 17–20 were new compounds. Bioassay results revealed that Compounds 6 and 11 exhibited strong antibacterial activity against S. aureus, while Compound 3b showed extremely high selectively against plant-pathogenic fungus F. graminearum (MIC = 0.25 μg/mL). Compound 10 presented extremely surprising selectivity against pathogenic bacteria E. coli (MIC = 1 μg/mL). Compound 13 showed high selectivity against V. anguillarum and S. aureus, which is much better than chloramphenicol, a well-known commercial antibiotic. Compound 14 also exhibited higher antibacterial activity than chloramphenicol against V. anguillarum, E. coli and S. aureus. Compounds 15, 16 and 17 displayed higher antibacterial activity than chloramphenicol against V. anguillarum, A. hydrophila, V. parahaemolyticus, V. harveyi, E. coli and S. aureus. Taken together, Compounds 15, 16 and 17 were demonstrated to possess the most potent inhibition of the bacteria tested in the present study, especially for marine Vibrio spp. (V. anguillarum, V. harveyi and V. parahaemolyticus), and might have potential as antibiotics or lead compounds.
Acknowledgments
This work was financially supported by programs from the Ministry of Science and Technology of China (2013AA092901 and 2010CB833800).
Supplementary Files
Supplementary Information (PDF, 5145 KB)
Author Contributions
Jian-Chun Zhao performed experiments for the synthesis, structure confirmation, and antimicrobial evaluation of penicimonoterpene (±)-1 and its derivatives and prepared the manuscript; Xiao-Ming Li performed the NMR measurments; James B. Gloer contributed to scientific discussion and corrected the manuscript; Bin-Gui Wang supervised the research work and revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Blunt J.W., Copp B.R., Keyzers R.A., Munro M.H.G., Prinsep M.R. Marine natural products. Nat. Prod. Rep. 2013;30:237–323. doi: 10.1039/c2np20112g. [DOI] [PubMed] [Google Scholar]
- 2.Demain A.L., Vaishnav P. Natural products for cancer chemotherapy. Microb. Biotechnol. 2011;4:687–699. doi: 10.1111/j.1751-7915.2010.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sawadogo W.R., Schumacher M., Teiten M.H., Cerella C., Dicato M., Diederich M. A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2011. Molecules. 2013;18:3641–3673. doi: 10.3390/molecules18043641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia A., Bocanegra-Garcia V., Palma-Nicolas J.P., Rivera G. Recent advances in antitubercular natural products. Eur. J. Med. Chem. 2012;49:1–23. doi: 10.1016/j.ejmech.2011.12.029. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y., Li X.M., Shang Z., Li C.S., Ji N.Y., Wang B.G. Meroterpenoid and diphenyl ether derivatives from Penicillium sp. MA-37, a fungus isolated from marine mangrove rhizospheric soil. J. Nat. Prod. 2012;75:1888–1895. doi: 10.1021/np300377b. [DOI] [PubMed] [Google Scholar]
- 6.An C.Y., Li X.M., Li C.S., Wang M.H., Xu G.M., Wang B.G. Aniquinazolines A–D, four new quinazolinone alkaloids from marine-derived endophytic fungus Aspergillus nidulans. Mar. Drugs. 2013;11:2682–2694. doi: 10.3390/md11072682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M.H., Li X.M., Li C.S., Ji N.Y., Wang B.G. Secondary metabolites from Penicillium pinophilum SD-272, a marine sediment-derived fungus. Mar. Drugs. 2013;11:2230–2238. doi: 10.3390/md11062230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun H.F., Li X.M., Meng L.H., Cui C.M., Gao S.S., Li C.S., Wang B.G. Two new secoanthraquinone derivatives from the marine-derived endophytic fungus Aspergillus wentii EN-48. Helv. Chim. Acta. 2013;96:458–462. doi: 10.1002/hlca.201200201. [DOI] [Google Scholar]
- 9.Li X., Li X.M., Xu G.M., Li C.S., Wang B.G. Antioxidant metabolites from marine alga-derived fungus Aspergillus wentii EN-48. Phytochem. Lett. 2014;7:120–123. doi: 10.1016/j.phytol.2013.11.008. [DOI] [Google Scholar]
- 10.Wright A.D., Wang H.Q., Gurrath M., Konig G.M., Kocak G., Neumann G., Loria P., Foley M., Tilley L. Inhibition of heme detoxification processes underlies the antimalarial activity of terpene isonitrile compounds from marine sponges. J. Med. Chem. 2001;44:873–885. doi: 10.1021/jm0010724. [DOI] [PubMed] [Google Scholar]
- 11.Ntalli N.G., Ferrari F., Giannakou I., Menkissoglu-Spiroudi U. Phytochemistry and nematicidal activity of the essential oils from 8 Greek lamiaceae aromatic plants and 13 terpene components. J. Agric. Food Chem. 2010;58:7856–7863. doi: 10.1021/jf100797m. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez-Burgos E., Gomez-Serranillos M.P. Terpene compounds in nature: A review of their potential antioxidant activity. Curr. Med. Chem. 2012;19:5319–5341. doi: 10.2174/092986712803833335. [DOI] [PubMed] [Google Scholar]
- 13.Gordaliza M. Cytotoxic terpene quinones from marine sponges. Mar. Drugs. 2010;8:2849–2870. doi: 10.3390/md8122849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gershenzon J., Dudareva N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 2007;3:408–414. doi: 10.1038/nchembio.2007.5. [DOI] [PubMed] [Google Scholar]
- 15.Gao S.S., Li X.M., Du F.Y., Li C.S., Proksch P., Wang B.G. Secondary metabolites from a marine-derived endophytic fungus Penicillium chrysogenum QEN-24S. Mar. Drugs. 2011;9:59–70. doi: 10.3390/md9010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J.X., Zhao L., Hu Y., Guo Q.L., Zhang L., Wang X.J., Li N.G., You Q.D. Studies on chemical structure modification and biology of a natural product, gambogic acid (I): Synthesis and biological evaluation of oxidized analogues of gambogic acid. Eur. J. Med. Chem. 2009;44:2611–2620. doi: 10.1016/j.ejmech.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 17.Wada A., Wang F., Suhara Y., Yamano Y., Okitsu T., Nakagawa K., Okano T. Efficient synthesis and biological evaluation of demethyl geranylgeranoic acid derivatives. Bioorg. Med. Chem. 2010;18:5795–5806. doi: 10.1016/j.bmc.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Perez-Meseguer J., Del Olmo E., Alanis-Garza B., Escarcena R., Garza-Gonzalez E., Salazar-Aranda R., Feliciano A.S., de Torres N.W. Synthesis of leubethanol derivatives and evaluation against Mycobacterium tuberculosis. Bioorg. Med. Chem. 2012;20:4155–4163. doi: 10.1016/j.bmc.2012.04.059. [DOI] [PubMed] [Google Scholar]
- 19.Singh J., Sharma M., Kad G.L., Chhabra B.R. Selective oxidation of allylic methyl groups over a solid support under microwave irradiation. J. Chem. Res. (S) 1997:264–265. doi: 10.1039/A700758B. [DOI] [Google Scholar]
- 20.Bhalerao U.T., Rapoport H. Stereochemistry of allylic oxidation with selenium dioxide. Stereospecific oxidation of gem-dimethyl olefins. J. Am. Chem. Soc. 1971;93:4835–4840. doi: 10.1021/ja00748a028. [DOI] [Google Scholar]
- 21.Arigoni D., Vasella A., Sharpless K.B., Jensen H.P. Selenium dioxide oxidations of olefins. trapping of allylic seleninic acid intermediate as a seleninolactone. J. Am. Chem. Soc. 1973;95:7917–7919. doi: 10.1021/ja00804a087. [DOI] [Google Scholar]
- 22.Fkyerat A., Burki N., Tabacchi R. Enantioselective synthesis of 3-hydroxycitronellic acid isolated from Ceratocystis fimbriata sp. platani. Tetrahedron. 1996;7:2023–2028. doi: 10.1016/0957-4166(96)00244-3. [DOI] [Google Scholar]
- 23.Sabitha G., Nayak S., Bhikshapathi M., Yadav J.S. Palladium hydroxide catalyzed isomerization of primary allylic alcohols to aldehydes: Application to the formal synthesis of (−)-brevisamide. Org. Lett. 2011;13:382–385. doi: 10.1021/ol102658d. [DOI] [PubMed] [Google Scholar]
- 24.Perrin D.D., Armarego W.L.F. Purification of Laboratory Chemicals. 3rd ed. Pergamon Press; Oxford, UK: 1988. p. 391. [Google Scholar]
- 25.Al-Burtamani S.K.S., Fatope M.O., Marwah R.G., Onifade A.K., Al-Saidi S.H. Chemical composition, antibacterial and antifungal activities of the essential oil of Haplophyllum tuberculatum from Oman. J. Ethnopharmacol. 2005;96:107–112. doi: 10.1016/j.jep.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 26.Carpinella M.C., Ferrayoli C.G., Palacios S.M. Antifungal synergistic effect of scopoletin, a hydroxycoumarin isolated from Melia azedarach L. fruits. J. Agric. Food Chem. 2005;53:2922–2927. doi: 10.1021/jf0482461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information (PDF, 5145 KB)