

Abstract
Diabetic retinopathy (DR), commonly classified as a microvascular complication of diabetes, is now recognized as a neurovascular complication or sensory neuropathy resulting from disruption of the neurovascular unit. Current therapies for DR target the vascular complications of the disease process, including neovascularization and diabetic macular edema. Since neurodegeneration is an early event in the pathogenesis of DR, it will be important to unravel the mechanisms that contribute to neural apoptosis in order to develop novel treatments for the early stages of DR. In this review we comment on how inflammation, the metabolic derangements associated with diabetes, loss of neuroprotective factors, and dysregulated glutamate metabolism might contribute to retinal neurodegeneration during diabetes. Promising potential therapies based on these specific aspects of DR pathophysiology are also discussed. Finally, we stress the importance of developing and validating new markers of visual function that can be used to shorten the duration of clinical trials and accelerate the delivery of novel treatments for DR to the public.
Keywords: Diabetes, Diabetic Retinopathy, Mechanisms, Neurodegeneration, Neurovascular, Pathogenesis, Treatment
INTRODUCTION
Diabetic retinopathy (DR), the leading cause of blindness in working age individuals in developed countries, has been viewed traditionally as a microvascular complication of diabetes. Indeed, the clinical classification system for diabetic retinopathy is based solely on structural changes to the retinal microvasculature[1, 2] due to the fact that the microvasculature is visible during ophthalmoscopy, but the neuroretina is transparent. Thus, changes to the neuroretina in diabetic retinopathy were not recognized until the 1960s when Wolter[3] and Bloodworth[4] identified degenerating neurons in the retinas of post-mortem diabetic patients. Since that time, evidence for the role of neurodegeneration in DR has accumulated to such an extent that therapies designed to ameliorate neuroretinal damage from diabetes have moved recently to clinical trials.[5]
The early emphasis on the vascular pathology in DR led to treatments to reduce vision loss related to neovascularization and diabetic macular edema (DME), with little consideration for the role of the neural retina in these processes. Panretinal photocoagulation (PRP) has been the mainstay of treatment for proliferative diabetic retinopathy (PDR) for five decades. The effects of reducing neovascularization and macular edema are achieved by coagulating the neuroretina to decrease the volume of remaining tissue and metabolic activity, thus reducing the demand for oxygen and expression of angiogenic factors such as vascular endothelial growth factor (VEGF). As such, the effects of photocoagulation on vascular pathology are indirect. This treatment is highly effective in preserving central visual acuity[6] but reduces peripheral visual fields[7–9] and night vision[10–12]. PRP is thus akin to amputation in that the peripheral retina is sacrificed so that central vision may be preserved, in much the same way that a foot might be sacrificed to preserve a lower extremity in a patient with a diabetic foot ulcer. These side effects of laser surgery have stimulated research to develop pharmacologic means to ameliorate diabetic retinopathy.
Before the advent of intravitreal injections, DME was similarly treated with focal laser in the central macula. Intravitreal injections of anti-VEGF agents such as bevacizumab and ranibizumab are an improvement over focal laser photocoagulation for the treatment of DME in terms of both enhanced visual acuity and slower overall progression of vascular lesions.[13] However, these agents lead to substantial (i.e. two to three lines of vision) improvements in visual acuity in 30 – 50% of patients with DME, [13–15] suggesting that events other than the upregulation of VEGF contribute to the pathogenesis of DR. Treatments designed to protect the entire retina by slowing the progression of DR at its earliest stages could benefit a broader range of persons with DR and warrant further investigation.[16]
In this review, we begin with an examination of the retina as a neurovascular unit and explore a concept of how diabetes alters the structure and function of the neurovascular retina. We then review the pathophysiology of the neuroretinal alterations of diabetes, including adaptations and maladaptions that may evolve nonlinearly over time. The therapeutic implications associated with these various pathologic mechanisms are subsequently discussed. Finally, we examine the important role that surrogate endpoints for visual acuity should play in future clinical trials evaluating therapies for DR.
THE NEUROVASCULAR UNIT OF THE RETINA
Neurons, glial and microglial cells, and blood vessels throughout the nervous system are organized into neurovascular units based on intimate physical contact and functional integration that facilitate physiologic adaptations in response to varying conditions (Figure 1). The neurovascular units coordinate metabolic demand, synaptic activity, blood delivery, and waste removal as coordinated by glutamate, nitric oxide, oxygen, adenosine, and the arachidonic acid metabolites, epoxyeicosatrienoic acids (EETs) and 20-hydroxyeicosatetraenoic.[17, 18]
Figure 1.
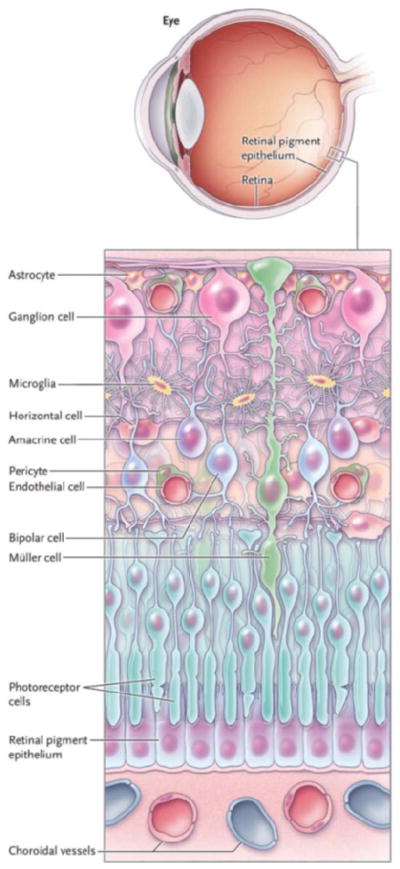
Neurons, glia, and vascular cells comprise the neurovascular unit of the retina. The immense number of physiologic and anatomical connections between these cells permit vision (Figure courtesy of Thomas Gardner, adapted from N Engl J Med 2012;366:1232)
Broadly speaking, neurons are the ultimate effectors of the nervous system and their responses depend on blood vessels to receive nutrients and eliminate waste products of tissue metabolism. Müller cells coordinate vascular responses to meet the metabolic demand of neurons, interchange metabolites, recycle neurotransmitters, and help to establish the extracellular chemical environments for membrane potentials and electrical activity. Astrocytes synapse on blood vessels to maintain autoregulation.[19] Microglial cells monitor local cellular and synaptic activity[20] and dispose of dying cells. Coordinated activity of neurons, glial and microglial cells, and the microvasculature is essential for normal vision. While many of the finer details of the connections between cells in the neurovascular retina remain to be determined, much work has been done to characterize the physical relationships among these cells. Neurons communicate through chemical synapses that utilize neurotransmitters such as glutamate, dopamine, gamma aminobutyric acid, acetylcholine or glycine, and electrical synapses (gap junctions) comprised of connexin protein assemblies that conduct ions between adjacent cells.[21] Vascular cells communicate with each other via tight and adherens junctions.[22] These complex connections reveal why early anatomists called this tissue the retina, literally a network of cells.
DIABETES-INDUCED ALTERATIONS OF THE NEUROVASCULAR UNIT
The specific time course and series of events leading to the onset of DR has yet to be clearly defined, but assessments of individuals with diabetes and no overt vascular retinopathy indicate that alterations of neuroretinal structure and function precede the clinically observable lesions traditionally associated with DR, such as microaneurysms, hemorrhages, and lipid exudates. Neurovascular unit physiology is similarly altered in brain degenerations such as stroke, [23] Alzheimer’s, and Parkinson’s diseases[24]. In persons with type 1 or type 2 diabetes who have no clinical signs of overt retinopathy, these alterations manifest as reduced vasoconstriction in response to breathing 100% oxygen and decreased vasodilation in response to flickering light stimulation.[25–28] These changes appear to signify early impairment of normal regulatory mechanisms throughout the neurovascular complex of the retina, rather than isolated vascular or neuroglial alterations.[29]
Further disturbances in the preclinical phase of retinopathy include reduced multifocal electroretinogram (mfERG) implicit time that predicts the onset of vascular lesions.[30, 31] In addition, reduced contrast sensitivity, [32] dark adaptation, [33, 34] frequency doubling technology perimetry, [34, 35] and optical coherence tomography measures of inner retinal thickness[36, 37] also occur in the preclinical period. Together, these studies show that diabetes has early deleterious effects on retinal neurovascular structure and function and that identifying subclinical alterations in neurovascular unit function could help to identify persons at risk for future vision loss. Some of these events may be physiologic adaptations to the steady state of diabetes that allow the retina to survive and maintain vision in the face of diabetes. The clinical features of retinopathy likely represent the point when the adaptations fail to maintain retinal viability, and/or maladaptive changes ensue as a result of additional insults such as hypertension or inflammation.[38] In the clinical phases of diabetic retinopathy, progressive disruption of the neurovascular unit and loss of autoregulation is evident as venous tortuosity and dilation that is most apparent with the onset of macular edema and proliferative retinopathy.[39–41] Thus, retinal neurovascular unit alterations in diabetes are part of the larger spectrum of nervous system effects of diabetes that includes sensory and autonomic neuropathies and cognitive impairment.[42, 43]
MOLECULAR MECHANISMS OF NEURODEGENERATION IN DR
The ocular and systemic factors that contribute to the development and progression of diabetic retinopathy remain complex and are difficult to fully unravel due to lack of affordable animal models that recapitulate the human phenotype. Nonetheless, considerable insights have been inferred from studying rodents in parallel with patients in an integrated manner.[44]
Metabolic alterations
At its core, diabetes is a metabolic disorder characterized by hyperglycemia secondary to a deficiency in insulin production (type 1 diabetes) or reduced insulin sensitivity (type 2 diabetes). Insulin insufficiency also develops from pancreatic failure in persons with type 2 diabetes, [45] and insulin resistance is a feature of type 1 diabetes, [46] particularly among those who are overweight.[47] Thus, while neuroinflammation, apoptosis, glutamate excitotoxicity, and/or a deficiency of neuroprotective factors may all contribute to retinal dysfunction in DR, it is important to evaluate these mechanisms through the lens of the systemic abnormalities in metabolism intrinsic to diabetes.
Insulin receptors in the retina, unlike those in skeletal muscle, stimulate neuronal development, growth, survival and anabolic synthesis rather than solely mediating glucose transport into the cell.[48–50] Retinal insulin receptors are also distinct in that light activates insulin receptors in rod and cone photoreceptors[51, 52] where they also mediate survival. Perhaps not surprisingly, retinal neurons, and in particular ganglion cells, begin to die by apoptosis within weeks of the onset of diabetes.[53, 54] However, diabetic rats treated with systemic insulin are protected from diabetes-induced apoptosis of retinal neurons.[54] To better understand whether the correction of systemic hyperglycemia or the addition of insulin was responsible for this reduction in neural cell death, Fort and colleagues[55] administered subconjunctival insulin injections or systemic phloridzin (a sodium-linked glucose transporter inhibitor) to streptozotocin-induced diabetic rats. The low dose insulin restores retinal insulin receptor activity without changing blood glucose levels, and phloridzin normalizes blood glucose levels without affecting systemic or local insulin levels. Interestingly, both phloridzin and local insulin were found to protect retinal cells from undergoing apoptosis by modulating growth factor signaling and inflammatory pathways, respectively. These findings suggest that neurodegeneration in DR is a consequence of both reduced insulin receptor signaling and systemic hyperglycemia and have direct implications for how the effects of intensive metabolic control impact the course of retinopathy.
Since hyperglycemia is the metabolic derangement that defines diabetes mellitus, many attempts to explain the pathogenesis of DR incorporate hyperglycemia as a causative factor in one form or another. For example, activation of protein kinase C (PKC), [56–58] formation of advanced glycation end products (AGEs), [59] increased flux through the polyol pathway, [60] and increased oxidative stress[61] are some of the proposed mechanisms through which hyperglycemia exerts its deleterious effects on the retina.
Among the mechanisms listed above, oxidative stress and the formation of AGEs have been specifically implicated in neurodegeneration during DR. Oxidative stress refers to a state where there are excess levels of reactive oxygen species (ROS), or oxygen atoms with unpaired electrons. ROS can alter the structure of other compounds, such as deoxyribonucleic acid (DNA), which can in turn lead to cellular and tissue level dysfunction. One theory for how ROS may increase during diabetes proposes that high glucose levels stimulate increased flux through the glycolytic and tricarboxylic acid (TCA) cycle pathways, thus flooding mitochondria with electrons.[61] Markers of increased oxidative stress have been associated with retinal neurodegeneration in experimental diabetes, [62] and treatment with a ROS scavenger has been shown to reduce the neurodegenerative changes in ganglion cells of diabetic rat retinas.[63] However, the issue of ROS in a chronic disease such as diabetic retinopathy hinges on the degree to which oxidative stress exceeds physiologic adaptations, since ROS also mediate neuroprotective effects via the TrkB receptor.[64] Therefore, additional work is needed to unravel the relative deleterious and beneficial effects of ROS in diabetes to understand how to manipulate them for therapeutic benefit.
AGEs are formed non-enzymatically when glucose molecules bond to amine residues on proteins, lipids, or nucleic acids. They may interact with a receptor for AGE (RAGE), which, along with glial fibrillary acidic protein (GFAP) is upregulated in Müller cells during experimental models of diabetes.[65] Stimulation of Müller cell RAGEs by circulating AGEs causes release of inflammatory cytokines such as VEGF and monocyte chemoattractant protein-1 (MCP-1).[65] Recent work shows that inhibition of AGE formation derived from methylglyoxal prevents retinal neuroglial and vascular pathology.[66]
Clearly, hyperglycemia is an important component of the metabolic dysregulation of diabetes, but there is not, in our opinion, definitive evidence showing that hyperglycemia alone is the primary instigator of retinopathy in humans. Better understanding of the role of other aspects of metabolism, including lipids, glucagon and prolactin[67] will be required to have a full appreciation of the mechanisms of diabetic retinopathy.
Inflammation in diabetic retinopathy
It has been long debated whether hyperglycemia or any specific metabolic disturbance is necessary and sufficient to cause DR. Over the past decade there has been greater appreciation for the role of the innate immune system as an adaptive response to obesity and diabetes.[68, 69] In this paradigm nutrient excess, such as a high level of saturated fatty acids, is sensed by adipose cells with release of pro-inflammatory mediators from macrophages, including lipopolysaccharide, interleukin-1 beta (IL-1β) and tumor necrosis factor alpha (TNF-α), resulting in systemic inflammation and, ultimately, cardiovascular disease and cancers.
Within the retina, endothelial cells increase expression of intercellular adhesion molecule 1 (ICAM-1) and P-selectin early in the course of diabetes, which causes leukocytes to adhere to vascular walls.[70] Such leukostasis contributes to disruption of the blood-retinal barrier and death of endothelial cells, perpetuating an inflammatory cascade within the retina. An important question is whether leukostasis also creates areas of capillary nonperfusion within the retina, which could rob neuronal tissue of essential nutrients and thereby contribute to neurodegeneration in DR.[71] It is also possible that degeneration of the neurosensory cells in the periphery leads or contributes to secondary vascular closure within the retina. The exact sequence of events leading to leukostasis in DR is not yet clearly defined, though Huang et al[72] recently showed that the absence of TNF-α, a pro-inflammatory cytokine upregulated during diabetes, prevents retinal leukostasis and apoptosis of vascular and neural cells in diabetic mice. This finding suggests that TNF-α may play a critical role in the progression of DR and that leukostasis might be linked to neurodegeneration in DR through a TNF dependent mechanism.
Along with TNF-α, [73] vitreous levels of other inflammatory cytokines, such as MCP-1, [74] IL-6, [75] and IL-1β, [73] are increased in patients with DR compared to controls without diabetes. Upregulation of these or other cytokines may contribute to glial activation in early DR, whereby normally quiescent microglia begin to secrete cytotoxic substances that contribute to neural cell death. Microglial activation in human DR has been demonstrated by Zeng and colleagues, [76] and inhibiting such glial cell activation may be a strategy to prevent neurodegeneration in DR. Much more work is needed to define the adaptive and maladaptive roles of microglia in neurodegenerative disorders.[77]
The cause of the ocular inflammation in persons with diabetes remains uncertain. A recent study found an association between non-healing ulcers and nephropathy, in addition to worse metabolic control, in the progression of nonproliferative to proliferative retinopathy.[78] Foot ulcers are also associated with increased circulating TNF-α and IL-1β, suggesting the possibility that part of the ocular inflammatory response derives from systemic sources and that treating systemic inflammation from foot ulcers or gingivitis may reduce the risk of progressive retinopathy.
The relationship between leukostasis and glial activation in DR appears to be mediated by inflammatory cytokines such as TNF-α and IL-1β, but the temporal relationship between these events and the upregulation of inflammatory cytokines requires further exploration. Ideally, therapies aimed at slowing the progression of DR would target the earliest pathogenic steps, so defining the temporal relationship among cytokine upregulation, leukostasis, and glial cell activation will be instrumental to devising treatments aimed at preventing the neurodegeneration associated with DR.
Glutamate
The neurotransmitter glutamate is essential for effective cell to cell communication between neurons. However, excessive pre-synaptic levels of glutamate in the central nervous system can lead to excitotoxicity, which has been implicated in the pathogenesis of neurodegenerative disorders such as Parkinson’s[79] and Alzheimer’s[80] diseases. In this paradigm, abnormally high levels of glutamate in the synaptic cleft lead to increased calcium influx into post-synaptic neurons[81] which can in turn initiate pro-apoptotic signaling cascades through both caspase-dependent and caspase-independent mechanisms.[82] Such events likely contribute to the observed increase in apoptotic neurons within the retina during diabetes.
Diabetes alters the equilibrium of glutamate and glutamine between glial cells and neurons via several potential mechanisms. First, diabetes reduces the activity of the enzyme glutamine synthetase in Müller cells, which hinders the ability of these cells to convert excess glutamate to glutamine.[83] Second, glutamate oxidation to α-ketoglutarate is impaired.[84] Third, glutamate uptake by Müller cells is decreased, which leads to an extracellular accumulation of glutamate in the neuroretina.[85] While there is clearly an increase in glutamate levels within the vitreous of patients with advanced DR, [86, 87] it remains unknown as to which of these mechanisms is the most important contributor to glutamate excitotoxicity.
Role of Neuroprotective Factors
The survival of retinal neurons in a hostile environment depends on the local availability of growth factors and neurotrophins, which are peptides that promote neuronal differentiation and survival. In diabetes, the efficacy and/or concentrations of such molecules are reduced, leading many to postulate that neuroprotective factors may have a therapeutic role in preventing vision loss associated with DR.
Local factors that exhibit neuroprotective properties in experimental models of diabetes include pigment epithelial derived factor (PEDF), brain derived neurotrophic factor (BDNF) and nerve growth factor (NGF). PEDF inhibits angiogenesis and neurodegeneration in diabetes[88] by reducing oxidative stress in the retina[89] or by increasing the expression of glutamine synthase, thus affording protection against glutamate excitotoxicity[90]. PEDF peptide eye drops were recently shown to decrease microglial activation, ganglion cell death, and vascular leakage in diabetic rats, [91] suggesting that exogenous PEDF may be a potential treatment for early DR. Like PEDF, retinal levels of BDNF are reduced in animal models of diabetes, [92] and this reduction in BDNF is correlated with amacrine cell degeneration. Intravitreal administration of BDNF to diabetic rats reverses amacrine cell degeneration, [92] and overexpression of BDNF in diabetic rats enhances ganglion cell survival and function[93]. NGF was implicated in the pathogenesis of DR in 1995, when Hammes et al[53] showed that treatment of diabetic rats with NGF prevents ganglion cell apoptosis. In diabetes, levels of all three growth factors (PEDF, [89] BDNF, [92] and NGF[94]) are reduced in association with retinal neurodegeneration, implying that these molecules may be useful as potential therapies for DR.
Like the neurotrophic factors PEDF, BDNF, and NGF, the retinal levels of erythropoietin, VEGF and insulin-like growth factor-1 (IGF-1) decrease shortly after diabetes onset. Unlike the aforementioned neurotrophins, however, VEGF, IGF-1, and erythropoietin all increase with the development of proliferative retinopathy, but the nature of this biphasic response remains uncertain. Other factors thought to play a role in protecting the retina from diabetes-induced neurodegeneration include somatostatin[95] and ciliary neurotrophic factor[96] though further work is needed to fully understand the therapeutic potential of such compounds.
To date, most studies have examined vitreous or retinal concentrations of various peptide growth factors that exert neurotrophic effects. However, the ultimate determinant of cell viability is the activity of the cognate receptors rather than the ligand levels. For example, experimental diabetes increases tyrosine nitration and decreases phosphorylation of retinal nerve growth factor TrkA, and these effects are reversed by neutralizing peroxynitrite.[97] Experimental diabetes also impairs activity of the retinal insulin receptor, an effect that is reversed by reducing systemic and ocular glucose concentrations with phloridzin or by adding very low dose subconjunctival insulin.[55] Thus, diabetes appears to induce a form of growth factor resistance in the retina, analogous to sepsis-induced resistance in liver and skeletal muscle.[98, 99]
THERAPEUTIC IMPLICATIONS
Metabolic Considerations
The Diabetes Control and Complications Trial (DCCT)[100] and United Kingdom Prospective Diabetes Study (UKPDS)[101] showed that controlling diabetes, as measured by hemoglobin A1c and blood glucose levels, reduces the chances of developing complications such as DR. However, maintaining intensive control can be difficult and even dangerous for some patients, as the Action to Control Cardiovascular Risk in Diabetes (ACCORD) study revealed. In this study, the group of patients with type 2 diabetes who underwent intensive glycemic reduction (median hemoglobin A1c = 6.4%) exhibited a slightly increased risk for cardiovascular mortality compared to the standard therapy group (median hemoglobin A1c = 7.5%).[102] Furthermore, while hyperglycemia is clearly implicated in the pathogenesis of DR, post-hoc analysis of DCCT data revealed that only 11% of the risk in retinopathy development could be attributed to hemoglobin A1c, suggesting that factors other than prolonged hyperglycemia may contribute to DR development.[103] Beyond hemoglobin A1c values, the fluctuations in glucose concentration over time are now a subject of interest in relation to complications. Current technologies to achieve intensive control are limited by the risk of hypoglycemia, weight gain, and expense of the devices. Continuous glucose monitors improve metabolic control and shorten durations of hypoglycemia in persons with type 1 diabetes.[104, 105] Emerging technologies that integrate continuous glucose sensors with insulin pumps (closed loop systems) are showing promise, [106, 107] but their widespread adoption may also be constrained by financial barriers. Nevertheless, ophthalmologists should be aware of these technologies and encourage patients to consider their potential benefits.
Anti-Apoptotic Agents
In diabetes, retinal neurons and vascular cells die by apoptosis, [53, 54, 108] a final common pathway for retinal neurodegeneration. Whether the triggers are hyperglycemia, glutamate excitotoxicity or neurotrophin deficiency, apoptosis links these potential mechanisms of neuronal injury and the ultimate death of cells. Therefore, inhibiting apoptosis could be a potential means of preventing neurodegeneration in DR. Apoptosis typically involves the activation of proteolytic enzymes which destroy cellular components that maintain the normal structure and function of the cell. Two such proteins that have been implicated in glutamate-induced apoptosis include caspase-3 and calpain.[82] Latanoprost, a prostaglandin analogue, was found to reduce neural and glial cell apoptosis in diabetic retinas when applied topically to rat eyes.[109] Caspase-3 inhibition is one proposed mechanism for this phenomenon, as treatment with latanoprost significantly reduced caspase-3 immunoreactive cells in the ganglion cell layer. In a separate model of retinal hypoxia, Nakajima and colleagues[110] found that calpain, but not caspase, was responsible for cellular damage observed during hypoxia in monkey retinal cells. In a more recent study, oral administration of a calpain inhibitor to diabetic mice prevented retinal ganglion cell death, again suggesting that calpain inhibition may be a viable neuroprotective strategy for treating DR.[111]
Glutamate Antagonists
Given the potential role of glutamate excitotoxicity in contributing to neurodegeneration in DR, blockade of glutamate receptors or augmentation of glutamate clearance/metabolism may be viable strategies for maintaining the integrity of the neuroretina during diabetes. There are two main types of glutamate receptors in the central nervous system: α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) and N-methyl-D-aspartate (NMDA) receptors.
Memantine is a non-competitive antagonist of NMDA receptors and is already approved by the Food and Drug Administration (FDA) for the treatment of Alzheimer’s disease, another well-known neurodegenerative condition. In diabetic rats, chronic memantine therapy has been shown to reduce retinal ganglion cell loss, improve retinal function, and decrease intraocular VEGF levels.[112] Memantine is FDA approved for Alzheimer’s disease and in light of the evidence for a role of glutamate excitotoxicity in diabetes-induced retinal neurodegeneration, memantine could be evaluated for the treatment of DR, but to our knowledge, no clinical trials are currently in progress.
Anti-Inflammatory Agents
Diabetes is associated with an upregulation of inflammatory cytokines, such as TNF-α, IL-1β, IL-6, and VEGF.[113] Furthermore, increased expression of ICAM-1 contributes to leukostasis in retinal blood vessels and may lead to ischemia of neuroretinal tissue, promoting neurodegeneration. Inflammatory cytokines released by immune cells during diabetes can also activate microglia, contributing to further neurodegeneration. Therefore, blocking the effects of these cytokines may be another approach to reducing the progression of DR.
The TNF-α inhibitor etanercept has shown promise in preventing retinal cell death and the upregulation of ICAM-1 (and thus leukostasis) in animal models of diabetes.[114, 115] However, a 2007 report of seven cases with severe diabetic macular edema who were treated with intravitreal etanercept indicated no improvement.[116] Moreover, intravitreal infliximab administration also had no discernible benefit in patients with refractory DME during a pilot study and instead promoted vitreous inflammation among the treated patients.[117] Therefore, despite the apparent role of TNF in promoting the disruption of the neurovascular unit in rodent models of diabetes, [72] there is currently little clinical evidence that its inhibition with monoclonal antibodies is beneficial.
Minocycline, a tetracycline antibiotic with anti-inflammatory properties, is also under investigation for the treatment of DR. In cultured retinal cells, minocycline inhibits microglial production of TNF-α, IL-1 β and nitric oxide after exposure to lipopolysaccharide, [118] and it reduces caspase-3 activation.[119] In a recent phase I/II clinical trial, oral minocycline improved visual acuity, central macular edema, and vascular leakage in five patients with DME, suggesting that inhibiting microglial activation may be part of a multi-tiered strategy for the treatment of DR.[120] The results from two recently completed trials of doxycycline (NCT00917553 and NCT00511875) may help to delineate the role that such agents could play in the treatment of patients with DR.
Observations of people with diabetes being treated for rheumatoid arthritis initially suggested that the non-steroidal anti-inflammatory drug (NSAID) aspirin could have therapeutic efficacy in the treatment of DR.[121] The Early Treatment Diabetic Retinopathy Study (ETDRS) subsequently tested the hypothesis that 650 mg/day of aspirin could slow the progression of DR and reduce vision loss but found oral aspirin to be ineffective for the treatment of DR.[122] However, in animal models of diabetes, the administration of salicylates reduced ganglion cell loss early in the disease course.[123] NSAIDs such as bromfenac are available as topical eye drops and could be evaluated more extensively as a potential therapy for early DR. Unpublished data from a recent clinical trial (NCT00782717) suggests that nepafenac drops reduce the risk of DME following cataract surgery.
Protein kinase C β inhibition with ruboxistaurin reduced progression of diabetic macular edema and the need for macular photocoagulation[124] but did not receive FDA approval. Antonetti et al[125] have recently shown that another protein kinase C isoform, PKC zeta, a member of the atypical PKC class, mediates both VEGF and TNF-induced retinal vascular permeability, and a PKC zeta inhibitor reduces VEGF-stimulated retinal vascular leakage in rats. This class of inhibitors is currently being tested for its effects on the neurovascular unit.
Local Neuroprotection
The findings that NGF, PEDF, and BDNF rescue dying neurons in animal models of diabetes and their levels within the retina are reduced in diabetes[53, 91, 92, 126] will hopefully spur further research into their effects on visual function in humans with diabetes. To this end, somatostatin, another potential neurotrophic agent, is currently being evaluated in a clinical trial to determine whether treatment with a topical formulation of the compound can slow the progression of DR.[5] Other neurotrophic agents, such as the peptide PEDF eye drops mentioned earlier, could be evaluated in a similar fashion to hasten the identification of clinically effective neuroprotective strategies for DR.
Insulin administration to the eye reduces the rate of cell death in diabetic animals and restores pro-survival signaling.[55] Misra et al[127] described insulin hydrogels that can be implanted subconjunctivally for long term insulin delivery to the eye and showed that these hydrogels were not associated with any adverse events in rats. Additional animal studies using non-invasive, local insulin delivery systems will be needed to better assess whether local insulin is more effective than systemic insulin at reducing neurodegeneration during DR.
CONCLUSION
Diabetic retinopathy is now understood to be a sensory neuropathy similar to other neuropathies that afflict persons with diabetes. Most therapies have been aimed at the advanced vascular sequelae of DR, and now neuroprotective strategies are underway to determine their ability to slow progressive visual dysfunction during diabetes. The molecular mechanisms involved in neurodegeneration during DR are complex and likely include a combination of ocular factors such as increased oxidative stress, loss of neuroprotective factors, increased inflammation, glutamate excitotoxicity, and systemic factors including hyperglycemia, dyslipidemia, and insulin deficiency. As new therapies are developed, it will be important to identify end points for clinical trials that accurately reflect visual function and that can be used as new markers of visual acuity. Since DR only leads to significant impairment in visual acuity during the advanced stages of the disease, the use of alternate endpoints for vision could shorten the duration of clinical trials and hasten the development of new therapies for DR. To this end, the European Consortium for the Early Treatment of Diabetic Retinopathy (EUROCONDOR) study group is using the mfERG implicit time as its primary endpoint to measure neuroretinal dysfunction, and other endpoints like the mfERG will need to be developed and standardized in order to assess the effectiveness of novel neuroprotective strategies for DR. Agents that reduce inflammation, restore insulin receptor signaling in the retina, inhibit NMDA receptors, or block apoptosis have great promise as novel therapies for early DR. However, these treatments must be evaluated in future clinical trials with the correct drug delivery method and the optimal end points before they can be used to treat the ever-increasing number of patients with DR.
The complexity of preventing and treating diabetic retinopathy is evident by the fact that only seven percent of persons with type 2 diabetes meet therapeutic goals for hemoglobin A1c level, blood pressure, and low-density lipoprotein cholesterol level[128] and only one-third of children with type 1 diabetes meet the American Diabetes Association goals for hemoglobin A1c level.[129] Therefore, adjunctive eye-specific treatments to minimize the risk of vision loss from diabetes will be critical in the near future, [130–132] along with more quantitative and sensitive means of detecting preclinical damage to the retinal neurovascular unit.[25, 34]
Acknowledgments
Financial Support: American Diabetes Association-Merck Clinical/Translational Postdoctoral Fellowship Award (MSS); JDRF, EY20582, DK094292, a Research to Prevent Blindness Physician-Scientist Award and the A. Alfred Taubman Medical Research Institute (TWG)
Footnotes
Penn State University holds intellectual property rights for the use of periocular insulin and protein kinase C zeta inhibitors for the treatment of diabetic retinopathy.
The sponsor or funding organization had no role in the design or conduct of this research.
References
- 1.Aiello LM. Perspectives on diabetic retinopathy. Am J Ophthalmol. 2003;136(1):122–135. doi: 10.1016/s0002-9394(03)00219-8. [DOI] [PubMed] [Google Scholar]
- 2.Wilkinson CP, Ferris FL, 3rd, Klein RE, Lee PP, Agardh CD, Davis M, Dills D, Kampik A, Pararajasegaram R, Verdaguer JT. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology. 2003;110(9):1677–1682. doi: 10.1016/S0161-6420(03)00475-5. [DOI] [PubMed] [Google Scholar]
- 3.Wolter JR. Diabetic retinopathy. Am J Ophthalmol. 1961;51:1123–1141. doi: 10.1016/0002-9394(61)91802-5. [DOI] [PubMed] [Google Scholar]
- 4.Bloodworth JM., Jr Diabetic retinopathy. Diabetes. 1962;11:1–22. [PubMed] [Google Scholar]
- 5.Cunha-Vaz J. Neurodegeneration as an early event in the pathogenesis of Diabetic Retinopathy: A multicentric, prospective, phase II–III, randomised controlled trial to assess the efficacy of neuroprotective drugs administered topically to prevent or arrest Diabetic Retinopathy. EUROCONDOR – EU FP7 Project. Acta Ophthalmol. 2012;90(249):0. [Google Scholar]
- 6.Preliminary report on effects of photocoagulation therapy. The Diabetic Retinopathy Study Research Group. . Am J Ophthalmol. 1976;81(4):383–396. doi: 10.1016/0002-9394(76)90292-0. [DOI] [PubMed] [Google Scholar]
- 7.Doft BH, Blankenship GW. Single versus multiple treatment sessions of argon laser panretinal photocoagulation for proliferative diabetic retinopathy. Ophthalmology. 1982;89(7):772–779. doi: 10.1016/s0161-6420(82)34734-x. [DOI] [PubMed] [Google Scholar]
- 8.Pahor D. Visual field loss after argon laser panretinal photocoagulation in diabetic retinopathy: full- versus mild-scatter coagulation. Int Ophthalmol. 1998;22(5):313–319. doi: 10.1023/a:1006367029134. [DOI] [PubMed] [Google Scholar]
- 9.Early photocoagulation for diabetic retinopathy. ETDRS report number 9. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology. 1991;98(5):766–785. [PubMed] [Google Scholar]
- 10.Seiberth V, Alexandridis E, Feng W. Function of the diabetic retina after panretinal argon laser coagulation. Graefes Arch Clin Exp Ophthalmol. 1987;225(6):385–390. doi: 10.1007/BF02334163. [DOI] [PubMed] [Google Scholar]
- 11.Russell PW, Sekuler R, Fetkenhour C. Visual function after pan-retinal photocoagulation: a survey. Diabetes Care. 1985;8(1):57–63. doi: 10.2337/diacare.8.1.57. [DOI] [PubMed] [Google Scholar]
- 12.Pender PM, Benson WE, Compton H, Cox GB. The effects of panretinal photocoagulation on dark adaptation in diabetics with proliferative retinopathy. Ophthalmology. 1981;88(7):635–638. doi: 10.1016/s0161-6420(81)34977-x. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen QD, Shah SM, Heier JS, Do DV, Lim J, Boyer D, Abraham P, Campochiaro PA. Primary End Point (Six Months) Results of the Ranibizumab for Edema of the mAcula in diabetes (READ-2) study. Ophthalmology. 2009;116(11):2175–2181. e1. doi: 10.1016/j.ophtha.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen QD, Brown DM, Marcus DM, Boyer DS, Patel S, Feiner L, Gibson A, Sy J, Rundle AC, Hopkins JJ, Rubio RG, Ehrlich JS. Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology. 2012;119(4):789–801. doi: 10.1016/j.ophtha.2011.12.039. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell P, Bandello F, Schmidt-Erfurth U, Lang GE, Massin P, Schlingemann RO, Sutter F, Simader C, Burian G, Gerstner O, Weichselberger A. The RESTORE study: ranibizumab monotherapy or combined with laser versus laser monotherapy for diabetic macular edema. Ophthalmology. 2011;118(4):615–625. doi: 10.1016/j.ophtha.2011.01.031. [DOI] [PubMed] [Google Scholar]
- 16.Simo R, Hernandez C. Neurodegeneration is an early event in diabetic retinopathy: therapeutic implications. Br J Ophthalmol. 2012;96(10):1285–1290. doi: 10.1136/bjophthalmol-2012-302005. [DOI] [PubMed] [Google Scholar]
- 17.Metea MR, Newman EA. Signalling within the neurovascular unit in the mammalian retina. Exp Physiol. 2007;92(4):635–640. doi: 10.1113/expphysiol.2006.036376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kur J, Newman EA, Chan-Ling T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog Retin Eye Res. 2012;31(5):377–406. doi: 10.1016/j.preteyeres.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron. 2011;71(5):782–797. doi: 10.1016/j.neuron.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Volgyi B, Kovacs-Oller T, Atlasz T, Wilhelm M, Gabriel R. Gap junctional coupling in the vertebrate retina: Variations on one theme? Prog Retin Eye Res. 2013 doi: 10.1016/j.preteyeres.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Runkle EA, Antonetti DA. The blood-retinal barrier: structure and functional significance. Methods Mol Biol. 2011;686:133–148. doi: 10.1007/978-1-60761-938-3_5. [DOI] [PubMed] [Google Scholar]
- 23.del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267(2):156–171. doi: 10.1111/j.1365-2796.2009.02199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Lott ME, Slocomb JE, Shivkumar V, Smith B, Gabbay RA, Quillen D, Gardner TW, Bettermann K. Comparison of retinal vasodilator and constrictor responses in type 2 diabetes. Acta Ophthalmol. 2012;90(6):e434–441. doi: 10.1111/j.1755-3768.2012.02445.x. [DOI] [PubMed] [Google Scholar]
- 26.Bek T, Hajari J, Jeppesen P. Interaction between flicker-induced vasodilatation and pressure autoregulation in early retinopathy of type 2 diabetes. Graefes Arch Clin Exp Ophthalmol. 2008;246(5):763–769. doi: 10.1007/s00417-008-0766-y. [DOI] [PubMed] [Google Scholar]
- 27.Garhofer G, Zawinka C, Resch H, Kothy P, Schmetterer L, Dorner GT. Reduced response of retinal vessel diameters to flicker stimulation in patients with diabetes. Br J Ophthalmol. 2004;88(7):887–891. doi: 10.1136/bjo.2003.033548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lasta M, Pemp B, Schmidl D, Boltz A, Kaya S, Palkovits S, Werkmeister R, Howorka K, Popa-Cherecheanu A, Garhofer G, Schmetterer L. Neurovascular dysfunction precedes neural dysfunction in the retina of patients with type 1 diabetes. Invest Ophthalmol Vis Sci. 2013 doi: 10.1167/iovs.12-10873. [DOI] [PubMed] [Google Scholar]
- 29.Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med. 2012;366(13):1227–1239. doi: 10.1056/NEJMra1005073. [DOI] [PubMed] [Google Scholar]
- 30.Harrison WW, Bearse MA, Jr, Ng JS, Jewell NP, Barez S, Burger D, Schneck ME, Adams AJ. Multifocal electroretinograms predict onset of diabetic retinopathy in adult patients with diabetes. Invest Ophthalmol Vis Sci. 2011;52(2):772–777. doi: 10.1167/iovs.10-5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ng JS, Bearse MA, Jr, Schneck ME, Barez S, Adams AJ. Local diabetic retinopathy prediction by multifocal ERG delays over 3 years. Invest Ophthalmol Vis Sci. 2008;49(4):1622–1628. doi: 10.1167/iovs.07-1157. [DOI] [PubMed] [Google Scholar]
- 32.Gualtieri M, Bandeira M, Hamer RD, Damico FM, Moura AL, Ventura DF. Contrast sensitivity mediated by inferred magno- and parvocellular pathways in type 2 diabetics with and without nonproliferative retinopathy. Invest Ophthalmol Vis Sci. 2011;52(2):1151–1155. doi: 10.1167/iovs.09-3705. [DOI] [PubMed] [Google Scholar]
- 33.Greenstein VC, Thomas SR, Blaustein H, Koenig K, Carr RE. Effects of early diabetic retinopathy on rod system sensitivity. Optom Vis Sci. 1993;70(1):18–23. doi: 10.1097/00006324-199301000-00005. [DOI] [PubMed] [Google Scholar]
- 34.Jackson GR, Scott IU, Quillen DA, Walter LE, Gardner TW. Inner retinal visual dysfunction is a sensitive marker of non-proliferative diabetic retinopathy. Br J Ophthalmol. 2012;96(5):699–703. doi: 10.1136/bjophthalmol-2011-300467. [DOI] [PubMed] [Google Scholar]
- 35.Parravano M, Oddone F, Mineo D, Centofanti M, Borboni P, Lauro R, Tanga L, Manni G. The role of Humphrey Matrix testing in the early diagnosis of retinopathy in type 1 diabetes. Br J Ophthalmol. 2008;92(12):1656–1660. doi: 10.1136/bjo.2008.143057. [DOI] [PubMed] [Google Scholar]
- 36.van Dijk HW, Verbraak FD, Kok PH, Garvin MK, Sonka M, Lee K, Devries JH, Michels RP, van Velthoven ME, Schlingemann RO, Abramoff MD. Decreased retinal ganglion cell layer thickness in patients with type 1 diabetes. Invest Ophthalmol Vis Sci. 2010;51(7):3660–3665. doi: 10.1167/iovs.09-5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park HY, Kim IT, Park CK. Early diabetic changes in the nerve fibre layer at the macula detected by spectral domain optical coherence tomography. Br J Ophthalmol. 2011;95(9):1223–1228. doi: 10.1136/bjo.2010.191841. [DOI] [PubMed] [Google Scholar]
- 38.Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, Kester M, Kimball SR, Krady JK, LaNoue KF, Norbury CC, Quinn PG, Sandirasegarane L, Simpson IA. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;5(9):2401–2411. doi: 10.2337/db05-1635. [DOI] [PubMed] [Google Scholar]
- 39.Sinclair SH, Grunwald JE, Riva CE, Braunstein SN, Nichols CW, Schwartz SS. Retinal vascular autoregulation in diabetes mellitus. Ophthalmology. 1982;89(7):748–750. doi: 10.1016/s0161-6420(82)34720-x. [DOI] [PubMed] [Google Scholar]
- 40.Kylstra JA, Wierzbicki T, Wolbarsht ML, Landers MB, 3rd, Stefansson E. The relationship between retinal vessel tortuosity, diameter, and transmural pressure. Graefes Arch Clin Exp Ophthalmol. 1986;224(5):477–480. doi: 10.1007/BF02173368. [DOI] [PubMed] [Google Scholar]
- 41.Kristinsson JK, Gottfredsdottir MS, Stefansson E. Retinal vessel dilatation and elongation precedes diabetic macular oedema. Br J Ophthalmol. 1997;81(4):274–278. doi: 10.1136/bjo.81.4.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Umegaki H. Neurodegeneration in diabetes mellitus. Adv Exp Med Biol. 2012;724:258–265. doi: 10.1007/978-1-4614-0653-2_19. [DOI] [PubMed] [Google Scholar]
- 43.Reijmer YD, Brundel M, de Bresser J, Kappelle LJ, Leemans A, Biessels GJ. Microstructural White Matter Abnormalities and Cognitive Functioning in Type 2 Diabetes: A diffusion tensor imaging study. Diabetes Care. 2013;36(1):137–144. doi: 10.2337/dc12-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gardner TW, Abcouwer SF, Barber AJ, Jackson GR. An integrated approach to diabetic retinopathy research. Arch Ophthalmol. 2011;129(2):230–235. doi: 10.1001/archophthalmol.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang-Chen KJ, Mullur R, Bernal-Mizrachi E. Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord. 2008;9(4):329–343. doi: 10.1007/s11154-008-9101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nadeau KJ, Regensteiner JG, Bauer TA, Brown MS, Dorosz JL, Hull A, Zeitler P, Draznin B, Reusch JE. Insulin resistance in adolescents with type 1 diabetes and its relationship to cardiovascular function. J Clin Endocrinol Metab. 2010;95(2):513–521. doi: 10.1210/jc.2009-1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pambianco G, Costacou T, Orchard TJ. The prediction of major outcomes of type 1 diabetes: a 12-year prospective evaluation of three separate definitions of the metabolic syndrome and their components and estimated glucose disposal rate: the Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes Care. 2007;30(5):1248–1254. doi: 10.2337/dc06-2053. [DOI] [PubMed] [Google Scholar]
- 48.Rajala RV, Anderson RE. Rhodopsin-regulated insulin receptor signaling pathway in rod photoreceptor neurons. Mol Neurobiol. 2010;42(1):39–47. doi: 10.1007/s12035-010-8130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajala RV. Phosphoinositide 3-kinase signaling in the vertebrate retina. J Lipid Res. 2010;51(1):4–22. doi: 10.1194/jlr.R000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reiter CE, Gardner TW. Functions of insulin and insulin receptor signaling in retina: possible implications for diabetic retinopathy. Prog Retin Eye Res. 2003;22(4):545–562. doi: 10.1016/s1350-9462(03)00035-1. [DOI] [PubMed] [Google Scholar]
- 51.Rajala RV, McClellan ME, Ash JD, Anderson RE. In vivo regulation of phosphoinositide 3-kinase in retina through light-induced tyrosine phosphorylation of the insulin receptor beta-subunit. J Biol Chem. 2002;277(45):43319–43326. doi: 10.1074/jbc.M206355200. [DOI] [PubMed] [Google Scholar]
- 52.Huang Z, Anderson RE, Cao W, Wiechmann AF, Rajala RV. Light-induced tyrosine phosphorylation of rod outer segment membrane proteins regulate the translocation, membrane binding and activation of type II alpha phosphatidylinositol-5-phosphate 4-kinase. Neurochem Res. 2011;36(4):627–635. doi: 10.1007/s11064-010-0146-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hammes HP, Federoff HJ, Brownlee M. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol Med. 1995;1(5):527–534. [PMC free article] [PubMed] [Google Scholar]
- 54.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102(4):783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fort PE, Losiewicz MK, Reiter CE, Singh RS, Nakamura M, Abcouwer SF, Barber AJ, Gardner TW. Differential roles of hyperglycemia and hypoinsulinemia in diabetes induced retinal cell death: evidence for retinal insulin resistance. PLoS One. 2011;6(10):e26498. doi: 10.1371/journal.pone.0026498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. 2007;55(6):498–510. doi: 10.1016/j.phrs.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 57.Way KJ, Katai N, King GL. Protein kinase C and the development of diabetic vascular complications. Diabet Med. 2001;18(12):945–959. doi: 10.1046/j.0742-3071.2001.00638.x. [DOI] [PubMed] [Google Scholar]
- 58.Aiello LP, Vignati L, Sheetz MJ, Zhi X, Girach A, Davis MD, Wolka AM, Shahri N, Milton RC. Oral protein kinase c beta inhibition using ruboxistaurin: efficacy, safety, and causes of vision loss among 813 patients (1,392 eyes) with diabetic retinopathy in the Protein Kinase C beta Inhibitor-Diabetic Retinopathy Study and the Protein Kinase C beta Inhibitor-Diabetic Retinopathy Study 2. Retina. 2011;31(10):2084–2094. doi: 10.1097/IAE.0b013e3182111669. [DOI] [PubMed] [Google Scholar]
- 59.Zong H, Ward M, Stitt AW. AGEs, RAGE, and diabetic retinopathy. Curr Diabetes Rep. 2011;11(4):244–252. doi: 10.1007/s11892-011-0198-7. [DOI] [PubMed] [Google Scholar]
- 60.Suzen S, Buyukbingol E. Recent studies of aldose reductase enzyme inhibition for diabetic complications. Curr Med Chem. 2003;10(15):1329–1352. doi: 10.2174/0929867033457377. [DOI] [PubMed] [Google Scholar]
- 61.Madsen-Bouterse SA, Kowluru RA. Oxidative stress and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Rev Endocr Metab Disord. 2008;9(4):315–327. doi: 10.1007/s11154-008-9090-4. [DOI] [PubMed] [Google Scholar]
- 62.Silva KC, Rosales MA, Biswas SK, Lopes de Faria JB, Lopes de Faria JM. Diabetic retinal neurodegeneration is associated with mitochondrial oxidative stress and is improved by an angiotensin receptor blocker in a model combining hypertension and diabetes. Diabetes. 2009;58(6):1382–1390. doi: 10.2337/db09-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang H, Zheng Z, Gong Y, Zhu B, Xu X. U83836E inhibits retinal neurodegeneration in early-stage streptozotocin-induced diabetic rats. Ophthalmic Res. 2011;46(1):19–24. doi: 10.1159/000321952. [DOI] [PubMed] [Google Scholar]
- 64.Huang YZ, McNamara JO. Neuroprotective effects of reactive oxygen species mediated by BDNF-independent activation of TrkB. J Neurosci. 2012;32(44):15521–15532. doi: 10.1523/JNEUROSCI.0755-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zong H, Ward M, Madden A, Yong PH, Limb GA, Curtis TM, Stitt AW. Hyperglycaemia-induced pro-inflammatory responses by retinal Muller glia are regulated by the receptor for advanced glycation end-products (RAGE) Diabetologia. 2010;53(12):2656–26566. doi: 10.1007/s00125-010-1900-z. [DOI] [PubMed] [Google Scholar]
- 66.Berner AK, Brouwers O, Pringle R, Klaassen I, Colhoun L, McVicar C, Brockbank S, Curry JW, Miyata T, Brownlee M, Schlingemann RO, Schalkwijk C, Stitt AW. Protection against methylglyoxal-derived AGEs by regulation of glyoxalase 1 prevents retinal neuroglial and vasodegenerative pathology. Diabetologia. 2012;55(3):845–854. doi: 10.1007/s00125-011-2393-0. [DOI] [PubMed] [Google Scholar]
- 67.Arnold E, Rivera JC, Thebault S, Moreno-Paramo D, Quiroz-Mercado H, Quintanar-Stephano A, Binart N, Martinez de la Escalera G, Clapp C. High levels of serum prolactin protect against diabetic retinopathy by increasing ocular vasoinhibins. Diabetes. 2010;59(12):3192–3197. doi: 10.2337/db10-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 69.Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339(6116):172–177. doi: 10.1126/science.1230721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147(3):642–653. [PMC free article] [PubMed] [Google Scholar]
- 71.Miyamoto K, Khosrof S, Bursell SE, Rohan R, Murata T, Clermont AC, Aiello LP, Ogura Y, Adamis AP. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A. 1999;96(19):10836–10841. doi: 10.1073/pnas.96.19.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang H, Gandhi JK, Zhong X, Wei Y, Gong J, Duh EJ, Vinores SA. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Invest Ophth Vis Sci. 2011;52(3):1336–1344. doi: 10.1167/iovs.10-5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou J, Wang S, Xia X. Role of intravitreal inflammatory cytokines and angiogenic factors in proliferative diabetic retinopathy. Curr Eye Res. 2012;37(5):416–420. doi: 10.3109/02713683.2012.661114. [DOI] [PubMed] [Google Scholar]
- 74.Hernandez C, Segura RM, Fonollosa A, Carrasco E, Francisco G, Simo R. Interleukin-8, monocyte chemoattractant protein-1 and IL-10 in the vitreous fluid of patients with proliferative diabetic retinopathy. Diabet Med. 2005;22(6):719–722. doi: 10.1111/j.1464-5491.2005.01538.x. [DOI] [PubMed] [Google Scholar]
- 75.Gustavsson C, Agardh CD, Agardh E. Profile of intraocular tumour necrosis factor-alpha and interleukin-6 in diabetic subjects with different degrees of diabetic retinopathy. Acta Ophthalmol. 2012 doi: 10.1111/j.1755-3768.2012.02430.x. [DOI] [PubMed] [Google Scholar]
- 76.Zeng HY, Green WR, Tso MO. Microglial activation in human diabetic retinopathy. Arch Ophthalmol. 2008;126(2):227–232. doi: 10.1001/archophthalmol.2007.65. [DOI] [PubMed] [Google Scholar]
- 77.Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339(6116):156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nwanyanwu KH, Talwar N, Gardner TW, Wrobel JS, Herman WH, Stein JD. Predicting Development of Proliferative Diabetic Retinopathy. Diabetes Care. 2013 doi: 10.2337/dc12-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blandini F. An update on the potential role of excitotoxicity in the pathogenesis of Parkinson’s disease. Funct Neurol. 2010;25(2):65–71. [PubMed] [Google Scholar]
- 80.Hu NW, Ondrejcak T, Rowan MJ. Glutamate receptors in preclinical research on Alzheimer’s disease: update on recent advances. Pharmacol Biochem Behav. 2012;100(4):855–862. doi: 10.1016/j.pbb.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 81.Pulido JE, Pulido JS, Erie JC, Arroyo J, Bertram K, Lu MJ, Shippy SA. A role for excitatory amino acids in diabetic eye disease. Exp Diabetes Res. 2007;2007:36150. doi: 10.1155/2007/36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang Y, Bhavnani BR. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006;7:49. doi: 10.1186/1471-2202-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lieth E, Barber AJ, Xu B, Dice C, Ratz MJ, Tanase D, Strother JM. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes. 1998;47(5):815–820. doi: 10.2337/diabetes.47.5.815. [DOI] [PubMed] [Google Scholar]
- 84.Lieth E, LaNoue KF, Antonetti DA, Ratz M. Diabetes reduces glutamate oxidation and glutamine synthesis in the retina. The Penn State Retina Research Group. Exp Eye Res. 2000;70(6):723–730. doi: 10.1006/exer.2000.0840. [DOI] [PubMed] [Google Scholar]
- 85.Li Q, Puro DG. Diabetes-induced dysfunction of the glutamate transporter in retinal Muller cells. Invest Ophthalmol Vis Sci. 2002;43(9):3109–3116. [PubMed] [Google Scholar]
- 86.Ambati J, Chalam KV, Chawla DK, D’Angio CT, Guillet EG, Rose SJ, Vanderlinde RE, Ambati BK. Elevated gamma-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol. 1997;115(9):1161–1166. doi: 10.1001/archopht.1997.01100160331011. [DOI] [PubMed] [Google Scholar]
- 87.Lu MJ, Pulido JS, McCannel CA, Pulido JE, Hatfield RM, Dundervill RF, 3rd, Shippy SA. Detection of elevated signaling amino acids in human diabetic vitreous by rapid capillary electrophoresis. Exp Diabetes Res. 2007;2007:39765. doi: 10.1155/2007/39765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barnstable CJ, Tombran-Tink J. Neuroprotective and antiangiogenic actions of PEDF in the eye: molecular targets and therapeutic potential. Prog Retin Eye Res. 2004;23(5):561–577. doi: 10.1016/j.preteyeres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 89.Yoshida Y, Yamagishi S, Matsui T, Jinnouchi Y, Fukami K, Imaizumi T, Yamakawa R. Protective role of pigment epithelium-derived factor (PEDF) in early phase of experimental diabetic retinopathy. Diabetes Metab Res Rev. 2009;25(7):678–686. doi: 10.1002/dmrr.1007. [DOI] [PubMed] [Google Scholar]
- 90.Shen X, Xie B, Cheng Y, Jiao Q, Zhong Y. Effect of pigment epithelium derived factor on the expression of glutamine synthetase in early phase of experimental diabetic retinopathy. Ocul Immunol Inflamm. 2011;19(4):246–254. doi: 10.3109/09273948.2011.580073. [DOI] [PubMed] [Google Scholar]
- 91.Liu Y, Leo LF, McGregor C, Grivitishvili A, Barnstable CJ, Tombran-Tink J. PEDF peptide eye drops reduce inflammation, cell death, and vascular leakage in diabetic retinopathy in the Ins2akita mice. Mol Med. 2012 doi: 10.2119/molmed.2012.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Seki M, Tanaka T, Nawa H, Usui T, Fukuchi T, Ikeda K, Abe H, Takei N. Involvement of brain-derived neurotrophic factor in early retinal neuropathy of streptozotocin-induced diabetes in rats: therapeutic potential of brain-derived neurotrophic factor for dopaminergic amacrine cells. Diabetes. 2004;53(9):2412–2419. doi: 10.2337/diabetes.53.9.2412. [DOI] [PubMed] [Google Scholar]
- 93.Gong Y, Chang ZP, Ren RT, Wei SH, Zhou HF, Chen XF, Hou BK, Jin X, Zhang MN. Protective Effects of Adeno-associated Virus Mediated Brain-derived Neurotrophic Factor Expression on Retinal Ganglion Cells in Diabetic Rats. Cell Mol Neurobiol. 2012 doi: 10.1007/s10571-011-9779-x. [DOI] [PubMed] [Google Scholar]
- 94.Ali TK, Al-Gayyar MM, Matragoon S, Pillai BA, Abdelsaid MA, Nussbaum JJ, El-Remessy AB. Diabetes-induced peroxynitrite impairs the balance of pro-nerve growth factor and nerve growth factor, and causes neurovascular injury. Diabetologia. 2011;54(3):657–668. doi: 10.1007/s00125-010-1935-1. [DOI] [PubMed] [Google Scholar]
- 95.Kiagiadaki F, Savvaki M, Thermos K. Activation of somatostatin receptor (sst 5) protects the rat retina from AMPA-induced neurotoxicity. Neuropharmacology. 2010;58(1):297–303. doi: 10.1016/j.neuropharm.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 96.Aizu Y, Katayama H, Takahama S, Hu J, Nakagawa H, Oyanagi K. Topical instillation of ciliary neurotrophic factor inhibits retinal degeneration in streptozotocin-induced diabetic rats. Neuroreport. 2003;14(16):2067–2071. doi: 10.1097/00001756-200311140-00012. [DOI] [PubMed] [Google Scholar]
- 97.Ali TK, Matragoon S, Pillai BA, Liou GI, El-Remessy AB. Peroxynitrite mediates retinal neurodegeneration by inhibiting nerve growth factor survival signaling in experimental and human diabetes. Diabetes. 2008;57(4):889–898. doi: 10.2337/db07-1669. [DOI] [PubMed] [Google Scholar]
- 98.Ahmed T, Yumet G, Shumate M, Lang CH, Rotwein P, Cooney RN. Tumor necrosis factor inhibits growth hormone-mediated gene expression in hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2006;291(1):G35–44. doi: 10.1152/ajpgi.00550.2005. [DOI] [PubMed] [Google Scholar]
- 99.Wei Y, Chen K, Whaley-Connell AT, Stump CS, Ibdah JA, Sowers JR. Skeletal muscle insulin resistance: role of inflammatory cytokines and reactive oxygen species. Am J Physiol Regul Integr Comp Physiol. 2008;294(3):R673–680. doi: 10.1152/ajpregu.00561.2007. [DOI] [PubMed] [Google Scholar]
- 100.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329(14):977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 101.Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352(9131):837–853. [PubMed] [Google Scholar]
- 102.Gerstein HC, Miller ME, Byington RP, Goff DC, Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH, Jr, Probstfield JL, Simons-Morton DG, Friedewald WT. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358(24):2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lachin JM, Genuth S, Nathan DM, Zinman B, Rutledge BN. Effect of glycemic exposure on the risk of microvascular complications in the diabetes control and complications trial--revisited. Diabetes. 2008;57(4):995–1001. doi: 10.2337/db07-1618. [DOI] [PubMed] [Google Scholar]
- 104.Floyd B, Chandra P, Hall S, Phillips C, Alema-Mensah E, Strayhorn G, Ofili EO, Umpierrez GE. Comparative analysis of the efficacy of continuous glucose monitoring and self-monitoring of blood glucose in type 1 diabetes mellitus. J Diabetes Sci Technol. 2012;6(5):1094–1102. doi: 10.1177/193229681200600513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Battelino T, Conget I, Olsen B, Schutz-Fuhrmann I, Hommel E, Hoogma R, Schierloh U, Sulli N, Bolinder J. The use and efficacy of continuous glucose monitoring in type 1 diabetes treated with insulin pump therapy: a randomised controlled trial. Diabetologia. 2012;55(12):3155–3162. doi: 10.1007/s00125-012-2708-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Breton M, Farret A, Bruttomesso D, Anderson S, Magni L, Patek S, Dalla Man C, Place J, Demartini S, Del Favero S, Toffanin C, Hughes-Karvetski C, Dassau E, Zisser H, Doyle FJ, 3rd, De Nicolao G, Avogaro A, Cobelli C, Renard E, Kovatchev B. Fully integrated artificial pancreas in type 1 diabetes: modular closed-loop glucose control maintains near normoglycemia. Diabetes. 2012;61(9):2230–2237. doi: 10.2337/db11-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Thabit H, Hovorka R. Closed-loop insulin delivery in type 1 diabetes. Endocrinol Metab Clin North Am. 2012;41(1):105–117. doi: 10.1016/j.ecl.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52(2):1156–1163. doi: 10.1167/iovs.10-6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nakanishi Y, Nakamura M, Mukuno H, Kanamori A, Seigel GM, Negi A. Latanoprost rescues retinal neuro-glial cells from apoptosis by inhibiting caspase-3, which is mediated by p44/p42 mitogen-activated protein kinase. Exp Eye Res. 2006;83(5):1108–1117. doi: 10.1016/j.exer.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 110.Nakajima E, Hammond KB, Rosales JL, Shearer TR, Azuma M. Calpain, not caspase, is the causative protease for hypoxic damage in cultured monkey retinal cells. Invest Ophth Vis Sci. 2011;52(10):7059–7067. doi: 10.1167/iovs.11-7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shanab AY, Nakazawa T, Ryu M, Tanaka Y, Himori N, Taguchi K, Yasuda M, Watanabe R, Takano J, Saido T, Minegishi N, Miyata T, Abe T, Yamamoto M. Metabolic stress response implicated in diabetic retinopathy: the role of calpain, and the therapeutic impact of calpain inhibitor. Neurobiol Dis. 2012;48(3):556–567. doi: 10.1016/j.nbd.2012.07.025. [DOI] [PubMed] [Google Scholar]
- 112.Kusari J, Zhou S, Padillo E, Clarke KG, Gil DW. Effect of memantine on neuroretinal function and retinal vascular changes of streptozotocin-induced diabetic rats. Invest Ophth Vis Sci. 2007;48(11):5152–5159. doi: 10.1167/iovs.07-0427. [DOI] [PubMed] [Google Scholar]
- 113.Koleva-Georgieva DN, Sivkova NP, Terzieva D. Serum inflammatory cytokines IL-1beta, IL-6, TNF-alpha and VEGF have influence on the development of diabetic retinopathy. Folia Med (Plovdiv) 2011;53(2):44–50. doi: 10.2478/v10153-010-0036-8. [DOI] [PubMed] [Google Scholar]
- 114.Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Dohmen S, Adamis AP. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. Faseb J. 2002;16(3):438–440. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 115.Joussen AM, Doehmen S, Le ML, Koizumi K, Radetzky S, Krohne TU, Poulaki V, Semkova I, Kociok N. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009;15:1418–1428. [PMC free article] [PubMed] [Google Scholar]
- 116.Tsilimbaris MK, Panagiotoglou TD, Charisis SK, Anastasakis A, Krikonis TS, Christodoulakis E. The use of intravitreal etanercept in diabetic macular oedema. Semin Ophthalmol. 2007;22(2):75–79. doi: 10.1080/08820530701418243. [DOI] [PubMed] [Google Scholar]
- 117.Wu L, Hernandez-Bogantes E, Roca JA, Arevalo JF, Barraza K, Lasave AF. intravitreal tumor necrosis factor inhibitors in the treatment of refractory diabetic macular edema: a pilot study from the Pan-American Collaborative Retina Study Group. Retina. 2011;31(2):298–303. doi: 10.1097/IAE.0b013e3181eac7a6. [DOI] [PubMed] [Google Scholar]
- 118.Wang AL, Yu AC, Lau LT, Lee C, Wu le M, Zhu X, Tso MO. Minocycline inhibits LPS-induced retinal microglia activation. Neurochem Int. 2005;47(1–2):152–158. doi: 10.1016/j.neuint.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 119.Krady JK, Basu A, Allen CM, Xu Y, LaNoue KF, Gardner TW, Levison SW. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes. 2005;54(5):1559–1565. doi: 10.2337/diabetes.54.5.1559. [DOI] [PubMed] [Google Scholar]
- 120.Cukras CA, Petrou P, Chew EY, Meyerle CB, Wong WT. Oral minocycline for the treatment of diabetic macular edema (DME): results of a phase I/II clinical study. Invest Ophth Vis Sci. 2012;53(7):3865–3874. doi: 10.1167/iovs.11-9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Powell ED, Field RA. Diabetic Retinopathy and Rheumatoid Arthritis. Lancet. 1964;2(7349):17–18. doi: 10.1016/s0140-6736(64)90008-x. [DOI] [PubMed] [Google Scholar]
- 122.Effects of aspirin treatment on diabetic retinopathy. ETDRS report number 8. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology. 1991;98(5):757–865. [PubMed] [Google Scholar]
- 123.Zheng L, Howell SJ, Hatala DA, Huang K, Kern TS. Salicylate-based anti-inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes. 2007;56(2):337–345. doi: 10.2337/db06-0789. [DOI] [PubMed] [Google Scholar]
- 124.Aiello LP, Davis MD, Girach A, Kles KA, Milton RC, Sheetz MJ, Vignati L, Zhi XE. Effect of ruboxistaurin on visual loss in patients with diabetic retinopathy. Ophthalmology. 2006;113(12):2221–2230. doi: 10.1016/j.ophtha.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 125.Titchenell PM, Lin CM, Keil JM, Sundstrom JM, Smith CD, Antonetti DA. Novel atypical PKC inhibitors prevent vascular endothelial growth factor-induced blood-retinal barrier dysfunction. Biochem J. 2012;446(3):455–467. doi: 10.1042/BJ20111961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ola MS, Nawaz MI, El-Asrar AA, Abouammoh M, Alhomida AS. Reduced Levels of Brain Derived Neurotrophic Factor (BDNF) in the Serum of Diabetic Retinopathy Patients and in the Retina of Diabetic Rats. Cell Mol Neurobiol. 2012 doi: 10.1007/s10571-012-9901-8. [DOI] [PubMed] [Google Scholar]
- 127.Misra GP, Singh RS, Aleman TS, Jacobson SG, Gardner TW, Lowe TL. Subconjunctivally implantable hydrogels with degradable and thermoresponsive properties for sustained release of insulin to the retina. Biomaterial’s. 2009;30(33):6541–6547. doi: 10.1016/j.biomaterials.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Saydah SH, Fradkin J, Cowie CC. Poor control of risk factors for vascular disease among adults with previously diagnosed diabetes. JAMA - J Am Med Assoc. 2004;291(3):335–342. doi: 10.1001/jama.291.3.335. [DOI] [PubMed] [Google Scholar]
- 129.Wood JR, Miller KM, Maahs DM, Beck RW, Dimeglio LA, Libman IM, Quinn M, Tamborlane WV, Woerner SE. Most Youth With Type 1 Diabetes in the T1D Exchange Clinic Registry Do Not Meet American Diabetes Association or International Society for Pediatric and Adolescent Diabetes Clinical Guidelines. Diabetes Care. 2013 doi: 10.2337/dc12-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ko F, Vitale S, Chou CF, Cotch MF, Saaddine J, Friedman DS. Prevalence of nonrefractive visual impairment in US adults and associated risk factors, 1999–2002 and 2005–2008. JAMA - J Am Med Assoc. 2012;308(22):2361–2368. doi: 10.1001/jama.2012.85685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhang X, Saaddine JB, Chou CF, Cotch MF, Cheng YJ, Geiss LS, Gregg EW, Albright AL, Klein BE, Klein R. Prevalence of diabetic retinopathy in the United States, 2005–2008. JAMA - J Am Med Assoc. 2010;304(6):649–656. doi: 10.1001/jama.2010.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Saaddine JB, Honeycutt AA, Narayan KM, Zhang X, Klein R, Boyle JP. Projection of diabetic retinopathy and other major eye diseases among people with diabetes mellitus: United States, 2005–2050. Arch Ophthalmol. 2008;126(12):1740–1747. doi: 10.1001/archopht.126.12.1740. [DOI] [PubMed] [Google Scholar]