

Abstract
DNA is the blueprint upon which life is based and transmitted, yet the manner in which chromatin, the dynamic complex of nucleic acids and proteins, is packaged and behaves within the cellular nucleus has only begun to be investigated. The packaging and modifications around the genome have been shown to exert significant influence on cellular behaviour and in turn, human development and disease. However, conventional techniques for studying epigenetic or conformational modifications of chromosomes have inherent limitations, and therefore, new methods based on micro- and nanoscale devices have been sought. Here, we review the development of these devices and explore their use in the study of DNA and chromatin modifications and higher order chromatin structure.
Keywords: Microfluidics, Nanofluidics, Optical Mapping, Nanopores, Sequencing, Nucleosomes, Histones, Methylation
In the 20th century, the role of DNA as the molecular basis for heredity and genetics was recognized. More recently, it has become clear that the much more complex and dynamic physical and chemical conditions of the chromosomes and structures that contain the DNA are as instrumental in regulating cellular processes. While the operating definitions of epigenetics1 and epigenomics are nearly as dynamic as chromatin remodeling, it is clear that the information encoded “on top of” genetics that does not include modifications to the underlying sequence, is a new level of quantifiable information that is of vital importance for understanding biology and disease2. In this review, we first introduce the different layers of the epigenome and highlight their importance in development and disease. Next, current assays to probe these layers are discussed and several limitations of the technologies are examined. New micro and nanoscale devices that have been used to ameliorate some of these limitations are then introduced. We look at the drive behind these new technologies, introduce how these tools are being applied to probe the different epigenetic layers and conclude with potential paths forward for performing multiplexed epigenetic profiling from small cell numbers.
Layers of the epigenome
Eukaryotic organisms package nearly two metres of DNA into a small nuclear compartment using a series of hierarchical layers. The temporal structural and chemical alteration of these layers influence gene activity and various cellular programs. The lowest layer of compaction occurs through wrapping of DNA into a nucleosome, which is formed from ∼146 base pairs (bp) of DNA wrapped around a protein octamer3. The protein octamer is composed of eight sub-units called histones (H2A, H2B, H3, H4) that assemble as one H3-H4 tetramer and two H2A-H2B dimers (Figure 1). An individual nucleosome is separated from an adjacent nucleosome by linker DNA, whose length can range from several kbp to as small as several base pairs4. The underlying DNA and histones can be subjected to modifications that alter DNA accessibility for transcription factors and other molecules such as RNA polymerase5. Modifications to the underlying DNA, such as cytosine methylation, which occurs on the fifth carbon residue (5mC) and typically on cytosine-guanine dinucleotides (CpG), are distributed across the genome6 and into discrete patterns7. CpG dinucleotides are generally underrepresented in eukaryotic genomes but most (60-80%) are methylated. The modifications conferred onto histones8 primarily occur on the protruding amino-terminal tails9, which include methylation, acetylation, phosphorylation, ubiquitylation, sumoylation and others as well as their different forms (mono, di, trimethylation for example)10. The histones can also be exchanged with variants (such as H2A.X, H2A.Z, H3.3) and the modifications and variants have both been associated with many functional responses11.
Figure 1.
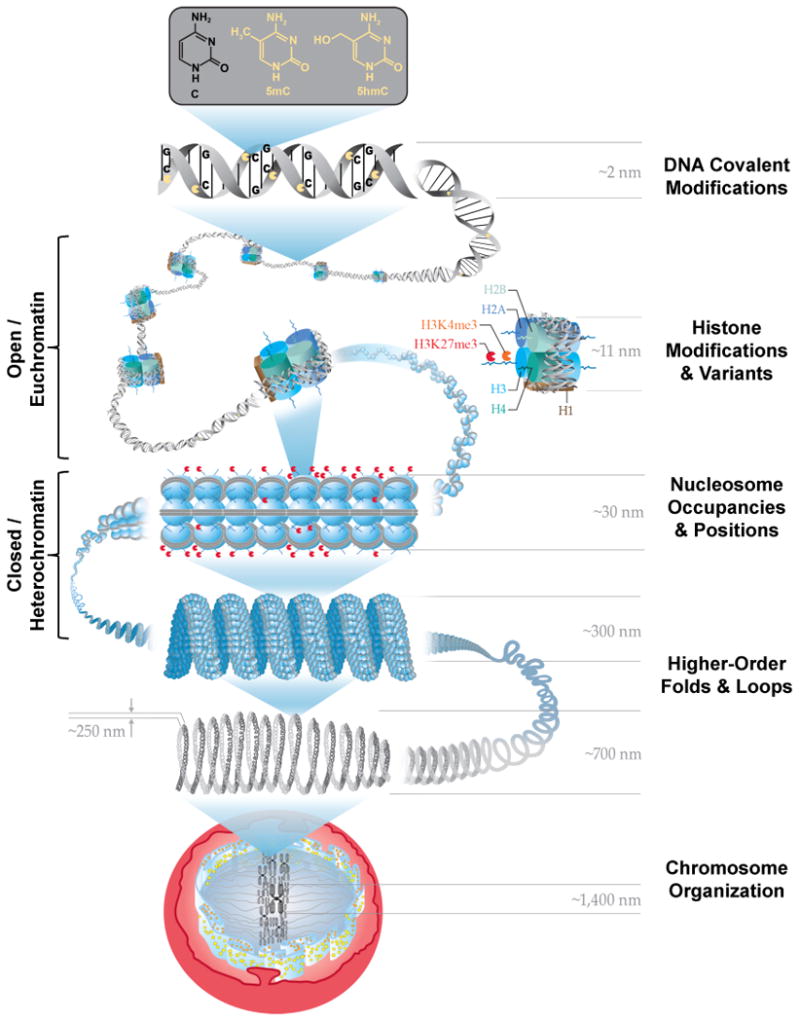
Overview of epigenetic layers and corresponding size scales. The root layer is the DNA sequence and covalent modifications such as cytosine methylation (5mC) and hydroxymethylcytosine (5hmC). The DNA is then wrapped around octameric histone proteins into nucleosomes and into chromatin. The nucleosomal histones H2A, H2B, H3 and H4 form pairs with one H3-H4 tetramer and two H2A-H2B dimers and can be exchanged with variants or chemically modified on their protruding tails such as histone 3 - lysine 27 - trimethylation (me3): H3K27me3. The structure of the chromatin is mediated by the nucleosome packing with open / euchromatin having less nucleosomes positioned than closed / heterochromatin. The condensed heterochromatin has been shown to possess a unique solenoid structure and higher-order loops and folds also exist to further compact the chromatin into chromosomes. The various layers and modifications establish whether the gene and the regulatory components (promoter, enhancer) are accessible and transcribed or inactive. DNA cytosine methylation and histone modifications such as H3K27me3 are broadly associated with inactive genes as to where hydroxymethylated cytosine bases and histone modifications such as H3K4me3 are nominally associated with active genes and regulatory elements.
The location and timing of the modifications to the DNA and histones work together to define the shape of the chromatin and epigenetic state. Local patterns of chromatin are typically described in two forms (open/euchromatin or closed/heterochromatin), and each has a unique scale. Euchromatin is commonly described with a “beads on a string” model, with the DNA (string) having a diameter of ∼2nm and nucleosomes (beads) having a diameter of ∼11nm. In this open form, DNA is much more accessible for DNA-binding proteins and polymerases and consequently, gene-rich areas tend to be packaged into euchromatin. Chromatin can also be packaged into a more condensed fibre called heterochromatin, which can contain many repetitive sequences and fewer genes and is frequently found near the nuclear lamina. Heterochromatin is packaged into a larger diameter fibre (∼30nm) and is aided by the H1 linker histone protein, which tightens the folding of the DNA around the nucleosome12 by attaching where the DNA enters and leaves the nucleosome. The epigenetic state can be described by chromatin packaging and transcriptional activity using several DNA and chromatin modifications. For example, the modification of Histone 3 on the Lysine 9 residue by trimethylation, H3K9me3, has been associated with heterochromatin and transcriptional inactivity. In contrast, cytosine bases that are hydroxymethylated (hydroxymethylation, 5hmC) and the modification of Histone 3 on the Lysine 36 residue by trimethylation, H3K36me3, have both been associated with transcriptional activity10. Chromatin is not static; rather DNA packaging is dynamically adjustable based on cues from environmental stimuli and activated cellular pathways13. As local chromatin packaging changes, global patterns of chromatin organization also change through higher-level loops and folds14, which in turn influence intra and inter-chromosomal interactions15 as well as nuclear compartmentalization.
Motivations for monitoring epigenetic phenomena
The transient activation or repression of gene expression and various cellular programs is partially controlled via epigenetic modifications. A model of this regulation is mammalian development, where changes in chromatin accessibility at key loci are mediated by DNA cytosine methylation16 and several histone modifications17. These epigenetic modifications and chromatin landscapes enhance or prevent transcription of developmental genes that define how a single genome can produce multiple cell types during embryonic development and preserve each cellular identity throughout life18. An example of this regulation is a bivalent domain17, which is two histone modifications that are located in the same region that demarcate different chromatin states (one modification is associated with active gene promoters: Histone 3 – Lysine 4 – Trimethylation, H3K4me3, and the other is associated with inactive genes: Histone 3 – Lysine 27 – Trimethylation, H3K27me3). The gain or loss of one histone modification in a bivalent domain in an embryonic stem cell influences chromatin structure and transcription at that locus, further nudging the cell to differentiate into a specific cell type. These epigenetic patterns also influence deleterious cellular processes such as cancer19. For example, profiling DNA cytosine methylation from different types of breast cancer tumour biopsies showed each tumour could be categorized by metastatic risk and the patterns of hypermethylation were also shared by glioma and colon cancer20. Regions with CG-dense sequences, also known as CpG islands, which are found near or on gene promoters, are largely unmethylated but hypermethylation of CpG islands has been found in nearly every tumour type19. Histone modification patterns also show differences between normal and cancer cells21,22, with multiple tumour suppressor genes undergoing chromatin remodeling that can lead to aberrant gene expression and tumourigenesis. Because DNA and histone modifications influence the 3-D chromatin landscape and how different loops and folds spatially contact one another, it is not surprising that a link between chromosomal alterations that are a hallmark of cancer and chromatin organization has also recently been shown23.
Early development and diseases such as cancer emphasize how the different epigenetic layers work in combinations across length scales and the genome. To characterize these important epigenetic modifications, high-throughput sequencing technologies have primarily been utilized. There are two technologies used to generate most epigenetic maps. The first is bi-sulfite conversion followed by sequencing (BSC-Seq), and the second is chromatin immunoprecipitation followed by sequencing (ChIP-Seq). BSC-Seq is used to study DNA cytosine methylation and operates by treatment of purified DNA with bisulphite, which converts all cytosine residues to uracil except those that are methylated, followed by sequencing the products to reveal the genomic location of the residual untransformed cytosines24. There are numerous other modifications of bases that are not revealed by BSC-Seq, for which techniques are evolving. ChIP-Seq uses antibodies to capture chromatin fragments bearing specific modifications. Release and sequencing of the DNA from the captured chromatin fragments identifies the genetic sequence associated with the selected modification25 and alignment to the known reference sequence, permits mapping of where the histone modifications existed throughout the genome. Both strategies construct detailed landscapes of epigenetic modifications but primarily rely on a large number of cells to determine statistically significant enrichments from background. They typically interrogate one mark at a time (rather than combinations) and ChIP-Seq cannot determine the absolute level of modifications (just presence). For example, if two copies of a histone are modified within the same nucleosome, ChIP-Seq cannot determine if there are one or two. Moreover, because chromatin patterns and remodeling processes are dynamic and vary across time and cellular types26, ensemble measurements may be confounded by sample inhomogeneity and average out effects such as noise27 that can potentially contribute to phenotype and disease. Accordingly, one of the primary research goals of the new methods is to reduce the required number of cells from thousands to single cells and to simultaneously quantify multiple epigenetic modifications28. This goal is particularly relevant for rare cell types such as circulating tumour cells29 and tissue biopsies30, which are often extracted with a mixture of cells and conventional analysis will only provide chromatin signatures of the most abundant type. While reducing the cell requirement will abate the heterogeneity of the signal, the transient dynamics of the chromatin fibre and remodeling processes are not captured. Therefore, new technologies are required that can dynamically track multiple epigenetic marks through intermediate states and scale to look at the interplay between local composition and global organization as well as operate from a single cell.
The critical sizes of the molecular machines that drive and organize the epigenetic layers are at the scale of micro and nanodevices (0.1-20,000 nm). Advances in micro and nanofabrication31,32 have facilitated construction of devices that can utilize novel physical phenomena to enable study of single cells and molecules with high precision. The coupling of nano and micro technologies also permits a path for interfacing to smaller sample sizes, microfluidic automation and chip-based parallel processing33. Sets of micro and nanotechnologies are presented and examined in the following sections and grouped by the type of epigenetic layer each has been utilized to probe. Lastly, technologies that have been used to investigate structural dynamics of chromatin and higher-order folding principles are examined.
Mapping DNA modifications
The placement and timing of covalent modifications to DNA participates in many cellular processes including initiation of diseases such as rheumatoid arthritis34 and cancer35. There are multiple technologies to detect and map DNA cytosine methylation but the most popular uses bisulfite-conversion of the DNA, which converts unmethylated cytosine bases to uracil, followed by sequencing36. While this approach possesses single-nucleotide resolution and can scale to the full genome, this technique also has several limitations, which were previously described.
One relatively new approach with single-nucleotide resolution involves single-molecule DNA sequencing. In this approach, individual DNA molecules are analyzed by observation of individual polymerase enzymes using an array of nanostructures to isolate the molecules for optical monitoring. A nanometre-scale aperture, smaller than the illumination wavelength, reduces the optical interrogation volume such that enzymatic incorporation of individual nucleotides by a single DNA polymerase can be monitored in real-time37. The observation of one of four different coloured fluorescent labels identifies the incorporated base, and from the temporal order of incorporation, the sequence of the individual molecule is derived38. The reaction kinetics for base incorporation depends on the chemical modifications of the bases in the DNA strand being sequenced. In some cases, these modifications can be identified by characteristic time differences between base incorporation events while the identity of the base is determined by the colour of the fluorescence. In this way both the genetic sequence and the chemical base modifications are determined. Detection of 5mC39, hydroxymethylcytosine (5hmC)40 and 6-methyladenine (6mA)41 have been accomplished in this way. As discussed above, 5mC plays a critical role in development and cancer, and 5hmC, which is an oxidized product of 5mC, is enriched in the brain and slightly less in other cell types and tissues42. In contrast to the cytosine modifications, 6mA is not abundant in mammalian cells, but is found in prokaryotes and has implications for metagenomic studies and regulation of pathogenicity43. Figure 2a shows a schematic of a covalently modified base on a DNA strand entering the polymerase, which is tethered to the base of the aperture. The traces to the right of these panels in Figure 2a illustrate the different times between base incorporations as a result of the chemical modifications. While this approach gives reading of the precise genomic location of the modifications in relatively long (≥1kbp) single DNA molecules, the incorporation rates observed may not unambiguously identify the chemical nature of the modifications. Additionally, there are computational challenges36,44 associated with mapping millions or billions of reads in a time-efficient manner and interpreting the context with which the identified modification exists (CpG island, promoter, phenotype, environment, etc.).
Figure 2.
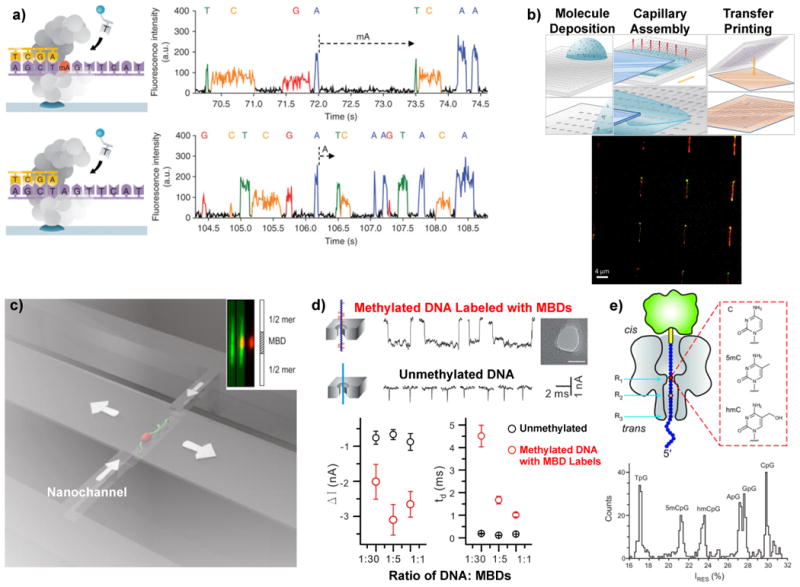
Micro and nano methods for mapping DNA covalent modifications. a) Detection of DNA covalent modifications using single molecule sequencing in a nanophotonic structure39. The right panels demonstrate detection of a methylated adenine base compared to a typical adenine. b) The top panel shows the capillary assembly protocol for optical mapping of large-scale arrays of single molecules71. The bottom panel shows fluorescently tagged methylation sites (green) on elongated single DNA molecules (red) using capillary assembly51. c) Optical mapping fluorescently-tagged methylation sites (red) on single DNA molecules (green) moving through a nanochannel50. The inset shows detection of a methylated region along a single molecule. d) Electrical differentiation of DNA with labeled cytosine modifications using a solid-state nanopore (MBDs – methyl binding domains)58. The bottom panel shows current blockades and translocation times for unmodified DNA molecules (black circles) compared to DNA labeled with varied amounts of cytosine methylation-specific labels (red circles). e) Electrical detection of DNA base modifications using a protein nanopore (α-hemolysin) where the residual current through the nanopore (bottom panel) is unique for each modification54.
Another strategy being investigated is to profile an individual DNA molecule by direct imaging of fluorescently tagged base modifications. This set of approaches can produce a “fingerprint” pattern of the location of selected modifications but they do not have the ability to provide the exact sequence position of the modifications. This is because DNA molecules in physiological solutions are an ensemble of random coils that are in a perpetual state of movement from Brownian agitation and chemical interactions with proteins45. To obtain a positional map of the DNA modifications, the molecules must be oriented so that the positions can be easily determined. One strategy to overcome the random conformations is to orient and stretch the molecule to approach its contour length. Molecular combing of DNA is a technique that stretches DNA on a surface using a moving liquid meniscus46 and has been used for genetic mapping using fluorescent hybridization probes. Capillary assembly47 is a related technique that uses a microstructured piece of silicone rubber and a single liquid droplet that is dragged over the topography (Figure 2b). The receding liquid (which contains DNA molecules) gets trapped in the topography and any molecule within the meniscus gets pulled apart and aligned in the direction of the capillary forces. The moving meniscus leaves behind the stretched molecules, which can be transferred to a substrate and optically mapped or barcoded for cytosine methylation sites. Figure 2b shows an array of single stretched DNA molecules after capillary assembly where fluorescent markers (DNA-red, methylation-sensitive peptide tag-green) are visible.
Another novel method to unfold a DNA or chromatin molecule is to use nanoconfinement48, in a channel with width and depth smaller than the DNA persistence length49 (∼50nm for physiological conditions). Figure 2c shows a schematic of a single DNA fragment moving through a nanochannel50 with two types of fluorescent tags (DNA-green and a methyl-CpG-binding domain-red) and the inset shows the locations of detected methylated regions. Capillary assembly and nanochannel profiling can be highly parallelized51 for simultaneously imaging thousands of molecules in a compact form factor. One limitation to conventional optical mapping of DNA modifications is diffraction-limited resolution52 (∼200nm or ∼588 bp), which is insufficient to resolve modifications that are in close proximity such as those found in CpG islands53. This limitation may be ameliorated with super-resolution microscopy or other high-resolution techniques such as electron microscopy.
Nanopore-based sensing offers a method for profiling covalent DNA modifications on a single molecule54,55. Nanopore sensing is based on a pore in a dielectric membrane that separates two electrolyte solutions, where one solution contains a concentration of DNA molecules. A voltage bias across the membrane drives an ionic current through the nanopore that a DNA molecule partially blocks as it passes through the pore56. Characterizing the translocation time and current blockade provides structural and compositional information about the molecule moving through. For example, recently single DNA molecules with various cytosine modifications (5mC, 5hmC) were translocated through a solid-state nanopore57. While this approach could determine the relative proportion of some modifications on a single molecule, mapping the position of the modifications was not achieved. This limitation was in part due to the speed of the molecule translocating through the nanopore. To enhance the detectability of methylated cytosines during translocation through the nanopores, another approach58 labeled methylated cytosines using methyl binding domain proteins (MBD1). This method was able to coarsely determine the number of methylation sites per molecule from the fraction of bound proteins (Figure 2d). The identification and differentiation of base modifications without a label remains an issue with solid-state nanopore profiling, but biological nanopores have been used to resolve individual bases along a single DNA fragment59,60 with substantially lower translocation speeds. Figure 2e illustrates the signal from a DNA strand being pulled through a protein nanopore (α-hemolysin). This approach is at an early stage, but it may enable protocols for simultaneously reading genomic61 and epigenomic maps with electronic detection.
There is clearly a need for richer information about the complex patterns of covalent DNA modifications and interest in obtaining this information more rapidly from smaller numbers of cells continues to grow. The discussion above involved the analysis of relatively durable chemical modifications that can survive relatively harsh treatment and techniques for stretching and orienting the DNA molecules. This is not the case for chromatin with its more delicate histone protein structure and different approaches and devices are required to analyze histone modifications and chromatin structure.
Mapping histone modifications and nucleosomes
The positions of nucleosomes and numerous modifications of their individual histones as well as their variants also regulate transcription and cellular fate. Determination of the presence and location of chromatin modifications is typically performed using ChIP-Seq techniques62, which use an immunoprecipitation step and a sequencing step. ChIP begins with cross-linking the proteins to the DNA, lysing open the cells, extraction of the nuclei and shearing into smaller fragments. The sheared chromatin is then immunoprecipitated, followed by reversal of the cross-links and disassociation of the protein-DNA complexes and DNA isolated for sequencing. While this approach can be somewhat automated with liquid-handling robots, the number of manual steps take time, require large numbers of cells and can result in significant user-variability. Moreover, antibody specificity continues to remain an issue that limits reproducibility of ChIP-Seq experiments.
Microfluidic automation has been applied to improve the speed of ChIP and reduce sample size requirements63,64 (Figure 3a). These microfluidic ChIP devices contain a network of small valves and chambers that are fabricated in silicone rubber65. The integrated valves permit small sample volumes to be controllably introduced and processed in miniature chambers, which can enhance antibody-target interactions and reduce incubation times. Rather than manual pipetting and multistep protocols with significant sample losses, miniature-interconnecting chambers and channels manipulate samples and fluids in a repeatable, rapid manner that reduces sample consumption. This level of integration can also be coupled with on-chip sonication66 and fragmentation67 to further automate the ChIP-Seq workflow. The microfluidic-ChIP devices have recently been used to screen antibody efficacy68 and used to gain insights into dynamic chromatin-remodeling processes. While these experiments still require an antibody and indirect assessment of the presence of a particular histone modification, other new approaches may provide direct mapping of histone modifications.
Figure 3.
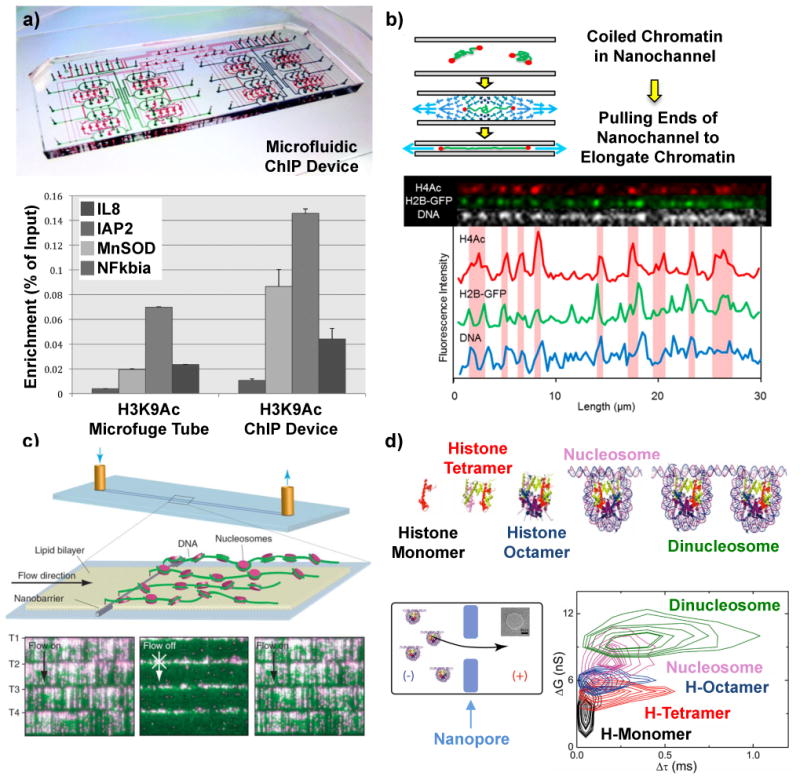
Micro and nano methods for mapping histone modifications and nucleosome arrays. a) Microfluidic device to perform automated chromatin immunoprecipitation (ChIP) reactions with higher efficiencies than conventional protocols (bottom panel)68. (b) Optical mapping of fluorescently tagged histone modifications on single unfolded chromatin molecules in nanochannels70. c) DNA curtains assay where single DNA molecules are anchored at one end and stretched by fluid flow to image DNA (green) and position quantum dot labeled nucleosomes (magenta)73. (d) Histone oligomers and nucleosomes moving through a solid-state nanopore block different amounts of current (ΔG corresponds to conductance change) and translocate through the nanopore with different times (Δτ)80.
Direct identification and quantification of histone modifications has recently been accomplished on single chromatin fragments using optical methods. One approach profiled histone modifications on single chromatin fragments moving through a nanochannel69 with multi-colour fluorescence microscopy. This technique labeled DNA, nucleosomal proteins and cytosine methylation with different colour fluorescent probes and was able to detect single molecules with relatively high throughput (10 Mbp/min). Another approach used nanoscale “squeezing” of single chromatin molecules to elongate fragments and profile histone modifications70. In this method, a triangular nanochannel is used to trap a single chromatin molecule. Pulling the ends of nanochannel and decreasing the height stretches the trapped chromatin molecule out to allow mapping of fluorescently labeled histone modifications (Figure 3b). Nanochannel-based techniques offer high parallelization (1000s of channels per chip) and potentially high throughput as well as the ability to pair with microfluidic preparatory schemes. A different approach used capillary assembly71 to elongate chromatin fragments that were immunolabeled with fluorescent markers. While this approach possessed a spatial resolution of ∼940 bp, high-density arrays (250,000) of long chromatin fragments could be formed to contain roughly one human genome on a single chip. Another method of optical mapping called DNA curtains, has also recently been demonstrated72, which uses DNA molecules anchored at one end to a fluid lipid bilayer surface and fluid flow / hydrodynamic force to push the free end of the molecule toward the leading edge of patterned nanofabricated barriers (Figure 3c). The aligned DNA is then labeled and imaged with a total internal reflection fluorescence microscope. DNA curtains were used to map arrays of nucleosomes73 and compare the results with in silico predictions4 and also showed the effects of a histone variant, CenH3, and a nonhistone protein, Scm3, on chromatin organization. Optical mapping of histone modifications on single chromatin fragments has also permitted insights into the short and long-range interactions of transcription factors74,75 on chromatin structure. While experiments to optically map histone modifications on single chromatin molecules are preliminary and require an increase in spatial resolution to distinguish multiple histone modifications on a single nucleosome or neighboring nucleosomes, they can be combined with higher resolution microscopy techniques and include labels for DNA modifications for multi-dimensional chromatin profiling76. A new approach suggests the possibility of pairing multi-dimensional chromatin profiling with the addition of precise genetic location on the same chromosomes77. The optical identification of these molecules in micro or nanofluidic systems can also be combined with sorting78,79, to collect fragments carrying selected epigenetic marks or combinations of histone and base modifications. In an analogy to ChIP-Seq, the DNA of these collected fragments could be amplified and sequenced to provide a multi-dimensional genomic map of selected modifications. As with ChIP-Seq, the exact genetic location of the modifications is limited by the length of DNA in the analyzed fragment and is not limited by optical resolution.
Solid-state nanopore-based sensing offers a means to electrically detect nucleosomes and sub-nucleosomal structures80. Figure 3d depicts passage of five different molecular complexes (histone monomer, tetramer, and octamer, nucleosome and dinucleosome) through a ∼20nm nanopore where each translocating complex could be differentiated by molecular size (larger molecular sizes showed longer translocation times and deeper conductance blockades). This approach may be extensible to mapping the positions of nucleosomes on long chromatin fragments, as well as monitor the influence of other DNA-binding proteins81,82,83, RNA polymerase84 and nucleosome remodeling enzymes85 on the measured chromatin landscape. A key limitation preventing this technique from high-throughput chromatin profiling is the spatial resolution, which may be ameliorated by reducing the translocation speed86, using atomically thin nanopore membranes87 and performing multiple measurements on each molecule88,89 for signal averaging. Another restriction limiting these types of experiments is appropriate signal processing and characterization, where the complex topology of the chromatin and interaction with the pore manifests aperiodic signals surrounded by a noisy background. Accordingly, tools such as support vector machines90 and change detection91,92 algorithms may prove invaluable to these types of studies.
These device innovations and methods offer potential to significantly change chromatin analysis. Microfluidic devices stand to further automate the conventional multistep ChIP sample preparation and offer enhanced performance with lower sample loss and from fewer cells. As these microfluidic devices can also pair with several of the mapping and single molecule detection methods, the input cell number requirement may drop further to single cells. Because the single-molecule methods can directly profile DNA and histone modifications and nucleosome positions, the possibility for multi-dimensional chromatin profiling76 also becomes tangible. A key element to expand this analysis is to combine the measured histone and base modifications with sequence data77 for comprehensive views of genetic location and epigenetic state. A novel recent study paired proximity ligation with in-situ hybridization to determine the presence of histone modifications on specific loci in tissue cross-sections93. While this approach could detect a single locus, extension to multiple loci (> 100) with high spatial localization may be challenging, owing to overlapping signals within a diffraction limited spot. In contrast to conventional mapping technologies such as ChIP-Seq, where millions or billions of reads are mapped back to their sequences of origin and annotated for a single type of modification or transcription factor, these technologies aim to generate maps of multiple chromatin features in the same experiment.
Understanding chromatin dynamics and organisation
Conventional ensemble technologies such as BSC-Seq and ChIP-Seq have been successful at mapping some aspects of epigenetic modifications of chromosomes, but do not address the structural effects of their presence. To dissect these subtle but critical forces that influence chromatin structure and activity, techniques such as optical and magnetic tweezers and atomic force microscopy have been used94. These methods have provided mechanistic insights into forces and structures that influence dynamic chromatin processes such as nucleosome assembly95,96 chromatin remodeling97 and higher-order chromatin organization. Using these insights, novel mechanical models can be constructed to characterize how DNA is packaged into chromatin and forms loops and folds to accommodate transcription factors and chromatin remodeling enzymes and how these displacements propagate within the nucleus during different activities.
At a fundamental level, when DNA wraps around the histone octamer and forms a nucleosome, several points of contact form between the two. The locations of contact can be determined using crystallography but the strength of those contact points cannot be measured. Sensitive force spectroscopy techniques such as optical and magnetic tweezers can measure these contact locations and forces and gauge them dynamically98. Optical tweezers operate by trapping a small dielectric particle with a focused laser beam and monitoring its displacement using a second laser probe beam99. When a single chromatin fibre is attached to the bead and the other end is attached to another bead or surface such as a cover slide or micropipette, small displacements (and forces) imposed on or by the fibre can be measured100,101. The high force-sensitivity (∼1 pN) of optical tweezers permits measurement of the strength and location of histone-DNA interactions on a single nucleosome102 and the associative forces between the histone octamer and DNA103 during interactions with RNA polymerase104 or a chromatin remodeler (SWI/SNF)105. Figure 4a shows an optical tweezers experiment where a single nucleosome was tracked on a short template of DNA as it cyclically wrapped and unwrapped106. This elegant study was able to track the path of an RNA Polymerase II complex as it transcribed DNA through nucleosomes containing modifications on the histone tails or histone-DNA contacts and showed differential effects for the nucleosomal barrier to transcription elongation.
Figure 4.
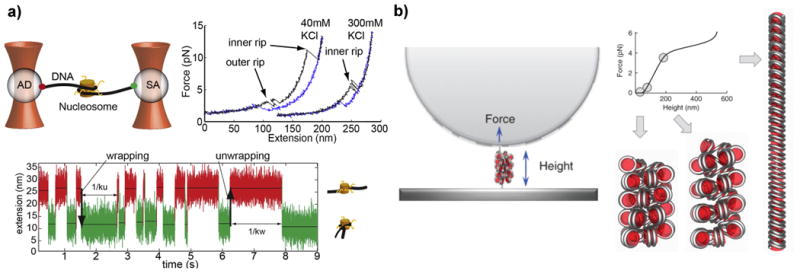
Micro and nano methods for understanding chromatin dynamics and chromosome organization. a) Optical tweezers experiment on a single nucleosome and force extension curves of the nucleosome in different salt concentrations106. The lower panel shows the nucleosome flipping between a wrapped and unwrapped state. b) Magnetic tweezers setup (left panel), where a single heterochromatin fibre can be pulled apart into different conformations (right panel)107. The different points on the force-extension curve show as the pulling force is increased, the fibre begins to unravel in a manner similar to a Hookian spring (solenoid shape), which keeps the DNA both condensed and accessible.
Magnetic tweezers employ a pair of macroscopic magnets to create a magnetic force gradient on a small magnetic bead that is tethered to a single molecule. The single molecule is attached at its end to a glass slide and manipulation of the position of the magnets induces a stretching force. Magnetic tweezers possess higher force sensitivity (10-2 pN) than optical tweezers and figure 4b illustrates an experiment where magnetic tweezers were used to stretch a single heterochromatin fibre107. By precisely displacing the chromatin fibre and measuring the resistive force, the chromatin fibre was observed to behave as a Hookian spring with distinct conformations for different forces. The compliance of the spring was found to be remarkably low, which facilitates high flexibility and extension (> 20%). The Hookian spring-like behavior offered several insights about chromatin structure, one of which is heterochromatin is organized into a solenoid shape, with stacks of nucleosomes in a helical structure that keep the DNA both highly compacted yet accessible. This result underscores how single molecule force spectroscopy assays can reveal insights into not only chromatin shape and packaging within the nucleus108, but also how these structural features dynamically evolve during interactions with chromatin remodeling complexes109,110.
Outlook
Recognition of the importance of epigenetic modifications is creating demand for new methods to rapidly obtain more complex information on the epigenetic state of cells. This demand is being met in part by the use and exploration of micro and nano devices. Nanometre scale devices and structures can access information at the molecular scale, and microdevices connect to the dimensions of cells and facilitate integration, miniaturization and automation. The technologies discussed above provide a pathway to more expeditiously obtain richer information about the epigenetic state of individual chromosomes and cells. This will enhance the ability to address dynamic processes such as acute or gradual cellular trends and decipher complex epigenetic relationships in biology. Coupled with technological advances to measure these complex chromatin topologies, new and innovative methods of statistical analysis111 are also required to interpret how these interdependent networks with varied types of interactions evolve through time.
The integration of micro and nano devices allows the scalability to perform epigenetic profiling from precisely selected samples and potentially from individual selected cells. Methods have been developed over the last decade to isolate single cells from an ensemble using miniaturized systems that take advantage of novel physical phenomena and intrinsic cellular properties112. These microfluidic isolation techniques can be combined with new approaches to perform extraction of intact chromosomes113,114 from a single cell115, partitioning of each chromosome into a small volume chamber116 and interrogation using one or several of the micro/nano-based techniques described above, followed by sorting specific molecules of interest78,79 and sequencing the extracted samples (Figure 5). Microfluidic integration also permits streamlined assays with precisely controlled inputs such as concentration gradients, temperature changes and serial (or parallel) introduction of stimuli. One may be able to select particular cells from controlled environments or precisely sample portions of tissues or tumours. This could herald new approaches to further dissect the roles of the large number of interacting elements117 that participate in biological function and provide mechanistic understanding of how chromatin interacts with these elements during events such as metastasis, inflammation and infection, which would greatly enhance our ability to identify, track and treat diseases.
Figure 5.
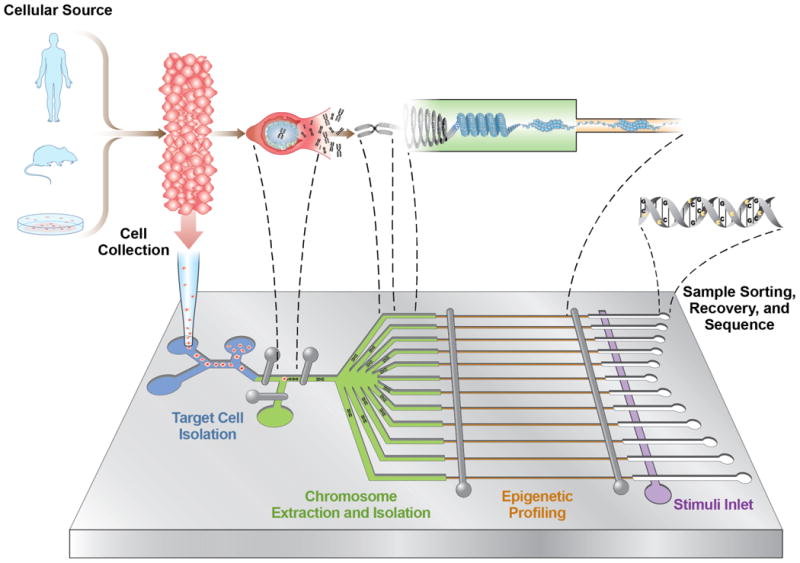
Possible hybrid micro and nanodevice architecture to perform multiplexed epigenomic measurements from a single cell. In the first stage of the device (blue channels), specific cells of interest are microfluidically isolated112 from a population and a single cell is trapped in a small-volume chamber. Next, in-tact chromosomes are extracted from the cell113,114,115 and partitioned (green channels) into chambers where each is profiled for multiple epigenetic modifications using one or several of the micro/nano-based techniques (orange channels). Once profiled, specific molecules of interest can then be sorted78,79 and recovered (white channels) for amplification or sequencing. The integrated architecture can also include a stimulus inlet (purple channel) to introduce factors such as chromatin remodelers to track how different molecules interact with and modulate the chromosome architecture.
One of the emerging themes of epigenetic studies is that specific types and patterns of modifications do not work locally and in isolation, but rather globally and in cooperation. For example, cytosine methylation patterns have been found to depend on the nucleosome architecture118 and histone modifications119, indicating potential cross-talk120 between enzymes that chemically modify DNA and chromatin, chromatin structure and the underlying DNA sequences. This unique combinatory schema enables the cell to exert coarse and fine temporal and spatial control over gene expression, whereby past, current or future transcription is precisely mediated. Thus, an individual cell can ‘prime’ the chromatin into a ‘poised’ state in anticipation of an event (such as differentiation121 or progression into a disease state) as well as tune or maintain transcriptional output using multiple epigenetic modifications. As more precise understanding of the functions and roles of the epigenetic layers advances, our ability to predict, read and manipulate122 cellular decisions and memories through infection123, disease124 and various environmental stimuli125, will ultimately provide foundations to discover and engineer new diagnostics and therapeutics.
Acknowledgments
The authors would like to thank Rohit Karnik, James Harper, Casey Gifford and Alexander Meissner for insightful discussions regarding this manuscript and Newton Taylor for assistance with artwork. H. G. C. acknowledges support from the National Institutes of Health grants R01 HG006850-01, U54 CA143876-03 and R01 DA030329-03. C.A.A. acknowledges support and sponsorhip by the Department of the Air Force under Air Force Contract #FA8721-05-C-0002. Opinions, interpretations, recommendations and conclusions are those of the authors and are not necessarily endorsed by the United States Government.
Footnotes
Competing Financial Interests: H.G.C. is a co-founder of Pacific Biosciences, Inc. and Odyssey Scientific, Inc.
Summary of Paper for Website: This article reviews recent advances in the use of micro- and nanoscale devices for studying epigenetic modifications, including covalent DNA modifications, differences in histone proteins and higher-order chromatin structures.
Contributor Information
Carlos A. Aguilar, Email: carlos.aguilar@ll.mit.edu.
Harold G. Craighead, Email: hgc1@cornell.edu.
References
- 1.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 2.Portela A, Esteller M. Epigenetic modifications and human disease. Nature Biotech. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 3.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 4.Segal E, et al. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schones D, Zhao K. Genome-wide approaches to studying chromatin modifications. Nature Rev Genet. 2008;9:179–191. doi: 10.1038/nrg2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 9.Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103:263–271. doi: 10.1016/s0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 10.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bednar J, et al. Nucleosomes, linker DNA and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc Nat'l Acad Sci USA. 1998;95:14173–14178. doi: 10.1073/pnas.95.24.14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohammad HP, Baylin SB. Linking cell signaling and the epigenetic machinery. Nature Biotech. 2010;28:1033–1038. doi: 10.1038/nbt1010-1033. [DOI] [PubMed] [Google Scholar]
- 14.Simonis M, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nature Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Z, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nature Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 16.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nature Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 17.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 18.Zhu J, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang F, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;3:75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chi P, Allis CD, Wang GG. Covalent histone modifications – miswritten, misinterpreted and mis-erased in human cancers. Nature Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fraga MF, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 23.Fudenberg G, Getz G, Meyerson M, Mirny L. High order chromatin architecture shapes the landscape of chromosomal alterations in cancer. Nature Biotech. 2011;29:1109–1113. doi: 10.1038/nbt.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gu H, et al. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nature Meth. 2010;7:133–136. doi: 10.1038/nmeth.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park PJ. ChIP-Seq: advantages and challenges of a maturing technology. Nature Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pujadas E, Feinberg AP. Regulated noise in the epigenetic landscape of development and disease. Cell. 2012;148:1123–1131. doi: 10.1016/j.cell.2012.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goren A, et al. Chromatin profiling by directly sequencing small quantities of immunoprecipitated DNA. Nature Meth. 2010;7:47–49. doi: 10.1038/nmeth.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stott SL, et al. Isolation and characterization of circulating tumor cells from patients with localized metastatic prostate cancer. Sci Transl Med. 2010;2:25ra23. doi: 10.1126/scitranslmed.3000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fanelli M, et al. Pathology tissue-chromatin immunoprecipitation, coupled with high-throughput sequencing, allows the epigenetic profiling of patient samples. Proc Nat'l Acad Sci USA. 2010;107:21535–21540. doi: 10.1073/pnas.1007647107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quake SR, Scherer A. From micro- to nanofabrication with soft materials. Science. 2000;290:1536–1540. doi: 10.1126/science.290.5496.1536. [DOI] [PubMed] [Google Scholar]
- 32.Qin D, Xia Y, Whitesides GM. Soft lithography for micro- and nanoscale patterning. Nature Protoc. 2010;5:491–502. doi: 10.1038/nprot.2009.234. This article illustrates different techniques for fabricating various kinds of micro and nanoscale devices that can be used for evaluating different kinds of epigenetic modifications. [DOI] [PubMed] [Google Scholar]
- 33.Craighead H. Future lab-on-a-chip technologies for interrogating individual molecules. Nature. 2006;442:387–393. doi: 10.1038/nature05061. [DOI] [PubMed] [Google Scholar]
- 34.Liu Y, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nature Biotech. 2013;31:142–147. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robertson KD. DNA methylation and human disease. Nature Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 36.Laird PW. Principles and challenges of genome-wide DNA methylation analysis. Nature Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 37.Levene MJ, et al. Zero-mode waveguides for single-molecule analysis at high concentrations. Science. 2003;299:682–686. doi: 10.1126/science.1079700. [DOI] [PubMed] [Google Scholar]
- 38.Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 39.Flusberg BA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nature Meth. 2010;7:461–465. doi: 10.1038/nmeth.1459. This article describes direct detection of DNA methylation, without bisulfite conversion, by monitoring a single molecule sequencing reaction within a nanophotonic waveguide. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song CX, et al. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nature Meth. 2012;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang G, et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nature Biotech. 2012;30:1232–1239. doi: 10.1038/nbt.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ratel D, Ravanat JL, Berger F, Wion D. N6-methyladenine: the other methylated base of DNA. Bioessays. 2006;28:309–315. doi: 10.1002/bies.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krueger F, Kreck B, Franke A, Andrews SR. DNA methylome analysis using short bisulfite sequencing data. Nature Meth. 2012;9:145–151. doi: 10.1038/nmeth.1828. [DOI] [PubMed] [Google Scholar]
- 45.Austin RH, Brody JP, Cox EC, Duke T, Volkmuth W. Stretch genes. Phys Today. 1997;50:32–38. [Google Scholar]
- 46.Bensimon A, et al. Alignment and sensitive detection of DNA by a moving interface. Science. 1994;265:2096–2098. doi: 10.1126/science.7522347. [DOI] [PubMed] [Google Scholar]
- 47.Cerf A, Alava T, Barton RA, Craighead HG. Transfer-printing of single DNA molecule arrays on graphene for high-resolution electron imaging and analysis. Nano Lett. 2011;11:4232–4238. doi: 10.1021/nl202219w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Streng DE, Lim SF, Pan J, Karpusenka A, Riehn R. Stretching chromatin through confinement. Lab on a Chip. 2009;9:2772–2774. doi: 10.1039/b909217j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tegenfeldt JO, et al. The dynamics of genomic-length DNA molecules in 100-nm channels. Proc Nat'l Acad Sci USA. 2004;101:10979–10983. doi: 10.1073/pnas.0403849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim SF, et al. DNA methylation profiling in nanochannels. Biomicrofluidics. 2011;5:034106. doi: 10.1063/1.3613671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cerf A, Cipriany BR, Benitez JJ, Craighead HG. Single DNA molecule patterning for high-throughput epigenetic mapping. Anal Chem. 2011;83:8073–8077. doi: 10.1021/ac202506j. This article reports a technique to pattern and optically map large-scale arrays of single molecules in an extended form to detect and map DNA cytosine methylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y, Reinhart WF, Tree DR, Dorfman KD. Resolution limit for DNA barcodes in the Odijk regime. Biomicrofluidics. 2012;6:014101. doi: 10.1063/1.3672691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 54.Wallace EVB, et al. Identification of epigenetic DNA modifications with a protein nanopore. Chem Commun. 2010;46:8195–8197. doi: 10.1039/c0cc02864a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mirsaidov U, et al. Nanoelectromechanics of methylated DNA in a synthetic nanopore. Biophys J. 2009;96:L32–L34. doi: 10.1016/j.bpj.2008.12.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venkatesan BM, Bashir R. Nanopore sensors for nucleic acid analysis. Nature Nano. 2011;6:615–624. doi: 10.1038/nnano.2011.129. [DOI] [PubMed] [Google Scholar]
- 57.Wanunu M, et al. Discrimination of methylcytosine from hydroxmethylcytosine in DNA molecules. J Amer Chem Soc. 2011;133:486–492. doi: 10.1021/ja107836t. This article describes fabrication and testing of a solid-state nanopore device to differentiate several types of DNA covalent modifications in a label-free, rapid manner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shim J, et al. Detection and quantification of methylation in DNA using solid-state nanopores. Sci Rep. 2013;3:1389. doi: 10.1038/srep01389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cherf GM, et al. Automated forward and reverse ratcheting of DNA in a nanopore at 5-angstrom precision. Nature Biotech. 2012;30:344–348. doi: 10.1038/nbt.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manrao EA, et al. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nature Biotech. 2012;30:349–353. doi: 10.1038/nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Branton D, et al. The potential and challenges of nanopore sequencing. Nature Biotech. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goren RB, et al. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nature Protoc. 2013;8:539–554. doi: 10.1038/nprot.2013.023. [DOI] [PubMed] [Google Scholar]
- 63.Wu AR, et al. Automated microfluidic chromatin immunoprecipitation from 2,000 cells. Lab on a Chip. 2009;9:1365–1370. doi: 10.1039/b819648f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Geng T, et al. Histone modification analysis by chromatin immunoprecipitation from a low number of cells on a microfluidic platform. Lab on a Chip. 2011;11:2842–2848. doi: 10.1039/c1lc20253g. [DOI] [PubMed] [Google Scholar]
- 65.Thorsen T, Maerkl SJ, Quake SR. Microfluidic large-scale integration. Science. 2002;298:580–584. doi: 10.1126/science.1076996. [DOI] [PubMed] [Google Scholar]
- 66.Tseng Q, Lomonosov AM, Furlong EE, Merten CA. Fragmentation of DNA in a sub-microliter microfluidic sonication device. Lab on a Chip. 2012;12:4677–4682. doi: 10.1039/c2lc40595d. [DOI] [PubMed] [Google Scholar]
- 67.Shui L, Bomer JG, Jin M, Carlen ET, van den Berg A. Microfluidic DNA fragmentation for on-chip genomic analysis. Nanotech. 2011;22:494013. doi: 10.1088/0957-4484/22/49/494013. [DOI] [PubMed] [Google Scholar]
- 68.Wu AR, et al. High throughput automated chromatin immunoprecipitation as a platform for drug screening and antibody validation. Lab on a Chip. 2012;12:2190–2198. doi: 10.1039/c2lc21290k. This article describes construction of an automated microfluidic device to perform ChIP from small cell numbers and with high reproducibility in a fast manner (hrs) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cipriany BR, et al. Single molecule epigenetic analysis in a nanofluidic channel. Anal Chem. 2010;82:2480–2487. doi: 10.1021/ac9028642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsuoka T, et al. Nanoscale squeezing in elastomeric nanochannels for single chromatin linearization. Nano Lett. 2012;12:6480–6484. doi: 10.1021/nl304063f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cerf A, Tian HC, Craighead HG. Ordered arrays of native chromatin molecules for high-resolution imaging and analysis. ACS Nano. 2012;6:7928–7934. doi: 10.1021/nn3023624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fazio T, Visnapuu ML, Wind S, Greene EC. DNA curtains and nanoscale curtain rods: high-throughput tools for single molecule imaging. Langmuir. 2008;24:10524–10531. doi: 10.1021/la801762h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Visnapuu ML, Greene EC. Single-molecule imaging of DNA curtains reveals intrinsic energy landscapes for nucleosome deposition. Nature Struct & Molec Biol. 2009;16:1056–1062. doi: 10.1038/nsmb.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang YM, et al. Single-molecule studies of repressor-DNA interactions show long-range interactions. Proc Nat'l Acad Sci USA. 2005;102:9796–9801. doi: 10.1073/pnas.0502917102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang YM, Tegenfeldt JO, Sturm J, Austin RH. Long-range interactions between transcription factors. Nanotech. 2005;16:1993–1999. doi: 10.1088/0957-4484/16/10/003. [DOI] [PubMed] [Google Scholar]
- 76.Murphy PM, et al. Single-molecule analysis of combinatorial epigenomic states in normal and tumor cells. Proc Nat'l Acad Sci USA. 2013;110:7772–7777. doi: 10.1073/pnas.1218495110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marie R, et al. Integrated view of genome structure and sequence of a single DNA molecule in a nanofluidic device. Proc Nat'l Acad Sci USA. 2013;110:4893–4898. doi: 10.1073/pnas.1214570110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cipriany BR, et al. Real-time analysis and selection of methylated DNA by fluorescence-activated single molecule sorting in a nanofluidic channel. Proc Nat'l Acad Sci USA. 2012;109:8477–8482. doi: 10.1073/pnas.1117549109. This article reports the fabrication of a nanofluidic device that can identify different kinds of epigenetic modifications on single chromatin fragments and sort them into different compartments based on the bound modifications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yamamoto T, Fujii T. Nanofluidic single-molecule sorting of DNA: a new concept in separation and analysis of biomolecules towards ultimate level of performance. Nanotech. 2010;21:395502. doi: 10.1088/0957-4484/21/39/395502. [DOI] [PubMed] [Google Scholar]
- 80.Soni GV, Dekker C. Detection of nucleosomal substructures using solid-state nanopores. Nano Lett. 2012;12:3180–3186. doi: 10.1021/nl301163m. [DOI] [PubMed] [Google Scholar]
- 81.Kowalcyzk SW, Hall AR, Dekker C. Detection of local protein structures along DNA using solid-state nanopores. Nano Lett. 2010;10:324–328. doi: 10.1021/nl903631m. [DOI] [PubMed] [Google Scholar]
- 82.Hornblower B, et al. Single-molecule analysis of DNA-protein complexes using nanopores. Nature Meth. 2007;4:315–317. doi: 10.1038/nmeth1021. [DOI] [PubMed] [Google Scholar]
- 83.Venkatesan B, et al. Stacked graphene-Al2O3 nanopore sensors for sensitive detection of DNA and DNA-protein complexes. ACS Nano. 2012;6:441–450. doi: 10.1021/nn203769e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raillon C, et al. Nanopore detection of single molecule RNAP-DNA transcription complex. Nano Lett. 2012;12:1157–1164. doi: 10.1021/nl3002827. [DOI] [PubMed] [Google Scholar]
- 85.Struhl K, Segal E. Determinants of nucleosome positioning. Nature Struc & Molec Biol. 2013;20:267–273. doi: 10.1038/nsmb.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Plesa C, et al. Fast translocation of proteins through solid-state nanopores. Nano Lett. 2013;13:658–663. doi: 10.1021/nl3042678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Garaj S, et al. Graphene as a subnanometre trans-electrode. Nature. 2010;467:190–193. doi: 10.1038/nature09379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sen YH, Jain T, Aguilar CA, Karnik R. Enhanced discrimination of DNA molecules in nanofluidic channels through multiple measurements. Lab on a Chip. 2012;12:1094–1101. doi: 10.1039/c2lc20771k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gershow M, Golovchenko JA. Recapturing and trapping single molecules with a solid-state nanopore. Nature Nanotech. 2007;2:775–779. doi: 10.1038/nnano.2007.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Winters-Hilt S, et al. Highly accurate classification of Watson-crick basepairs on termini of single DNA molecules. Biophys J. 2003;84:967–976. doi: 10.1016/S0006-3495(03)74913-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Raillon C, Granjon P, Graf M, Steinbock LJ, Radenovic A. Fast and automatic processing of multi-level events in nanopore translocation experiments. Nanoscale. 2012;4:4916–4924. doi: 10.1039/c2nr30951c. [DOI] [PubMed] [Google Scholar]
- 92.Pedone D, Firnkes M, Rant U. Data analysis of translocation events in nanopore experiments. Anal Chem. 2009;81:9689–9694. doi: 10.1021/ac901877z. [DOI] [PubMed] [Google Scholar]
- 93.Gomez D, Shankman LS, Nguyen AT, Owens GK. Detection of histone modifications at specific gene loci in single cells in histological sections. Nature Meth. 2013;10:171–177. doi: 10.1038/nmeth.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Neuman KC, Nagy A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nature Meth. 2008;5:491–505. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Killian JL, Li M, Sheinin MY, Wang MD. Recent advances in single molecule studies of nucleosomes. Curr Op Struct Biol. 2012;22:80–87. doi: 10.1016/j.sbi.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Simon M, et al. Histone fold modifications control nucleosome unwrapping and disassembly. Proc Nat'l Acad Sci USA. 2011;108:12711–12716. doi: 10.1073/pnas.1106264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cairns BR. Chromatin remodeling: insights and intrigue from single molecule studies. Nature Struc Mol Biol. 2007;14:989–996. doi: 10.1038/nsmb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dulin D, Lipfert J, Moolman C, Dekker N. Studying genomic processes at the single-molecule level: introducing the tools and applications. Nature Rev Genet. 2013;14:9–22. doi: 10.1038/nrg3316. [DOI] [PubMed] [Google Scholar]
- 99.Fazal FM, Block SM. Optical tweezers study life under tension. Nature Photon. 2011;5:318–321. doi: 10.1038/nphoton.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cui Y, Bustamante C. Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc Nat'l Acad Sci USA. 2000;97:127–132. doi: 10.1073/pnas.97.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bennink ML, et al. Unfolding individual nucleosomes by stretching single chromatin fibers with optical tweezers. Nature Struc Mol Biol. 2001;8:606–610. doi: 10.1038/89646. [DOI] [PubMed] [Google Scholar]
- 102.Hall MA, et al. High resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nature Struc Mol Biol. 2009;16:124–129. doi: 10.1038/nsmb.1526. This article described the use of optical tweezers to generate a comprehensive map of histone-DNA interactions on a single nucleosome and showed a novel periodicity in contact strength with several broad regions of strong contact. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mihardja S, Spakowitz AJ, Zhang Y, Bustamante C. Effect of force on mononucleosomal dynamics. Proc Nat'l Acad Sci USA. 2006;103:15871–15876. doi: 10.1073/pnas.0607526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jin J, et al. Synergistic action of RNA polymerases in overcoming the nucleosomal barrier. Nature Struc Mol Biol. 2010;17:745–752. doi: 10.1038/nsmb.1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shundrovsky A, Smith CL, Lis JT, Peterson CL, Wang MD. Probing SWI/SNF remodeling of the nucleosome by unzipping single DNA molecules. Nature Struc Mol Biol. 2006;13:549–554. doi: 10.1038/nsmb1102. [DOI] [PubMed] [Google Scholar]
- 106.Bintu L, et al. Nucleosomal elements that control the topography of the barrier to transcription. Cell. 2012;151:738–749. doi: 10.1016/j.cell.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kruithof M, et al. Single-molecule force spectroscopy reveals a highly compliant helical folding for the 30-nm chromatin fiber. Nature Struc Mol Biol. 2009;16:534–540. doi: 10.1038/nsmb.1590. This article used magnetic tweezers to pull on a single heterochromatin fibre and determined the molecule behaved similar to a Hookian spring with low compliance, which indicated the DNA can be kept both highly compacted and accessible. [DOI] [PubMed] [Google Scholar]
- 108.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Das C, Tyler JK, Churchill ME. The histone shuffle: histone chaperones in an energetic dance. Trends Biochem Sci. 2010;35:476–489. doi: 10.1016/j.tibs.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vlijm R, Smitshuijzen JSJ, Lusser A, Dekker C. NAP1-assisted nucleosome assembly on DNA measured in real time by single-molecule magnetic tweezers. PLoS ONE. 2012;7:e46306. doi: 10.1371/journal.pone.0046306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nature Rev Genet. 2010;11:476–486. doi: 10.1038/nrg2795. This article describes strategies to analyze, visualize, manipulate and interpret data for different types of genomics experiments in an integrated way. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lenshof A, Laurell T. Continuous separation of cells and particles in microfluidic systems. Chem Soc Rev. 2010;39:1203–1217. doi: 10.1039/b915999c. [DOI] [PubMed] [Google Scholar]
- 113.Rasmussen KH, et al. A device for extraction, manipulation and stretching of DNA from single human chromosomes. Lab on a Chip. 2011;11:1431–1433. doi: 10.1039/c0lc00603c. [DOI] [PubMed] [Google Scholar]
- 114.Fan HC, Wang J, Potanina A, Quake SR. Whole-genome molecular haplotyping of single cells. Nature Biotech. 2011;29:51–57. doi: 10.1038/nbt.1739. This article describes a microfluidic device capable of trapping a single cell and isolating each chromosome in a small chamber for haplotyping. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Benitez JJ, et al. Microfluidic extraction, stretching and analysis of human chromosomal DNA from single cells. Lab on a Chip. 2012;12:4848–4854. doi: 10.1039/c2lc40955k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pelletier J, et al. Physical manipulation of the Escherichia coli chromosome reveals its soft nature. Proc Nat'l Acad Sci USA. 2012;109:E2649–2656. doi: 10.1073/pnas.1208689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ramakrishna RK, et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cedar H, Bergman Y. Linking DNA methylation and histone modifications: patterns and paradigms. Nature Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 120.Brinkman AB, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA-methylation cross-talk. Genome Res. 2012;22:1128–1138. doi: 10.1101/gr.133728.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gifford CA, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1–15. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Haynes KA, Silver PA. Synthetic reversal of epigenetic silencing. J Biol Chem. 2011;286:27176–27182. doi: 10.1074/jbc.C111.229567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hamon MA, Cossart P. Histone modifications and chromatin remodeling during bacterial infections. Cell Host & Microbe. 2008;4:100–109. doi: 10.1016/j.chom.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 124.Maunakea AK, Chepelev I, Zhao K. Epigenome mapping in normal and disease states. Circ Res. 2010;107:327–339. doi: 10.1161/CIRCRESAHA.110.222463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nature Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]