

SUMMARY
Tau, the microtubule‐associated protein, forms insoluble filaments that accumulate as neurofibrillary tangles in Alzheimer's disease (AD) and related tauopathies. Under physiological conditions, tau regulates the assembly and maintenance of the structural stability of microtubules. In the diseased brain, however, tau becomes abnormally hyperphosphorylated, which ultimately causes the microtubules to disassemble, and the free tau molecules aggregate into paired helical filaments. A large body of evidence suggests that tau hyperphosphorylation results from perturbation of cellular signaling, mainly through imbalance in the activities of different protein kinases and phosphatases. In AD, it appears that ß‐amyloid peptide (Aß) plays a pivotal role in triggering this imbalance. In this review, we summarize our current understanding of the role of tau in AD and other tauopathies, and highlight key issues that need to be addressed to improve the success of developing novel therapies.
Keywords: Alzheimer’s disease, ß‐Amyloid, Hyperphosphorylation, Kinase, Neurofibrillary tangles, Neuron, Phosphatase, Tau protein, Tauopathies
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder associated with memory loss, spatial disorientation, and gradual deterioration of intellectual capacity. Numerous pathological changes have been described in the postmortem brains of AD patients, including synaptic and neuronal loss, oxidative damage, activated inflammatory cells, amyloid plaques mainly composed of the ß‐amyloid peptide (Aß), and neurofibrillary tangles (NFTs) comprised of hyperphosphorylated aggregates of the microtubule‐associated protein tau, the latter two of which are considered the pathological hallmarks [1, 2, 3]. For several reasons, research on the involvement of Aß in AD has progressed more quickly than that on tau. The description of the “amyloid cascade hypothesis” based on the discovery of genetic mutations that cause autosomal familial AD centered the focus of research on Aß[4, 5]. Also, the biochemical studies of amyloid precursor protein (APP) and the presenilins have greatly enhanced the understanding of the molecular pathways leading to Aß generation [6]. These studies favored the systematic development of disease‐modifying therapies based on Aß pathway.
Many clinical trials are currently underway that are evaluating the efficacy of targeting some aspect of Aß biology (i.e., genesis, aggregation, clearance, etc.) in AD patients. Although it is too early to know the outcome of these trials, there is justifiable concern that targeting Aß in patients with AD, even in those with milder stages of the disease, may be insufficient because of the numerous pathways and resultant damage triggered by the accumulation of Aß. Eventually, the field will progress to the point that multiple/combination therapies are evaluated in clinical trials. In the meantime, a challenge for ongoing preclinical studies involving animal models is to identify the most promising therapeutic combinations.
Given its longstanding and prominent role in the molecular pathology of AD, there has been surprisingly scant attention focused on tau as a therapeutic target, which is reflected by the large disparity in clinical trials that target Aß versus tau (see http://www.clinicaltrials.gov). Advances over the past decade in the generating animal models with tau pathology are helping to erase this deficit (Figure 1). In this review, we will consider the existing evidence validating tau as a therapeutic target for AD.
Figure 1.

Key discoveries in tau protein research.
Molecular Biology of Protein Tau
The discovery that mutations in the microtubule‐associated protein tau (MAPT) gene cause fronto‐temporal dementia with Parkinsonism linked to chromosome 17 (FTLD‐17) was a watershed discovery for the field, and provided genetic evidence that established that dysfunction in tau was sufficient to trigger neurodegeneration, in the absence of Aß[7, 8, 9]. Several CNS disorders—AD, Pick's disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and argyrophilic grain disease—are characterized by the aberrant accumulation of tau [10]. Hence, developing targets against tau offers the added extrinsic benefit of not only being useful for AD, but also for the larger class of tauopathies.
In the adult human brain, six isoforms of tau are expressed, generated through alternative splicing of the MAPT gene located on chromosome 17q21 [11]. Alternative splicing of exons 2 and 3 yields variants containing zero (0N), one (1N), or two (2N) inserts at the N‐terminus. In addition, the absence or presence of exon 10 leads to tau proteins that contain either three (3R) or four (4R) C‐terminal microtubule‐binding domains, with 3R tau isoforms binding the microtubules less tightly than 4R tau [12]. Hence, six tau isoforms are possible: 3R0N, 3R1N, 3R2N, 4R0N, 4R1N, and 4R2N. In the normal human brain, the expression levels of 3R and 4R tau occur at a 1:1 ratio. Importantly, several tauopathies are characterized by the alteration in the ratio of 3R:4R tau. For instance, some FTDP‐17 families with mutations around exon 10 have an increased proportion of 4R tau, thereby exacerbating the ability of tau to interact with microtubules [7, 8, 9].
The classically described function of tau is as a neuronal microtubule‐associated protein, mainly found in axons. Under physiological conditions, tau exists as a highly soluble and natively unfolded protein that interacts with tubulin and promotes its assembly into microtubules, which helps to stabilize their structure [13]. Recent evidence points to additional functions for tau. For example, tau phosphorylation enables neurons to escape from an acute apoptotic death through stabilizing ß‐catenin [14]. Also, tau exerts an essential role in the balance of microtubule‐dependent axonal transport of organelles and biomolecules by modulating the anterograde transport by kinesin and the dynein‐driven retrograde transport [15, 16]. Mechanistic understanding of the role of tau, including the daunting challenge of elucidating the specific effects of phosphorylation at multiple sites on tau, will be extremely valuable for the future development of treatments for AD and other related disorders.
Tau is regulated during both normal homeostasis and in stress‐induced responses by an array of posttranslational modifications that includes glycosylation, ubiquitination, glycation, nitration, and oxidation. Of the posttranslational modifications, phosphorylation has been most extensively studied (Figure 2). In the healthy brain, 2–3 residues on tau are phosphorylated. In AD and other tauopathies, however, the phosphorylation level of tau is significantly higher, with approximately nine phosphates per molecule [17, 18]. Hyperphosphorylation of tau may occur at different putative serine, threonine, and tyrosine residues through a disruption in the equilibrium of tau kinases and tau phosphatases activity. The consequences of this phenomenon are still under investigation, but produces the outcome of lowering tau's affinity for the microtubules as well as increasing tau's resistance to calcium‐activated neutral proteases and its degradation by the ubiquitin‐proteosome pathway [19]. Ultimately, tau hyperphosphorylation leads to fibrillization and aggregation into NFTs [20, 21, 22, 23, 24]. The major tau kinases include glycogen‐synthase kinase‐3ß (GSK‐3ß), cyclin‐dependent protein kinase 5 (CDK5), cAMP‐dependent protein kinase (PKA), mitogen‐activated protein kinases (MAPK), calcium‐calmodulin‐dependent kinase‐II (CaMK II), and microtubule affinity‐regulating kinase (MARK) [25, 26, 27, 28, 29, 30]. Among the phosphatases, protein phosphatase 2 (PP2A) has been most implicated in the dephosphorylation of abnormal tau [31]. Notably, changes in the expression and/or activation of tau kinases and tau phosphatases have been well documented in AD and related disorders [32, 33, 34, 35]. Studies in transgenic mouse models of AD suggest that it is likely that multiple, overlapping processes contribute to abnormal hyperphosphorylation of tau, including Aß, impaired brain glucose metabolism, and inflammation [36, 37, 38, 39, 40, 41]. Understanding which cellular pathways mediate posttranslational modifications of tau is of high interest in the discovery of therapeutic targets.
Figure 2.
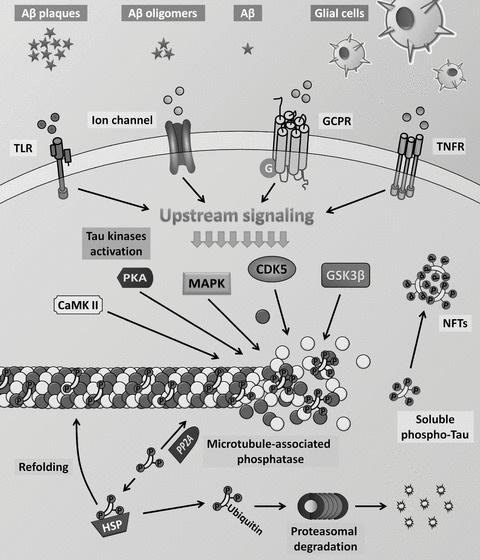
Molecular mechanisms of tau regulation.
Experimental Models of Tau
Since 1995, there has been significant progress in generating cellular and animal models, including using vertebrate and invertebrate species, to analyze and dissect the role of tau in AD and related tauopathies [42]. Because of space limitations, we will focus on transgenic models, and consider how tau transgenic approaches have contributed to the development of novel therapeutic strategies.
There are many studies addressing the pathological effects and functional consequences of human tau mutations, with the most commonly studied tau mutations being glycine residue 272 to valine (G272V), asparagine residue 279 to lysine (N279K), proline residue 301 to leucine (P301L), valine residue 337 to methionine (V337M), and arginine residue 406 to tryptophan (R406W) [43]. All these mutations markedly reduce the ability to promote microtubule assembly, presumably by promoting hyperphosphorylation of tau, followed by its assembly into filaments aggregates. The identification of key kinases and phosphatases that modulate pathological changes of tau in AD and related tauopathies has been another major focus of research. The majority of these studies concentrated on the role that GSK‐3ß and CDK5 play in phosphorylating tau. Double transgenic mouse models have been developed for mutant tau and GSK‐3ß or CDK5 [30, 44]. These bigenic models showed a dramatic increase in tau hyperphosphorylation and NFTs; moreover, the hippocampal atrophy observed in the GSK‐3ß single transgenic mice was accelerated in the tau/GSK‐3ß line [44]. Furthermore, crossing mutant PP2A and tau model resulted in a remarkable increase in the number of the hippocampal neurons expressing hyperphosphorylated tau and NFTs [45]. Together, these data highlight the critical role that certain kinases and phosphatases play in triggering key modifications in tau, and also suggest that these enzymes themselves may be a valuable therapeutic target. However, there are some major concerns about inhibiting kinases and phosphatases because of the plethora of targets they affect. Hence, convincing preclinical data will be required, including in higher order species like nonhuman primates, to ensure that adverse effects are minimized in human trials.
Because APP and/or presenilin mutant transgenic mice do not develop NFT pathology despite extensive plaque deposition, our laboratory adopted an aggressive genetic approach to produce a transgenic model with both plaques and tangles. These mice express the P301L mutation in tau as well as mutant APP and PS1 and are referred as triple transgenic model (3xTg‐AD) [46]. The 3xTg‐AD mice represent a critical resource for elucidating the relationship among Aß, tau, and synaptic dysfunction; they have also proven to be useful for evaluating the efficacy of novel therapeutic strategies in mitigating the neurodegenerative effects mediated by both AD signature lesions. These mice progressively develop Aß and tau pathology, with a temporal‐ and regional‐specific profile that closely mimics their development in the human AD brain [41]. Also, despite equivalent expression of the human APP and human tau transgenes, Aß deposition develops prior to the tangle pathology. Notably, the clearance of Aß using Aß‐specific antibodies reduces the tau burden in the 3xTg‐AD [41]. This is in accordance with studies from others that have shown that intracranial Aß injection in tau mutant mice [47] or coexpression of mutant APP and tau leads to tau pathology [48]. Such studies clearly indicate a link between Aß and tau pathology and suggest that Aß accumulation can exacerbate tau pathology in the course of AD.
The evidence that Aß induces tau phosphorylation has lead to the question about whether the modulation of tau would also influence Aß pathology. Recent study has shown that reduction of tau expression prevents the cognitive deficits in the mutant APP without changing the level of Aß[49]. Moreover, we have shown that genetically augmenting tau levels and hyperphosphorylation in the 3xTg‐AD mice has no effect on the onset and progression of Aß pathology [50]. Together, these studies suggest that the link between Aß and tau is predominantly if not exclusively unidirectional, which is consistent with the Aß cascade hypothesis and may explain why tauopathy‐only disorders are devoid of any Aß pathology.
Further important issues that were recently highlighted using different in vitro and in vivo models regard about how the different tau complexes (soluble oligomeric forms of phosphorylated tau vs. insoluble NFTs) and the extracellular tau may exert their toxicity in the brain. SantaCruz and colleagues [51] have shown that turning off expression of the mutant tau transgene, in a repressible tau transgenic mouse model, attenuates the neurodegeneration and improves memory, in spite of ongoing NFTs accumulation. Supporting this finding, it has been shown that inhibition of tau hyperphosphorylation in the tau P301L transgenic mouse model prevents severe motor deficits and reduces the amount of soluble tau aggregates without changes of NFTs [52]. Also, it has been shown that phosphorylation of soluble tau inhibits the assembly of microtubules by a process dependent on normal MAPs sequestration and results in dendrites and axonal degeneration [53, 54]. These studies strongly suggest that abnormal hyperphosphorylation of soluble forms of tau plays a critical role in the development of tauopathies and that accumulation of NFTs occur independent of memory loss as a possible neural protective mechanism. Concerning the accumulation of extracellular tau, it has been demonstrated by in vitro studies that tau aggregates can propagate a fibrillar, misfolded state from the outside to the inside of a cell [55]. The in vivo relevance of these data is still unknown, however, could represent a new component in the progression of tau dysfunction. Indeed, additional studies are necessary to access the mechanisms in which the different tau filaments and/or oligomeric forms as well as the extracellular tau aggregates affect the neuronal function and exert their toxicity. Strategies that neutralize the intracellular soluble tau forms and the extracellular tau aggregates may be an alternative therapeutic approach for AD and related tauopathies.
Numerous transgenic animal models of AD have been used as important tools for the characterization of abnormal APP processing and/or tau phosphorylation, as well as for the study of the mechanisms of cognitive impairment [42]. Although there is no doubt that these models have helped in unraveling disease processes and have boosted drug discovery and development, they may present an incomplete perspective of AD pathology since most of then employ mutant tau. At present, only a few studies have been developed using transgenic mouse models that express human wild‐type tau protein. Although these mice do not manifest NFTs, they reproduce some features of human pathology such as a strong somatodendritic hyperphosphorylated tau [56, 57, 58, 59, 60, 61]. In addition, Zilka and colleagues have shown that nonmutated truncated tau is an important upstream factor that induces neurofibrillary neurodegeneration of AD type in absence of tau mutation [62]. Future studies will be needed to identify conditions that are able to produce robust pathological states of tau pathology in human wild‐type tau transgenic models.
Tau As a Therapeutic Target
A prominent number of studies focusing on tau therapeutics are in progress. The general information about the principal therapeutics compounds used in the different strategies for tau is summarized in Table 1. Thus, the principal strategies targeting tau in neurodegenerative diseases are (1) to inhibit the abnormal tau hyperphosphorylation through modulation of specific protein kinases as GSK‐3ß, CDK5, casein kinase‐1 (CK‐1), PKA, CaMK II, and MAPK family (ERK1/2, p70S6 kinase, JNK, p38), (2) to induce disassembly of tau aggregates with compounds such as Methylene blue, Antharaquinones, etc., (3) to stimulate microtubule stabilizing molecule with Taxol and Taxol‐derived compounds (taxotere, paclitaxel), (4) to trigger intracellular clearance pathways such as the ubiquitin‐proteasome and/or autophagic system, (5) tau immunotherapy, and (6) antiinflammatory therapy.
Table 1.
Therapeutical tau progress
| Antiphosphorylation approaches | ||
|---|---|---|
| Cdk5 inhibitory peptide | Interferes with cdk5/p25 complex formation and inhibits abnormal tau phosphorylation in cortical neurons. | [92, 93] |
| Roscovitine | A cdk5 inhibitor that reduces tau phosphorylation and neurodegeneration in p25 transgenic mice. | [94, 95] |
| Lithium | A GSK3β inhibitor that prevents tau phosphorylation, aggregation and axonal degeneration in transgenic mice. Also, in culture cells lithium reduces tau phosphorylation and promotes microtubule assembly. | [96, 97, 98, 99, 100, 101] |
| AR‐A014418 | A GSK3β inhibitor that decreases insoluble and aggregate tau levels. | [102] |
| AF267B | A selective muscarinic M1 receptor agonist that decreases the level of tau phosphorylation through reduction of GSK3β activity. | [36] |
| A‐582941 | A selective α7‐nAChR agonist that stimulates phosphorylation of GSK3β in residue Ser‐9 and decreases tau phosphorylation in GSK3β‐sensitive and double tau‐APP model. | [103] |
| NP‐12 | A GSK3β inhibitor that reduces tau phosphorylation, neuronal loss in hippocampus and entorhinal cortex, improves spatial memory deficit, and reduces amyloid plaque load in mouse brain. | [104] |
| K252a | A serine/threonine protein kinase inhibitor that blocks PHF‐like tau hyperphosphorylation in culture of cells and rat brain slices. | [105] |
| SRN‐033–556 (K252a analog) | Prevents motor deficits and reduces soluble aggregated hyperphosphorylated tau in P301L and JNPL3 transgenic mouse model. | [52] |
| Memantine | Protects culture neurons against Aβ‐induced toxicity by attenuating tau phosphorylation and restores PP‐2A activity. Chronic memantine treatment reduces the accumulation and tau phosphorylation in the 3xTg‐AD mice. Also, P‐tau is reduced in CSF of AD patient after memantine treatment. | [63, 64, 106, 107] |
| Docosahexaenoic acid | Reduces the levels of early‐stage phospho‐tau epitopes through inhibition of c‐Jun N‐terminal kinase (JNK). | [69] |
| Antiaggregation studies | ||
| Methylene Blue | A noneuroleptic phenothiazine that reverses the proteolotic stability of tau aggregation and prevents the further propagation of tau capture in AD. | [70] |
| Anthraquinone | Inhibits tau aggregation and dissolves paired helical filaments (PHF) in vitro and culture assay. | [108] |
| Phenylthiazolyl‐Hydrazide | Inhibits tau aggregation and disassembles preformed aggregates. | [109] |
| Rhodanine | Inhibits tau aggregation and promotes paired helical filament (PHF) disassembly in cell model of tauopathy. | [110] |
| Microtubule‐stabilizing drugs | ||
| Taxol | Stimulates microtubule polymerization, stabilizes microtubules, and protects axonal microtubules from accumulating filaments. | [111] |
| Taxotere | Stimulates microtubule assembly and stabilization. Reverses the effect of tau on mitochondrial distribution. | [112] |
| Paclitaxel | Stabilizes microtubules and reverses fast axonal transport deficits in a tauopathy model. | [113] |
| NAP (AL‐108) | A neuronal tubulin‐preferring agent that reduces the levels of soluble and insoluble P‐tau and enhances cognitive function in 3xTg‐AD mice. | [72] |
| Tau degradation mechanisms | ||
| Rapamycin | Stimulates autophagy leading to decreased tau toxicity and decreases insoluble tau in a drosophila fruit model. | [114] |
| Puromycin‐sensitive aminopeptidase | Mediates amino tau‐degradation of soluble and insoluble (PHF) tau purified from AD brain in vitro studies. | [115] |
| Tau‐immunotherapy | ||
| Active immunization | Active immunization with a phosphorylated tau epitope, in P301L tangle model mice, reduces aggregated tau in the brain, and slows progression of the tangle‐related behavioral phenotype. | [116, 117, 118] |
| Antiinflammatory approaches | ||
| PMX205 | Acyclic hexapeptide C5a receptor antagonist reduces hyperphosphorylate tau in 3xTg‐AD mice. | [37] |
| Atorvastatin | A statin with antiinflammatory properties that reduced the level of neurofibrillary tangle (NFT) burden in DM‐Tau‐Tg. | [67] |
| Antioxidative studies | ||
| α‐tocopherol | Antioxidative agent that suppresses and/or delays the number of tau aggregate in T44‐Tg. | [68] |
One of the prominent tau strategies under intensive investigation is to attenuate the abnormal hyperphosphorylation of tau through inhibition of the different tau kinases or restoring or upregulating phosphatase activity, such as PP2A. Along these lines, a recent study showed that memantine (Namenda) restores PP2A activity and reduces abnormal hyperphosphorylation of tau [63, 64]. In addition, we have recently demonstrated that chronic memantine treatment markedly increased the inhibitory phosphorylation of GSK‐3ß and consequently reduce the accumulation and phosphorylation of tau in the 3xTgAD mice [65]. Finally, there is evidence that memantine reduces phospho‐tau species in the cerebrospinal fluid (CSF) of AD patients [63, 64, 66]. The therapeutical use of memantine has been approved only for patients with moderate‐to‐severe AD, as studies in mild‐to‐moderate AD have not consistently revealed significant benefits in this patient population. Further clinical studies are necessary to confirm the therapeutic potential of memantine as a disease‐modifying drug for AD.
A search of the National Institutes of Health (NIH) clinical trial database indicates that over 500 clinical trials are or have been conducted studying some aspect of different tauopathies (search for “tauopathy” in the http://www.clinicaltrials.gov). The majority of these studies, however, are primarily directed to find new AD treatments or diagnostics. A few new classes of drugs that have been previously tested in preclinical tau models are currently being tested in different clinical trial phases, including atorvastatin (NCT00151502) [67], α‐tocopherol (NCT00235716) [68], and docosahexaenoic acid (NCT00440050) [69]. Interesting, the only drugs that are being currently evaluated in clinical trials and that directly modulate tau are Methylene blue (Rember) [70] and the neuronal tubulin‐preferring agent (AL‐108) (NCT00505765) [71, 72]. Methylene blue (Rember) is a compound that belongs to the phenothiazine family, and it is believed that its mechanisms of action occurs by preventing tau aggregation, thereby blocking tau filament formation without an effect on the ability of tau to interact with microtubules. A phase II study has been completed with Rember and a significant improvement in the cognitive function was obtaining compared with placebo patients [73, 74]. A phase III study is planned to validate the efficacy and safety of this compound. The second component is a neuronal tubulin‐preferring agent NAP‐(AL‐108) that promotes microtubule assembly, reduces soluble and insoluble tau hyperphosphorylation, Aβ accumulation, and improved in cognitive memory in 3xTg‐AD [71, 72]. NAP is currently in a human AD phase II study [74, 75]. These studies emphasize the idea of multiple pathways control the progression of the disease and suggest that effective therapeutic approaches will depend on a synergistic combination of different agents to reduce the progression the global impact of these neurodegenerative diseases.
Conclusion Remarks
Over the past few years, evidence has accumulated that point to tau as being a feasible therapeutic target for AD and related disorders. Despite the setbacks associated with developing an effective AD treatment so far, there is reason to be optimistic, as we expand the potential targets to include other key elements of the disease such as tau pathology. The data summarized here indicate that there are several agents that are capable of effectively blocking tau phosphorylation or dysfunction in animal and cellular models. Our predication is that the most likely therapeutic regimen for AD will depend on the use of polypharmacy, that is, a combination of drugs that interfere with different targets in AD, such a blocking the effects of Aß or the subsequent oxidative and inflammatory damage. The successful combination will be assembled stepwise and driven by efficacy testing in one or more of the animal models described here as well as in new models of nonmutated APP and tau. Such studies could not only further our knowledge of tau in AD and related diseases, but also open new avenues for therapy of human dementia.
Conflict of Interest
The authors state that they have no conflict of interest pertaining to this manuscript.
Acknowledgments
Authors wish to thank the NIH for funding this work (AG‐027544, AG‐021982, and P50 AG16573).
References
- 1. Glenner GG, Wong CW. Alzheimer's disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984;120:885–890. [DOI] [PubMed] [Google Scholar]
- 2. Grundke‐Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule‐associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 1986;83:4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 2004;44:181–193. [DOI] [PubMed] [Google Scholar]
- 4. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 5. Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: A genetic perspective. Cell 2005;120:545–555. [DOI] [PubMed] [Google Scholar]
- 6. LaFerla FM, Oddo S. Alzheimer's disease: A beta, tau and synaptic dysfunction. Trends Mol Med 2005;11:170–176. [DOI] [PubMed] [Google Scholar]
- 7. Hutton M, Lendon CL, Rizzu P, et al Association of missense and 5’‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 1998;393:702–705. [DOI] [PubMed] [Google Scholar]
- 8. Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: A new group of tauopathies. Brain Pathol 1998;8:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poorkaj P, Bird TD, Wijsman E, et al Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 1998;43:815–825. [DOI] [PubMed] [Google Scholar]
- 10. Hernandez F, Avila J. Tauopathies. Cell Mol Life Sci 2007;64:2219–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule‐associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 1989;3:519–526. [DOI] [PubMed] [Google Scholar]
- 12. Panda D, Samuel JC, Massie M, Feinstein SC, Wilson L. Differential regulation of microtubule dynamics by three‐ and four‐repeat tau: Implications for the onset of neurodegenerative disease. Proc Natl Acad Sci U S A 2003;100:9548–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 1975;72:1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li HL, Wang HH, Liu SJ, et al Phosphorylation of tau antagonizes apoptosis by stabilizing beta‐catenin: A mechanism involved in Alzheimer's neurodegeneration. Proc Natl Acad Sci U S A 2007;104:3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stokin GB, Lillo C, Falzone TL, et al Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 2005;307:1282–1288. [DOI] [PubMed] [Google Scholar]
- 16. Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008;319:1086–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iqbal K, Grundke‐Iqbal I, Zaidi T, et al Defective brain microtubule assembly in Alzheimer's disease. Lancet 1986;2:421–426. [DOI] [PubMed] [Google Scholar]
- 18. Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke‐Iqbal I. Microtubule‐associated protein tau: Abnormal phosphorylation of a non‐paired helical filament pool in Alzheimer disease. J Biol Chem 1993;268:24374–24384. [PubMed] [Google Scholar]
- 19. Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke‐Iqbal I. Mechanisms of tau‐induced neurodegeneration. Acta Neuropathol 2009;118:53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alonso A, Zaidi T, Novak M, Grundke‐Iqbal I, Iqbal K. Hyperphosphorylation induces self‐assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A 2001;98:6923–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alonso AC, Grundke‐Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med 1996;2:783–787. [DOI] [PubMed] [Google Scholar]
- 22. Alonso Adel C, Mederlyova A, Novak M, Grundke‐Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem 2004;279:34873–34881. [DOI] [PubMed] [Google Scholar]
- 23. Grundke‐Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule‐associated protein tau: A component of Alzheimer paired helical filaments. J Biol Chem 1986;261:6084–6089. [PubMed] [Google Scholar]
- 24. Sengupta A, Kabat J, Novak M, Wu Q, Grundke‐Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys 1998;357:299–309. [DOI] [PubMed] [Google Scholar]
- 25. Baudier J, Cole RD. Interactions between the microtubule‐associated tau proteins and S100b regulate tau phosphorylation by the Ca2+/calmodulin‐dependent protein kinase II. J Biol Chem 1988;263:5876–5883. [PubMed] [Google Scholar]
- 26. Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule‐associated proteins and trigger microtubule disruption. Cell 1997;89:297–308. [DOI] [PubMed] [Google Scholar]
- 27. Drewes G, Lichtenberg‐Kraag B, Doring F, et al Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer‐like state. Embo J 1992;11:2131–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase‐3 induces Alzheimer's disease‐like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett 1992;147:58–62. [DOI] [PubMed] [Google Scholar]
- 29. Litersky JM, Johnson GV. Phosphorylation by cAMP‐dependent protein kinase inhibits the degradation of tau by calpain. J Biol Chem 1992;267:1563–1568. [PubMed] [Google Scholar]
- 30. Noble W, Olm V, Takata K, et al Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 2003;38:555–565. [DOI] [PubMed] [Google Scholar]
- 31. Gong CX, Grundke‐Iqbal I, Iqbal K. Dephosphorylation of Alzheimer's disease abnormally phosphorylated tau by protein phosphatase‐2A. Neuroscience 1994;61:765–772. [DOI] [PubMed] [Google Scholar]
- 32. Liu R, Zhou XW, Tanila H, et al Phosphorylated PP2A (tyrosine 307) is associated with Alzheimer neurofibrillary pathology. J Cell Mol Med 2008;12:241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pei JJ, An WL, Zhou XW, et al P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett 2006;580:107–114. [DOI] [PubMed] [Google Scholar]
- 34. Zhou XW, Gustafsson JA, Tanila H, Bjorkdahl C, Liu R, Winblad B, Pei JJ. Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiol Dis 2008;31:386–394. [DOI] [PubMed] [Google Scholar]
- 35. Zhou XW, Winblad B, Guan Z, Pei JJ. Interactions between glycogen synthase kinase 3beta, protein kinase b, and protein phosphatase 2a in tau phosphorylation in mouse n2a neuroblastoma cells. J Alzheimers Dis 2009;17:929–937. [DOI] [PubMed] [Google Scholar]
- 36. Caccamo A, Oddo S, Billings LM, Green KN, Martinez‐Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD‐like pathology in transgenic mice. Neuron 2006;49:671–682. [DOI] [PubMed] [Google Scholar]
- 37. Fonseca MI, Ager RR, Chu SH, et al Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol 2009;183:1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gong CX, Liu F, Grundke‐Iqbal I, Iqbal K. Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O‐GlcNAcylation. J Alzheimers Dis 2006;9:1–12. [DOI] [PubMed] [Google Scholar]
- 39. Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide‐induced inflammation exacerbates tau pathology by a cyclin‐dependent kinase 5‐mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci 2005;25:8843–8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu F, Shi J, Tanimukai H, et al Reduced O‐GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer's disease. Brain 2009;132:1820–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 2004;43:321–332. [DOI] [PubMed] [Google Scholar]
- 42. Gotz J, Gladbach A, Pennanen L, van Eersel J, Schild A, David D, Ittner LM. Animal models reveal role for tau phosphorylation in human disease. Biochim Biophys Acta 2009. doi: 10.1016/j.bbadis.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 43. Goedert M, Hasegawa M. The tauopathies: Toward an experimental animal model. Am J Pathol 1999;154:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Engel T, Lucas JJ, Gomez‐Ramos P, Moran MA, Avila J, Hernandez F. Cooexpression of FTDP‐17 tau and GSK‐3beta in transgenic mice induce tau polymerization and neurodegeneration. Neurobiol Aging 2006;27:1258–1268. [DOI] [PubMed] [Google Scholar]
- 45. Deters N, Ittner LM, Gotz J. Substrate‐specific reduction of PP2A activity exaggerates tau pathology. Biochem Biophys Res Commun 2009;379:400–405. [DOI] [PubMed] [Google Scholar]
- 46. Oddo S, Caccamo A, Shepherd JD, et al Triple‐transgenic model of Alzheimer's disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003;39:409–421. [DOI] [PubMed] [Google Scholar]
- 47. Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001;293:1491–1495. [DOI] [PubMed] [Google Scholar]
- 48. Lewis J, Dickson DW, Lin WL, et al Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001;293:1487–1491. [DOI] [PubMed] [Google Scholar]
- 49. Roberson ED, Scearce‐Levie K, Palop JJ, et al Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science 2007;316:750–754. [DOI] [PubMed] [Google Scholar]
- 50. Oddo S, Caccamo A, Cheng D, Jouleh B, Torp R, LaFerla FM. Genetically augmenting tau levels does not modulate the onset or progression of Abeta pathology in transgenic mice. J Neurochem 2007;102:1053–1063. [DOI] [PubMed] [Google Scholar]
- 51. Santacruz K, Lewis J, Spires T, et al Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005;309:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Le Corre S, Klafki HW, Plesnila N, et al An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci U S A 2006;103:9673–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alonso Adel C, Li B, Grundke‐Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A 2006;103:8864–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li B, Chohan MO, Grundke‐Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol 2007;113:501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 2009;284:12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM. Age‐dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 1999;24:751–762. [DOI] [PubMed] [Google Scholar]
- 57. Ishihara T, Zhang B, Higuchi M, Yoshiyama Y, Trojanowski JQ, Lee VM. Age‐dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. Am J Pathol 2001;158:555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Probst A, Gotz J, Wiederhold KH, et al Axonopathy and amyotrophy in mice transgenic for human four‐repeat tau protein. Acta Neuropathol 2000;99:469–481. [DOI] [PubMed] [Google Scholar]
- 59. Spittaels K, Van Den Haute C, Van Dorpe J, et al Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four‐repeat human tau protein. Am J Pathol 1999;155:2153–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gotz J, Probst A, Spillantini MG, Schafer T, Jakes R, Burki K, Goedert M. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. Embo J 1995;14:1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brion JP, Tremp G, Octave JN. Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer's disease. Am J Pathol 1999;154:255–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zilka N, Filipcik P, Koson P, et al Truncated tau from sporadic Alzheimer's disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett 2006;580:3582–3588. [DOI] [PubMed] [Google Scholar]
- 63. Chohan MO, Khatoon S, Iqbal IG, Iqbal K. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett 2006;580:3973–3979. [DOI] [PubMed] [Google Scholar]
- 64. Li L, Sengupta A, Haque N, Grundke‐Iqbal I, Iqbal K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett 2004;566:261–269. [DOI] [PubMed] [Google Scholar]
- 65. Martinez‐Coria H, Green KN, Billings LM, et al Memantine improves cognition and reduces Alzheimer's‐like neuropathology in transgenic mice. Am J Pathol 2010;176:870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Iqbal K, Grundke‐Iqbal I. Alzheimer neurofibrillary degeneration: Significance, etiopathogenesis, therapeutics and prevention. J Cell Mol Med 2008;12:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Boimel M, Grigoriadis N, Lourbopoulos A, Touloumi O, Rosenmann D, Abramsky O, Rosenmann H. Statins reduce the neurofibrillary tangle burden in a mouse model of tauopathy. J Neuropathol Exp Neurol 2009;68:314–325. [DOI] [PubMed] [Google Scholar]
- 68. Nakashima H, Ishihara T, Yokota O, Terada S, Trojanowski JQ, Lee VM, Kuroda S. Effects of alpha‐tocopherol on an animal model of tauopathies. Free Radic Biol Med 2004;37:176–186. [DOI] [PubMed] [Google Scholar]
- 69. Green KN, Martinez‐Coria H, Khashwji H, Hall EB, Yurko‐Mauro KA, Ellis L, LaFerla FM. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid‐beta and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci 2007;27:4385–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease‐like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A 1996;93:11213–11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Matsuoka Y, Gray AJ, Hirata‐Fukae C, et al Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer's disease at early pathological stage. J Mol Neurosci 2007;31:165–170. [DOI] [PubMed] [Google Scholar]
- 72. Matsuoka Y, Jouroukhin Y, Gray AJ, et al A neuronal microtubule‐interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer's disease. J Pharmacol Exp Ther 2008;325:146–153. [DOI] [PubMed] [Google Scholar]
- 73. Gura T. Hope in Alzheimer's fight emerges from unexpected places. Nat Med 2008;14:894. [DOI] [PubMed] [Google Scholar]
- 74. Rafii MS, Aisen PS. Recent developments in Alzheimer's disease therapeutics. BMC Med 2009;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Melnikova I. Therapies for Alzheimer's disease. Nat Rev Drug Discov 2007;6:341–342. [DOI] [PubMed] [Google Scholar]
- 76. Kidd M. Paired helical filaments in electron microscopy of Alzheimer's disease. Nature 1963;197:192–193. [DOI] [PubMed] [Google Scholar]
- 77. Terry RD. The fine structure of neurofibrillary tangles in Alzheimer's disease. J Neuropathol Exp Neurol 1963;22:629–642. [DOI] [PubMed] [Google Scholar]
- 78. Delacourte A, Defossez A. Biochemical characterization of an immune serum which specifically marks neurons in neurofibrillary degeneration in Alzheimer's disease. C R Acad Sci III 1986;303:439–444. [PubMed] [Google Scholar]
- 79. Kosik KS, Joachim CL, Selkoe DJ. Microtubule‐associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 1986;83:4044–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule‐associated protein tau. Proc Natl Acad Sci U S A 1988;85:4051–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wischik CM, Novak M, Thogersen HC, et al Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 1988;85:4506–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Harada A, Oguchi K, Okabe S, et al Altered microtubule organization in small‐calibre axons of mice lacking tau protein. Nature 1994;369:488–491. [DOI] [PubMed] [Google Scholar]
- 83. Alonso AC, Zaidi T, Grundke‐Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A 1994;91:5562–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Busciglio J, Lorenzo A, Yeh J, Yankner BA. Beta‐amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 1995;14:879–888. [DOI] [PubMed] [Google Scholar]
- 85. Mori H, Hosoda K, Matsubara E, et al Tau in cerebrospinal fluids: Establishment of the sandwich ELISA with antibody specific to the repeat sequence in tau. Neurosci Lett 1995;186:181–183. [DOI] [PubMed] [Google Scholar]
- 86. Jensen M, Basun H, Lannfelt L. Increased cerebrospinal fluid tau in patients with Alzheimer's disease. Neurosci Lett 1995;186:189–191. [DOI] [PubMed] [Google Scholar]
- 87. Smith MA, Siedlak SL, Richey PL, et al Tau protein directly interacts with the amyloid beta‐protein precursor: Implications for Alzheimer's disease. Nat Med 1995;1:365–369. [DOI] [PubMed] [Google Scholar]
- 88. Alonso AD, Grundke‐Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule‐associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A 1997;94:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta‐amyloid‐induced neurotoxicity. Proc Natl Acad Sci U S A 2002;99:6364–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging 2003;24:1063–1070. [DOI] [PubMed] [Google Scholar]
- 91. Bellucci A, Westwood AJ, Ingram E, Casamenti F, Goedert M, Spillantini MG. Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am J Pathol 2004;165:1643–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zheng YL, Kesavapany S, Gravell M, et al A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. Embo J 2005;24:209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Camins A, Verdaguer E, Folch J, Canudas AM, Pallas M. The role of CDK5/P25 formation/inhibition in neurodegeneration. Drug News Perspect 2006;19:453–460. [DOI] [PubMed] [Google Scholar]
- 94. Oumata N, Bettayeb K, Ferandin Y, et al Roscovitine‐derived, dual‐specificity inhibitors of cyclin‐dependent kinases and casein kinases 1. J Med Chem 2008;51:5229–5242. [DOI] [PubMed] [Google Scholar]
- 95. Schneider A, Mandelkow E. Tau‐based treatment strategies in neurodegenerative diseases. Neurotherapeutics 2008;5:443–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A 1996;93:8455–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase‐3. J Biol Chem 1997;272:25326–25332. [DOI] [PubMed] [Google Scholar]
- 98. Munoz‐Montano JR, Moreno FJ, Avila J, Diaz‐Nido J. Lithium inhibits Alzheimer's disease‐like tau protein phosphorylation in neurons. FEBS Lett 1997;411:183–188. [DOI] [PubMed] [Google Scholar]
- 99. Perez M, Hernandez F, Lim F, Diaz‐Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis 2003;5:301–308. [DOI] [PubMed] [Google Scholar]
- 100. Noble W, Planel E, Zehr C, et al Inhibition of glycogen synthase kinase‐3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A 2005;102:6990–6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Caccamo A, Oddo S, Tran LX, LaFerla FM. Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol 2007;170:1669–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bhat R, Xue Y, Berg S, et al Structural insights and biological effects of glycogen synthase kinase 3‐specific inhibitor AR‐A014418. J Biol Chem 2003;278:45937–45945. [DOI] [PubMed] [Google Scholar]
- 103. Bitner RS, Nikkel AL, Markosyan S, Otte S, Puttfarcken P, Gopalakrishnan M. Selective alpha7 nicotinic acetylcholine receptor activation regulates glycogen synthase kinase3beta and decreases tau phosphorylation in vivo. Brain Res 2009;1265:65–74. [DOI] [PubMed] [Google Scholar]
- 104. Panza F, Solfrizzi V, Frisardi V, et al Beyond the neurotransmitter‐focused approach in treating Alzheimer's disease: Drugs targeting beta‐amyloid and tau protein. Aging Clin Exp Res 2009;21:386–406. [DOI] [PubMed] [Google Scholar]
- 105. Hubinger G, Geis S, LeCorre S, et al Inhibition of PHF‐like tau hyperphosphorylation in SH‐SY5Y cells and rat brain slices by K252a. J Alzheimers Dis 2008;13:281–294. [DOI] [PubMed] [Google Scholar]
- 106. Degerman GM, Kilander L, Basun H, Lannfelt L. Reduction of phosphorylated tau during memantine treatment of Alzheimer's disease. Dement Geriatr Cogn Disord 2007;24:247–252. [DOI] [PubMed] [Google Scholar]
- 107. Song MS, Rauw G, Baker GB, Kar S. Memantine protects rat cortical cultured neurons against beta‐amyloid‐induced toxicity by attenuating tau phosphorylation. Eur J Neurosci 2008;28:1989–2002. [DOI] [PubMed] [Google Scholar]
- 108. Pickhardt M, Gazova Z, von Bergen M, et al Anthraquinones inhibit tau aggregation and dissolve Alzheimer's paired helical filaments in vitro and in cells. J Biol Chem 2005;280:3628–3635. [DOI] [PubMed] [Google Scholar]
- 109. Pickhardt M, Larbig G, Khlistunova I, et al Phenylthiazolyl‐hydrazide and its derivatives are potent inhibitors of tau aggregation and toxicity in vitro and in cells. Biochemistry 2007;46:10016–10023. [DOI] [PubMed] [Google Scholar]
- 110. Bulic B, Pickhardt M, Khlistunova I, Biernat J, Mandelkow EM, Mandelkow E, Waldmann H. Rhodanine‐based tau aggregation inhibitors in cell models of tauopathy. Angew Chem Int Ed Engl 2007;46:9215–9219. [DOI] [PubMed] [Google Scholar]
- 111. Jordan MA. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Med Chem Anticancer Agents 2002;2:1–17. [DOI] [PubMed] [Google Scholar]
- 112. Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin‐dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer's disease. J Cell Biol 1998;143:777–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhang B, Maiti A, Shively S, et al Microtubule‐binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A 2005;102:227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Berger Z, Ravikumar B, Menzies FM, et al Rapamycin alleviates toxicity of different aggregate‐prone proteins. Hum Mol Genet 2006;15:433–442. [DOI] [PubMed] [Google Scholar]
- 115. Sengupta S, Horowitz PM, Karsten SL, et al Degradation of tau protein by puromycin‐sensitive aminopeptidase in vitro. Biochemistry 2006;45:15111–15119. [DOI] [PubMed] [Google Scholar]
- 116. Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci 2007;27:9115–9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer's disease and related tauopathies. J Alzheimers Dis 2008;15:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sigurdsson EM. Tau‐focused immunotherapy for Alzheimer's disease and related tauopathies. Curr Alzheimer Res 2009;6:446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]