

Abstract
Bacteria associate with surfaces and one another by elaborating an extracellular matrix to encapsulate cells, creating communities termed biofilms. Biofilms are beneficial in some ecological niches, but also contribute to the pathogenesis of serious and chronic infectious diseases. New approaches and quantitative measurements are needed to define the composition and architecture of bacterial biofilms to help drive the development of strategies to interfere with biofilm assembly. Solid-state NMR is uniquely suited to the examination of insoluble and complex macromolecular and whole-cell systems. This article highlights three examples that implement solid-state NMR to deliver insights into bacterial biofilm composition and changes in cell-wall composition as cells transition to the biofilm lifestyle. Most recently, solid-state NMR measurements provided a total accounting of the protein and polysaccharide components in the extracellular matrix of an E. coli biofilm and transform our qualitative descriptions of matrix composition into chemical parameters that permit quantitative comparisons among samples. We present additional data for whole biofilm samples (cells plus the extracellular matrix) that complement matrix-only analyses. The study of bacterial biofilms by solid-state NMR is an exciting avenue ripe with many opportunities and we close the article by articulating some outstanding questions and future directions in this area.
Introduction
Bacteria are ubiquitous across nature and have evolved an arsenal of survival techniques that enable them to colonize and even thrive in an astonishing range of environments, including those with extreme temperature and pH [1, 2]. An important survival mechanism in niches ranging from within the human host to the extremely acidic microbial mats in Yellowstone, is the cohabitation of bacteria in multicellular communities termed biofilms. Biofilms are composed of bacteria encased within a self-produced, non-crystalline extracellular matrix (ECM) that can consist of proteins, polysaccharides, lipids, and other molecules. Although bacteria have traditionally been studied as planktonic organisms, it is now appreciated that the majority of bacteria exist and persist as biofilms in nature [1]. Biofilms provide protection to bacteria through physical, chemical, and physiological mechanisms. The ECM can serve as a physical barrier against assaults such as UV light and chemical and biological antibacterial agents. The biofilm also harbors persister cells, or populations of dormant bacteria that are not responsive to antibiotics [3]. Although bacterial biofilms are essential to ecological stability in diverse niches on our planet, biofilm formation is also implicated in a number of adverse consequences, including aquatic biofouling [4]; contamination of medical devices [2, 3] such as intravenous catheters, prosthetic heart valves, and artificial joints; and chronic bacterial infection [2, 5].
Extensive research efforts to better understand biofilms have revealed their complexity and built an extensive foundation of measured parameters that aid in connecting biofilm properties with function [6]. These include the identification of: genetic and molecular features that mediate and influence biofilm formation; patterns of nutrient flow within biofilm networks; and surface and mechanical properties of biofilm. However, biofilms pose a challenge to quantitative analysis by traditional biochemical techniques due to their insolubility and intractability [7]. Thus, while we have a basic parts list for some biofilms, including required molecular components, a complete accounting of the parts or atomistic structural detail of intact material has been lacking. New approaches are needed to transform biofilm descriptors into quantitative parameters of chemical and molecular composition. Some studies have employed FTIR and solid-state NMR to generally profile the types of chemical functionalities in biofilm samples, e.g. carbonyls and peptide bonds, aromatics, and aliphatics [8–10]. Within this article, we describe the unique ability to implement solid-state NMR approaches to deliver quantitative insights regarding composition and structure in biofilm systems. At the close of this article, we discuss some of the exciting and promising future avenues utilizing solid-state NMR to map the composition and structure of bacterial biofilms.
Solid-state NMR for Biological Macrosystems
The CPMAS Platform
Solid-state NMR is uniquely suited to the study of complex and insoluble systems such as bacterial biofilms [11–14]. It does not require homogeneous protein preparations or high quality crystals (x-ray crystallography), high tumbling rates in solution (solution NMR), chemical processing, or enzymatic digestion (HPLC-MS). Thus, solid-state NMR has been utilized to define composition, structure, and metabolism in a variety of otherwise intractable biological systems including bacterial whole cells and cell walls [15–18], amyloids [19–21], membrane proteins [22], and intact plant leaves [23–24]. Obtaining NMR spectra of such large and insoluble systems is not possible in solution-state NMR as the influence of dipolar couplings and chemical-shift anisotropy are not averaged out as they are in smaller, soluble, rapidly tumbling systems. Implementation of magic-angle spinning (MAS) in solid-state NMR experiments averages over the spatial coordinates in the dipolar coupling and chemical-shift anisotropy Hamiltonians and permits the acquisition of high-resolution spectra in solids [25–28]. Expressions for both Hamiltonians contain an angular dependence, 3cos2θ-1, where θ is the angle between the internuclear spin vector and the external magnetic field. Thus, by mechanically spinning the sample at the magic-angle (~54.74°), the “3cos2θ-1” term is averaged to zero and suppresses the influence of the dipolar coupling and CSA, yielding high-resolution NMR spectra harboring the isotropic chemical shifts plus associated spinning sidebands [26, 28]. A significant difference between MAS in solid-state NMR and molecular tumbling in solution-state NMR is that the spatial averaging is performed in a coherent manner, at a precise angle and frequency. Thus, since the dipolar coupling Hamiltonian, for example, depends on both space and spin coordinates, it is possible to manipulate the spin coordinates, interfere with the averaging and extract the dipolar coupling (as in the Rotational-Echo Double-Resonance (REDOR) experiment [29]). In standard solution-state NMR experiments, the dipolar couplings are inaccessible unless the molecules are prepared in ways to induce alignment or enhanced viscosity to restrict isotropic tumbling [30].
In most biological solids applications, MAS [25–28] is coupled with cross-polarization (CP) [31–33] to enhance the NMR signal of nuclei with a low gyromagnetic ratio (γ13C≈γ1H/4) by magnetization transfer from 1H nuclei by adjusting the radiofrequency pulse power to satisfy Hartmann-Hahn matching conditions. The resulting CPMAS experiment [34] is the platform for most biological solid-state NMR studies. Typical experiments employing CPMAS at low-to-modest spinning speeds (up to 15kHz) are also performed in the presence of high-power proton decoupling to effect rapid spin flips among the 1H nuclei to remove their couplings to other nuclei. These couplings can be re-introduced, if desired, by interrupting decoupling to distinguish between protonated and non-protonated carbons [35] or to estimate distances by spin diffusion [36], for example.
Solid-state NMR Insights into Bacterial Biofilm Composition
Amyloid-integrated biofilm formation in E. coli
We recently reported the first determination of the chemical composition of the intact ECM of a bacterial biofilm using solid-state NMR, biochemical analysis, and electron microscopy [37]. The NMR data provided the key quantitative parameters of the intact matrix--the percentages of matrix components by mass. The analysis was performed on an important human pathogenic bacterium, the uropathogenic E. coli (UPEC) strain UTI89. As introduced earlier, this work was motivated by the need for fundamental chemical and molecular descriptions of bacterial biofilms in order to develop effective strategies to prevent and treat biofilm-associated infections. Biofilms are implicated in urinary tract infection (UTI), which most often is caused by UPEC [38, 39]. UPEC can form biofilm-like intracellular bacterial communities [39] that evade host defenses and antibiotic treatment, and are implicated in recurrent and persistent infections, including those associated with catheter-associated UTI [40]. We employed the UPEC strain, UTI89, which is a prototypical clinical isolate used in most of the pioneering in vivo work demonstrating the coordinated genetic and molecular cascade that accompanies host-pathogen interactions during infections of the bladder and kidneys [41–43]. UTI89 is also one of many E. coli clinical isolates that produce functional amyloid fibers termed curli [44, 45]. Curli mediate adhesion to biotic and abiotic surfaces and, together with cellulose, serve as adhesive scaffolds and promote biofilm formation [46]. Although genetic and molecular analyses have determined that curli and cellulose, for example, can be co-produced in E. coli biofilms, the extent to which each one is present in the ECM had not been determined before the NMR work reviewed below [47–52]. Indeed, the general quantification of protein-to-polysaccharide ratios is difficult to determine by conventional analytical methods due to the resistance of the matrix to dissolution methods and the perturbation that accompanies harsh degradative methods to analyze biofilm parts.
Quantitative determination of the composition of the extracellular matrix in E. coli
To define the protein and polysaccharide contributions to the biofilm framework, we obtained electron micrographs, protein gels, and 13C CPMAS spectra of: (i) the complete extracellular matrix (ECM); (ii) the curli-free ECM produced by the curli mutant strain UTI89ΔcsgA; and (iii) purified curli. These spectra indicated that the biofilm matrix formed by curli-producing bacteria has two major components, curli and cellulose, each in a quantifiable amount. The bacteria were grown on YESCA nutrient agar, a medium on which UTI89 exhibits the hallmark wrinkled colony morphology that is associated with E. coli biofilm production [47]. Congo red is often used in microbiological assays to enhance visualization of this morphology, and growing the cells in the presence of this dye allowed us to visually track the presence of the ECM during the purification and to then precipitate the polysaccharide component of the ECM [47, 59, 63]. The nonperturbative purification procedure was based on the curli isolation protocol utilizing fluid shear forces to remove the ECM while leaving the cells intact and separable by low-speed centrifugation [45].
The natural abundance curli-only 13C spectrum harbors contributions from all of its amino acids and differs from the carbon spectrum that would be associated with proteins with different amino acid sequences (Figure 1A, top). Backbone and sidechain carbonyls contribute to the region around 175 ppm. Aromatic amino acids sidechains and also histidine and arginine sidechain carbons contribute to the region from 120–160 ppm and the aliphatic region from 10–70 ppm contains alpha carbons and other sidechain carbons. Each protein’s unique pool of carbons based on the protein composition can be profiled in this way by one-dimensional CPMAS. The spectrum of UTI89ΔcsgA ECM lacked a carbonyl peak, which indicated that there was no detectable protein in this sample (Figure 1A, top). Instead, the UTI89ΔcsgA spectrum contained contributions from both cellulose and Congo red, which were consistent with spectra of commercial cellulose and Congo red with the exception of an additional carbon peak at 40 ppm [37]. The 40-ppm carbon was hypothesized to be bonded to nitrogen, based on its chemical shift and the presence of a single nitrogen amine peak in the 15N CPMAS spectrum of a uniformly 15N biosynthetically labeled sample for which cells were grown in the presence of 15NH4Cl. This possibility was investigated by 13C{15N}Rotational-Echo Double Resonance (REDOR), a solid-state NMR experiment that enables the measurement of heteronuclear dipolar couplings between heteronuclear spin pairs in a sample [12, 13, 29]. In this case, a one-bond 13C{15N} REDOR filter revealed that the 40-ppm carbon was completely dephased and was directly bonded to a nitrogen. Together with the collective chemical shifts, the 40-ppm carbon was determined to be consistent with a hydroxyl-substituted O-linked 2-aminoethyl modification to cellulose [37, 54, 55]. By deconvolution of the 13C CPMAS spectra, the UTI89ΔcsgA sample was determined to contain 50% Congo red and 50% of the modified cellulose [37]. The wild-type UTI89 spectrum was a nearly perfect match for the spectral sum of the scaled spectra of the curli and UTI89ΔcsgA ECM (Figure 1A, bottom). The scaled sum was obtained by normalizing each spectrum by its mass and scaling the curli spectrum to 72% and the UTI89ΔcsgA to 28%. After accounting for the Congo red in the UTI89ΔcsgA sample, we found that the UTI89 insoluble ECM grown on YESCA nutrient agar is composed of a 6:1 ratio of curli to polysaccharide. This result was further confirmed by obtaining the 13C CPMAS spectrum of a physical mixture of biofilm parts within one rotor [37] and, together, has provided the first total accounting of the components of the intact ECM of a bacterial biofilm.
Figure 1.

NMR sum of the parts. (A) The spectral sum of the UTI89ΔcsgA extracellular material and purified curli (top) completely recapitulates the 13C CPMAS spectrum of the intact UTI89 ECM (bottom), each associated with transmission electron micrographs associated with the samples. MAS was performed at 7143 Hz, and 32,768 scans were obtained for each spectrum. (B) The 13C CPMAS spectrum of the whole biofilm sample contains carbon contributions from whole cells plus the ECM. MAS was performed at 7143. This figure is adapted from reference 37.
Previous biochemical studies that have examined the general production of curli have only profiled whether curli production is either increased or decreased relative to a reference sample as a function of bacterial strain or environmental conditions. These analyses do not provide an absolute concentration or the relative amount of curli with respect to other components. Some reviews assume that extracellular matrices would be primarily polysaccharide [56] (due to the appreciated ‘stickiness’ of polysaccharides), perhaps 80% polysaccharide and 20% curli in the case of E. coli. Thus, the development of this approach and the determination that the insoluble matrix is 85% curli and 15% polysaccharide in the specified growth conditions is the first determination of the composition of an extracellular matrix and provides a valuable first-of-kind strategy that can be recruited to answer many exciting questions regarding other biofilm formers and the influence of biofilm inhibitors.
Mapping of global carbon pools in the intact biofilm by solid-state NMR
Comparison of the 13C CPMAS spectrum of the intact biofilm, including the bacterial whole cells plus the ECM is also provided here in Figure 1B. There are an increased number of carbon contributions, as expected, from the cellular contents, including the abundant diversity of macromolecules, e.g. proteins and nucleic acids, as well as lipids, carbohydrates, metabolites, and small molecules. The spectrum harbors broad resonances that represent the vast array of chemical environments represented in the whole cells. Indeed, most of the mass of the whole biofilm in these preparations arises from the intact cells. Thus, in the dissection of the composition of the ECM (Figure 1A), it was invaluable to be able to extract the ECM from the biofilm for analysis by solid-state NMR. We employed a minimally perturbative method for extraction of the ECM from bacterial biofilms that relies upon homogenization with shear force followed by differential centrifugation to separate the ECM from the bacterial cells, a procedure that is useful for the extraction of ECM from biofilms formed by other strains and species of bacteria, although the ECM of some organisms is difficult to extract and alternate protocols are developed as needed to permit their isolation.
NMR comparison of UTI89 carbon pools in intact biofilms at the air-liquid interface
As shown above, solid-state NMR has the sensitivity and resolution to profile the global carbon composition of intact biofilms, containing cells plus the ECM. We utilized NMR to study the influence of the small molecule dimethyl sulfoxide (DMSO) on biofilm production, specifically biofilms formed at the air-liquid interface, referred to as pellicles [57]. E. coli exhibits a selective increase in curli protein expression (csgA and csgB mRNA expression increases over 10-fold) in response to DMSO resulting in an increase in curli production (Figure 2A) and a visually observable increase in film robustness, an acceleration and increase in the development of the elastic modulus of the film, and an enhancement in the ability to recover from strain [57, 58]. This effect is tunable as a function of DMSO concentration, ranging from 0.5% (v/v) to 4% (v/v). Given the relatively high concentration of DMSO in the nutrient broth, we examined whether the increases in curli production could be, in part, attributed to the utilization of DMSO as a carbon source. Thus, 13C CPMAS spectra of intact pellicles formed by UTI89 in the presence of DMSO or [13C2]DMSO were compared. If DMSO was used as a carbon source, an increase in peak intensity at a chemical shift corresponding to that of an end metabolic product in the [13C2]DMSO sample relative to that of the unlabeled DMSO growth conditions would have been observed. However, the only observable increase in peak intensity in the [13C2]DMSO sample appeared at 39 ppm, which corresponds to the DMSO carbon chemical shift, resulting from residual [13C2]-DMSO among the harvested cells (Figure 2B, black and red) [57]. These results indicated that the enhancements in biofilm formation observed in the presence of DMSO were not connected to any utilization of the DMSO as a carbon source, which would have lead to overall increases in some carbon-peak intensities. Overall, the two spectra were similar, also revealing the biological similarity of different biofilm preparations. It has been our experience with other whole-cell and cell-wall systems that significant fluctuations in carbon pools are not observed among such preparations, which leads to the power in comparisons when using CPMAS to examine complex bacterial systems. Here, we also include the non-DMSO treated pellicle for comparison (Figure 2, blue). The three spectra were scaled according to the carbonyl peak to enable the detection of relative differences in the carbon pools. Treatment with DMSO did yield increases in the 50–75 ppm chemical-shift range relative to the carbonyl peak height as compared to the sample without DMSO treatment and presents a whole-biofilm signature that is associated with increased curli and ECM production.
Figure 2.

Comparative analysis of carbon pools in intact pellicles. (A) Transmission electron micrographs of pellicles formed in YESCA nutrient broth in the presence and absence of 4% DMSO. (B) Natural-abundance 13C CPMAS NMR spectra provided a complete accounting of the carbon contributions in intact pellicles formed by UTI89 in the absence of DMSO and in the presence of DMSO and [13C2]DMSO. Treatment with DMSO increases curli production and is accompanied by a spectroscopic increase in carbons in the 50–75 ppm region, with respect to the carbonyl peak. Except for the enhancement in peak intensity at 39 ppm (the isotropic carbon chemical shift of DMSO), growth in medium supplemented with labeled [13C2]DMSO did not result in other carbon peak intensity increases as compared to treatment with unlabeled DMSO. This figure is adapted from reference 57.
Biofilm formation in Gram-positive bacteria
Cell surfaces and biofilms in Staphylococcus aureus
Gram-positive bacteria, such as S. aureus, differ from Gram-negative bacteria such as E. coli and Salmonella, in their cellular organization. Gram-negative bacteria harbor an inner membrane, periplasmic space containing a thin layer of peptidoglycan, and an outer membrane. The ECM in Gram-negative bacteria is produced beyond the outer membrane and ECM components must be secreted through the outer membrane. In contrast, Gram-positive bacteria harbor a single cell membrane surrounded by a thick layer of cell-wall material comprised primarily of peptidoglycan. Biofilm formation by Staphylococcus aureus is characterized by the production of an ECM outside its thick cell wall [49, 50] and, like E. coli, biofilm-associated S. aureus infections are also a significant concern in the clinic [59, 60]. We and others are beginning to study Gram-positive biofilm formation by solid-state NMR. We are interested in examining the S. aureus ECM framework, and the variability in the ECM as a function of strain or external stimuli such as antibiotic treatment. En route to studying the S. aureus ECM, we recently discovered that the S. aureus peptidoglycan is altered as cells progress through stationary phase, increasing in cell density, and depleting nutrients from the growth medium [37]. We believe that considerations of cell-wall composition and architecture will be important in fully understanding biofilm formation in S. aureus and other Gram-positive bacteria. There is precedence for this notion, as peptidoglycan breaks have been identified in lactococcal biofilm communities and are correlated with the ability to form biofilms [61]. Bacteria that lacked the proper peptidoglycan hydrolase could not alter their cell wall and were deficient in adhesion and unable to form biofilms [61]. Biofilm formation was restored by the addition of exogenous hydrolases to perturb the cell wall [61]. Similarly, based on biochemical data and functional phenotypes, peptidoglycan remodeling has been implicated in adhesion and biofilm formation in Pseudomonas aeruginosa and E. coli, potentially to allow for proper assembly of large macromolecular complexes involved in adhesion and biofilm assembly [62]. Thus, we describe our recent discovery of significant cell-wall alterations in bacterial cultures as a function of cell density and nutrient limitation.
Cell-wall compositional changes in S. aureus during nutrient limitation in the stationary phase
We have started to study the composition of the S. aureus ECM, but first made some discoveries regarding the cell-wall composition as cells reach stationary phase and deplete nutrients from the surrounding medium, events that promote biofilm formation in vivo and in the laboratory [63]. Ultimately, one wants to map and understand all the changes in the biofilm community, those that occur within the individual cells (in the cytoplasm and in the cell wall) and those that occur in the extracellular matrix. Solid-state NMR has been used extensively, particularly over the past ten years, to examine bacterial cell-wall composition and architecture in Gram-positive bacteria such as S. aureus and in defining the modes of action of cell-wall targeting antibiotics [17, 18, 64–66], although these studies are beyond the scope of this perspective. The major component of the cell wall is the peptidoglycan [67]. The peptidoglycan is a mesh-like network consisting of polymerized peptidoglycan subunits that are synthesized inside the cell and transported to the cell surface for polymerization via transglycosylation (linking of the sugars) and transpeptidation (linking of the peptide stems). In S. aureus, the peptidoglycan precursor consists of a disaccharide linked to a pentapeptide stem (L-Ala-D-isoGlutamine-L-Lys-D-Ala-D-Ala) that is normally functionalized with a pentaglycine bridge attached to the ε-amino group of the L-Lys of the pentapeptide. During transpeptidation, the pentaglycine bridge becomes cross-linked to the penultimate D-Ala of a neighboring stem, accompanied by the concomitant loss of the terminal D-Ala (Figure 3A). The highly cross-linked peptidoglycan of S. aureus cannot be fully digested by chemical or enzymatic hydrolysis and, thus, cannot be fully resolved by traditional biochemical techniques including HPLC-MS, emphasizing the necessity of a method such as solid-state NMR to characterize the intact peptidoglycan.
Figure 3.
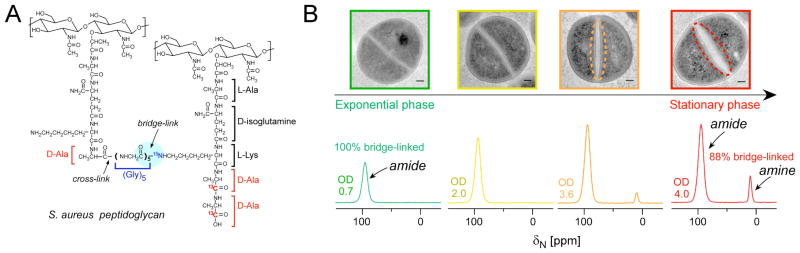
Alterations in the S. aureus cell wall during stationary phase and nutrient depletion. (A) Chemical structure of S. aureus peptidoglycan, which is cross-linked through an inter-peptide bridge consisting of five glycines to connect the ε-amino group of L-Lys in the third position of one stem (bridge-link, highlighted) to the D-Ala in the fourth position of the connected stem (cross-link) with the concomitant cleavage of the terminal D-Ala. (B, top) TEM images of ultrathin sections of S. aureus cells harvested at four different growth stages, where non-uniform thickening is indicated with dashed lines in the TEM images of cells harvested at OD 3.6 and OD 4. Scale bars are 100 nm. (B, bottom) 15N CPMAS spectra of [ε-15N]Lys labeled peptidoglycan isolated from S. aureus cells at OD 0.7, OD 2, OD 3.6 and OD 4. Spectra are normalized to the amide peak. The amine peak at 10 ppm appears at OD 2.0 (not obvious) and increases towards OD 4 (stationary phase). Spectra were acquired on a 500 MHz spectrometer with MAS at 8 kHz and were referenced to solid ammonium sulfate. This figure is adapted from reference 16.
Using 15N-CPMAS, we quantified populations of bridge-links in specifically labeled peptidoglycan across different growth stages as cells increase in density in the stationary phase [16]. Peptidoglycan was isolated from specifically L-[ε-15N] lysine labeled cells, where the L-[ε-15N]Lys label does not scramble as evidenced by the lack of 15N peaks corresponding to other amino acid sidechains [16]. In the exponential growth stage, only one peak was observed in the 15N CPMAS spectrum (Figure 3B), assigned to the amide form of the Lys ε-N in a bridge-link with the pentaglycine bridge attached (Figure 3A)[16]. In contrast, the spectrum of the peptidoglycan isolated from bacteria harvested in the stationary growth stage contained a second peak, which increased at later growth stages and was assigned to the amine form of the Lys ε-N, corresponding to stems without attached bridges. Interestingly, cell wall septa were also thicker in stationary phase (Figure 3B). The changes in nutrient levels in the broth medium during bacterial growth were monitored by solution NMR, and the emergence of the peptide stems without glycine bridges occurred concomitantly with the depletion of glycine from the nutrient medium[16]. Thus, the bacteria transported imperfect stems to the extracellular surface, rather than slowing down cell-wall synthesis until glycine production could increase and canonical cell-wall precursors could be assembled and transported to the cell surface for assembly. Pulse-chase labeling experiments confirmed that the emergence of peptidoglycan stems with the free lysyl amines resulted from new cell-wall synthesis rather than from the degradation of existing cell wall. Future work will explore whether the decreased crosslinking provides an advantage to the bacteria, perhaps by allowing increased porosity to transport large proteins beyond the cell wall as they assemble biofilms or, alternatively, if the production of the altered and less highly crosslinked cell wall renders the cells more sensitive to antibiotics that target cell wall assembly.
Conclusions and future avenues
We have recently launched solid-state NMR as a valuable approach to determine the composition of the bacterial ECM. We defined the composition of the E.coli ECM under amyloid-integrated biofilm growth conditions, transforming qualitative descriptions of the ECM into quantitative parameters based on molecular composition and spectroscopic signatures that can be compared across samples. Traditional forms of analyses have provided only qualitative identification of the components of biofilms without a quantitative understanding of their molecular connectivity and structural complexities. The understanding and methodology that we have developed will be valuable to the continued study of the fundamental chemical and molecular basis of biofilm structure involving a range of disciplines including: (i) microbiology, especially as new bacterial amyloids are discovered; and (ii) structural biology and biophysics, similar to the study of other large macromolecular complexes and machinery including bacterial cell walls, flagella, and ATP synthase. Thus far, we have successfully described and provided an accounting for the components of the ECM of a clinically relevant E. coli strain UTI89. Previous studies of curli-integrated biofilms only profiled whether curli production was either increased or decreased. Thus, the development of the integrated approach summarized here and the determination that the matrix is 85% curli and 15% polysaccharide in the specified growth conditions provides a first-of-kind strategy that can be recruited to answer many exciting questions regarding other biofilm formers. Key insights can emerge from unlabeled samples with only natural abundance carbon contributions. Thus, the approach is broadly applicable to examining biofilm production among other strains and species of bacteria, including organisms such as S. aureus, Pseudomonas aeruginosa, and Vibrio cholera.
There are many fundamental questions about biofilm formation that remain unanswered and can begin to be explored with solid-state NMR. We can explore the types and extent of contacts that exist between the biofilm components and begin to create atomistic maps of biofilm structure. Indeed, it should be possible to utilize biosynthetic labeling strategies and NMR-detection schemes beyond what we have discussed in this review to further refine our developing description of a biofilm and its corresponding ECM as an organized assembly of polymeric macromolecules, connecting composition, structure, and function. The lessons learned from Nature’s assembly of biofilms may even inspire the synthesis of materials with new properties. Another significant opportunity and scientific need is to examine the influence of biofilm inhibitors [5, 68]. The non-crystalline and insoluble nature of biofilms makes it difficult to determine the localization of small molecules in biofilms using conventional methods. However, solid-state NMR can be used to map the binding of small molecules to biofilm components and to assess the changes in biofilm composition and structure, helping to reveal the molecular-modes of action of biofilm inhibitors. This understanding can help to drive the development of new biofilm inhibitors needed to prevent undesired bacterial colonization and biofilm formation associated with disease.
Acknowledgments
L.C. holds a Career Award at the Scientific Interface from the Burroughs Welcome Fund. C.R. is the recipient of the Stanford Graduate Fellowship. We gratefully acknowledge support from the NIH Director’s New Innovator Award, Stanford University, the Stanford Terman Fellowship, and the Hellman Faculty Scholar Award.
References
- 1.Hall-Stoodley L, Costerton JW, Stoodley P. Nature reviews Microbiology. 2004;2 (2):95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 2.Donlan RM, Costerton JW. Clinical Microbiology Reviews. 2002;15 (2):167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.del Pozo JL, Patel R. Clinical pharmacology and therapeutics. 2007;82 (2):204–209. doi: 10.1038/sj.clpt.6100247. [DOI] [PubMed] [Google Scholar]
- 4.Flemming HC. Applied microbiology and biotechnology. 2002;59 (6):629–640. doi: 10.1007/s00253-002-1066-9. [DOI] [PubMed] [Google Scholar]
- 5.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. Nature reviews Microbiology. 2008;6 (1):17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flemming HC, Wingender J. Nature reviews Microbiology. 2010;8 (9):623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 7.Sutherland IW. Microbiology-Uk. 2001;147:7. [Google Scholar]
- 8.Ha J, Gelabert A, Spormann AM, Brown GE. Geochimica Et Cosmochimica Acta. 2010;74 (1):1–15. [Google Scholar]
- 9.Jiao YQ, Cody GD, Harding AK, Wilmes P, Schrenk M, Wheeler KE, Banfield JF, Thelen MP. Applied and Environmental Microbiology. 2010;76 (9):2916–2922. doi: 10.1128/AEM.02289-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim JY, May JM, Cegelski L. Applied and Environmental Microbiology. 2012;78 (9):3369–3378. doi: 10.1128/AEM.07743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kramer KJ, Hopkins TL, Schaefer J. 1998;707:14–33. [Google Scholar]
- 12.Grage SL, Watts A. 2006;60:191–228. [Google Scholar]
- 13.Gullion T. 2009;65:111–137. [Google Scholar]
- 14.Renault M, Cukkemane A, Baldus M. Angewandte Chemie International Edition. 49(45):8346–8357. doi: 10.1002/anie.201002823. [DOI] [PubMed] [Google Scholar]
- 15.Toke O, Cegelski L. eMagRes. John Wiley & Sons, Ltd; 2007. [Google Scholar]
- 16.Zhou X, Cegelski L. Biochemistry. 2012;51 (41):8143–8153. doi: 10.1021/bi3012115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SJ, Cegelski L, Sutdelska DR, O’Connor RD, Mehta AK, Schaefer J. Biochemistry. 2002;41:11. doi: 10.1021/bi0121407. [DOI] [PubMed] [Google Scholar]
- 18.Kim SJ, Cegelski L, Preobrazhenskaya M, Schaefer J. Biochemistry. 2006;45:16. doi: 10.1021/bi052660s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tycko R. Quarterly reviews of biophysics. 2006;39 (1):1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 20.Tycko R. Annual review of physical chemistry. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. Proceedings of the National Academy of Sciences of the United States of America. 2002;99 (26):16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDermott A. Annual Review of Biophysics. 2009;38 (1):385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 23.Cegelski L, Schaefer J. The Journal of biological chemistry. 2005;280 (47):39238–39245. doi: 10.1074/jbc.M507053200. [DOI] [PubMed] [Google Scholar]
- 24.Dirks RC, Singh M, Potter GS, Sobotka LG, Schaefer J. New Phytologist. 196(4):1109–1121. doi: 10.1111/j.1469-8137.2012.04333.x. [DOI] [PubMed] [Google Scholar]
- 25.Andrew ER, Bradbury A, Eades RG. Nature. 1958;182 (4650):1659–1659. [Google Scholar]
- 26.Andrew ER, Newing RA. Proceedings of the Physical Society of London. 1958;72 (468):959–972. [Google Scholar]
- 27.Andrew ER, Bradbury A, Eades RG. Nature. 1959;183 (4678):1802–1803. [Google Scholar]
- 28.Lowe IJ. Physical Review Letters. 1959;2 (7):285–287. [Google Scholar]
- 29.Gullion T, Schaefer J. Journal of magnetic resonance. 1989;81 doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Prestegard JH, Al-Hashimi HM, Tolman JR. Quarterly Reviews of Biophysics. 2000;33 (04):371–424. doi: 10.1017/s0033583500003656. [DOI] [PubMed] [Google Scholar]
- 31.Hartmann SR, Hahn EL. Physical Review. 1962;128(5):2042. [Google Scholar]
- 32.Lurie FM, Slichter CP. Physical Review a-General Physics. 1964;133(4A):1108. [Google Scholar]
- 33.Pines A, Gibby MG, Waugh JS. Journal of Chemical Physics. 1973;59 (2):569–590. [Google Scholar]
- 34.Schaefer J, Stejskal EO. Journal of the American Chemical Society. 1976;98 (4):1031–1032. [Google Scholar]
- 35.Opella SJ, Frey MH. Journal of the American Chemical Society. 1979;101 (19):5854–5856. [Google Scholar]
- 36.Demco DE, Johansson A, Tegenfeldt J. Solid State Nuclear Magnetic Resonance. 1995;4 (1):13–38. doi: 10.1016/0926-2040(94)00036-c. [DOI] [PubMed] [Google Scholar]
- 37.McCrate OA, Zhou X, Reichhardt C, Cegelski L. Journal of molecular biology. doi: 10.1016/j.jmb.2013.06.022. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson GG, Dodson KW, Hooton TM, Hultgren SJ. Trends in microbiology. 2004;12 (9):424–430. doi: 10.1016/j.tim.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 39.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Science. 2003;301 (5629):105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 40.Soto SM, Smithson A, Martinez JA, Horcajada JP, Mensa J, Vila J. Journal of Urology. 2007;177 (1):365–368. doi: 10.1016/j.juro.2006.08.081. [DOI] [PubMed] [Google Scholar]
- 41.Mulvey MA, Schilling JD, Hultgren SJ. Infection and Immunity. 2001;69 (7):4572–4579. doi: 10.1128/IAI.69.7.4572-4579.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Justice SS, Hung C, Theriot JA, Fletcher DA, Anderson GG, Footer MJ, Hultgren SJ. Proceedings of the National Academy of Sciences of the United States of America. 2004;101 (5):1333–1338. doi: 10.1073/pnas.0308125100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Science. 2003;301 (5629):105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 44.Olsen A, Jonsson A, Normark S. Nature. 1989;338 (6217):652–655. doi: 10.1038/338652a0. [DOI] [PubMed] [Google Scholar]
- 45.Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. Science. 2002;295 (5556):851–855. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnhart MM, Chapman MR. Annual review of microbiology. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zogaj X, Nimtz M, Rohde M, Bokranz W, Romling U. Mol Microbiol. 2001;39:12. doi: 10.1046/j.1365-2958.2001.02337.x. [DOI] [PubMed] [Google Scholar]
- 48.Beloin C, Roux A, Ghigo JM. Bacterial Biofilms. 2008;322:41. doi: 10.1007/978-3-540-75418-3_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zogaj X, Bokranz W, Nimtz M, Romling U. Infection and Immunity. 2003;71 (7):4151–4158. doi: 10.1128/IAI.71.7.4151-4158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Da Re S, Ghigo JM. Journal of bacteriology. 2006;188 (8):3073–3087. doi: 10.1128/JB.188.8.3073-3087.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gualdi L, Tagliabue L, Bertagnoli S, Ierano T, De Castro C, Landini P. Microbiology. 2008;154(Pt 7):2017–2024. doi: 10.1099/mic.0.2008/018093-0. [DOI] [PubMed] [Google Scholar]
- 52.Saldana Z, Xicohtencatl-Cortes J, Avelino F, Phillips AD, Kaper JB, Puente JL, Giron JA. Environmental microbiology. 2009;11 (4):992–1006. doi: 10.1111/j.1462-2920.2008.01824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCrate OA, Zhou X, Cegelski L. Chemical communications. 2013;49 (39):4193–4195. doi: 10.1039/c2cc37792f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kai A, Xu P, Horii F, HUSH Polymer. 1994;35:5. [Google Scholar]
- 55.Maunu S, Liitia T, Kauliomaki S, Hortling B, Sundquist J. Cellulose. 2000;7:13. [Google Scholar]
- 56.Lindhorst TK, Kötter S, Krallmann-Wenzel U, Ehlers S. Journal of the Chemical Society, Perkin Transactions. 2001;1 (8):823–831. [Google Scholar]
- 57.Lim JY, May JM, Cegelski L. Applied and environmental microbiology. 2012;78 (9):3369–3378. doi: 10.1128/AEM.07743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu C, Lim JY, Fuller GG, Cegelski L. Biophysical journal. 2012;103 (3):464–471. doi: 10.1016/j.bpj.2012.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Otto M. Annual Review of Medicine. 2013;64:175–188. doi: 10.1146/annurev-med-042711-140023. [DOI] [PubMed] [Google Scholar]
- 60.Kiedrowski MR, Horswill AR. Annals of the New York Academy of Sciences. 1241(1):104–121. doi: 10.1111/j.1749-6632.2011.06281.x. [DOI] [PubMed] [Google Scholar]
- 61.Mercier C, Durrieu C, Briandet R, Domakova E, Tremblay J, Buist G, Kulakauskas S. Molecular Microbiology. 2002;46 (1):235–243. doi: 10.1046/j.1365-2958.2002.03160.x. [DOI] [PubMed] [Google Scholar]
- 62.Gallant CV, Daniels C, Leung JM, Ghosh AS, Young KD, Kotra LP, Burrows LL. Molecular Microbiology. 2005;58 (4):1012–1024. doi: 10.1111/j.1365-2958.2005.04892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weigel LM, Donlan RM, Shin DH, Jensen B, Clark NC, McDougal LK, Zhu W, Musser KA, Thompson J, Kohlerschmidt D, Dumas N, Limberger RJ, Patel JB. Antimicrobial agents and chemotherapy. 2007;51 (1):231–238. doi: 10.1128/AAC.00576-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cegelski L, Kim SJ, Hing AW, Studelska DR, O’Connor RD, Mehta AK, Schaefer J. Biochemistry. 2002;41:6. doi: 10.1021/bi0202326. [DOI] [PubMed] [Google Scholar]
- 65.Sharif S, Singh M, Kim SJ, Schaefer J. J Am Chem Soc. 2009;131:8. doi: 10.1021/ja808971c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim SJ, Tanaka KSE, Dietrich E, Rafai Far A, Schaefer J. Biochemistry. 52(20):3405–3414. doi: 10.1021/bi400054p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vollmer W, Blanot D, de Pedro MA. FEMS microbiology reviews. 2008;32 (2):149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 68.Cegelski L, Pinkner JS, Hammer ND, Cusumano CK, Hung CS, Chorell E, Aberg V, Walker JN, Seed PC, Almqvist F, Chapman MR, Hultgren SJ. Nature chemical biology. 2009;5 (12):913–919. doi: 10.1038/nchembio.242. [DOI] [PMC free article] [PubMed] [Google Scholar]