

Abstract
The inheritance of the ε4 allele of the apolipoprotein E (apoE) gene is the major genetic risk factor for developing late‐onset Alzheimer disease. In transgenic mice overexpressing amyloid precursor protein (APP), replacing the endogenous mouse apoE gene with the human apolipoprotein E4 (apoE4) gene alters the distribution of amyloid‐β (Aβ) deposits from the brain parenchyma to the vasculature. However, the effects of this distribution on the onset and progression of tau pathology remain to be established. To address this issue, we used a genetic approach to replace the endogenous apoE gene with the human apoE4 allele in the 3xTg‐AD mice. We showed that changing Aβ distribution from the parenchyma to the vasculature drastically reduces the tau pathology. The 3xTg‐AD mice expressing the human apoE4 gene were virtually depleted of any somatodendritic tau deposits. These data strongly suggest that the somatodendritic tau accumulation is dependent on the parenchyma Aβ deposits.
Keywords: Alzheimer, apoE, tau, tangles
INTRODUCTION
Alzheimer disease (AD), the most common age‐related neurodegenerative disorder, is characterized by the accumulation of two insoluble protein aggregates: amyloid‐β (Aβ), containing deposits in the form of diffuse and fibrillar amyloid plaques (27), and tau‐laden neurofibrillary tangles 14, 24. Aβ is derived from the sequential cleavage of the amyloid precursor protein (APP), first by the β‐site APP cleavage enzyme (BACE1) (47) and then by γ‐secretase (41). It has been postulated that Aβ accumulation is central to the disease pathogenesis, whereas tau pathology and other neuropathologic changes are a downstream consequence of the pathologic accumulation of Aβ species (15).
Genetic studies have shown the apolipoprotein E (apoE) gene is a major risk factor for AD (39) and for cerebral amyloid angiopathy (CAA), an age‐dependent disorder characterized by vascular Aβ deposits. apoE is a polymorphic gene, for which three different allelic isoforms exist in humans: ε2, ε3 and ε4. The inheritance of the ε4 allele is associated with an earlier age of onset for AD 36, 39, brain atrophy (7), higher Aβ plaque burden 37, 39 and high vascular deposits of Aβ(5), suggesting that interactions between apoE and Aβ facilitate the development of pathology 4, 18. The introduction of human apolipoprotein E3 (apoE3) or apoE4 alleles into mutant APP transgenic mice markedly delays the buildup of Aβ deposition and is associated with a robust enhancement of CAA, with the greatest pathology occurring in mice expressing apoE4 8, 11, 20.
Given the critical role that Aβ plays in the pathogenesis of AD, we sought to understand its relationship to tau pathology. Using the 3xTg‐AD mice, which develop plaque and tangle pathology in an age‐dependent fashion 29, 30, 33, we previously showed that Aβ immunotherapy not only removes Aβ deposits, but is also sufficient to reduce early tau deposits 31, 33, 34. These results suggest that the tau pathology in the 3xTg‐AD mice is highly dependent on brain Aβ accumulation. The relationship between vascular Aβ deposits and tau pathology, however, is not well understood. To directly address this question, we genetically shifted Aβ accumulation from the parenchyma to the vasculature by replacing the endogenous mouse apoE gene with the human apoE4 gene. We reported that shifting the Aβ deposits from mainly parenchymal to mainly vascular delays the onset and progression of tau deposits. These data further support an upstream role of Aβ in triggering tau pathology and showed for the first time that tau pathology is dependent on the presence of parenchyma Aβ deposits.
MATERIAL AND METHODS
Mice
The derivation and characterization of the 3xTg‐AD mice has been described elsewhere (30). Briefly, two independent transgenes encoding human APPSwe and the human tauP301L (both under the control of the mouse Thy1.2 regulatory element) were co‐microinjected into single‐cell embryos harvested from homozygous mutant PS1M146V knockin (PS1‐KI) mice. The derivation and characterization of the apoE4‐KI mice has been previously described (23). Mice were given ad libitum access to food and water.
Antibodies
The following antibodies were used: anti‐Aβ 6E10, anti‐Aβ 1560, both raised against amino acids 1–17 of Aβ, anti‐Aβ1‐42, anti‐APP 22C11 (raised against amino acid 66–81 of APP), anti‐APP CT20 (raised against amino acids 751–770 of APP), anti‐Tau HT7 (raised against amino acids 159–163), AT8 (recognizes phosphorylated Ser202, Thr205), anti‐β‐actin, anti‐CDK5, which recognizes amino acids 268–283, anti‐GSK3βSer9, which is raised against phosphorylated GSK3β at Ser9, anti‐human apoE (Serotec, Raleigh, NC) and anti‐mouse apoE (Abcam, Cambridge, MA).
Protein extraction, immunohistochemistry and Western blot
Mice were sacrificed by CO2 asphyxiation and their brains were cut in half sagitally. One‐half of the brain was fixed for 48 h in 4% paraformaldehyde in PBS for immunohistochemical analysis. The other half was frozen in dry ice for biochemical analysis. Fifty‐micrometer‐thick free‐floating sections were obtained using a vibratome slicing system (Vibratome, Cambridge, MA). A detailed immunostaining procedure has been previously described (32). For confocal microscopy, after incubating the sections in the appropriate primary antibody, suitable Alexa Fluor secondary antibody (Invitrogen, Carlsbad, CA) was applied.
The primary antibodies were applied at the following dilutions: 1:1000 for 6E10, 1:3000 for 1560, 1:200 for Aβ42, 1:1000 for HT7, 1:200 for AT8, 1:1000 for anti‐human apoE and 1:1000 for anti‐mouse apoE.
For biochemical analysis, the brains were homogenized in tissue protein extraction reagent solution [(T‐PER) Pierce, Rockford, IL] in H2O containing 0.7 mg/mL pepstatin A supplemented with complete mini protease inhibitor tablet and phosphatase inhibitors (1:100). The homogenized mixes were sonicated to sheer the DNA and centrifuged at 4°C for 1 h at 100 000 g. The supernatant was stored as a soluble fraction. The pellet was rehomogenized in 70% formic acid (FA) and centrifuged as above. The supernatant was stored as the insoluble fraction. Proteins from the soluble fraction were resolved using standard Western blot techniques.
ELISA
Aβ1‐40 and Aβ 1‐42 levels were measured using a sensitive sandwich ELISA system. Proteins from the soluble fraction (see above) were loaded directly onto ELISA plates, and FA fractions were diluted (1:20) in neutralization buffer (1 M Tris base; 0.5 M NaH2PO dibasic) prior to loading. MaxiSorp immunoplates (Nalge Nunc International, Rochester, NY, USA) were coated with monoclonal antibody 20.1, a specific antibody against Aβ1‐16 (from Dr. W.E. Van Nostrand, Stony Brook University, NY, USA) in coating buffer (0.1 M NaCO3, pH 9.6), and blocked with 3% bovine serum albumin (BSA). Synthetic Aβ standards (Bachem, King of Prussia, PA, USA) were defibrillated by dissolving in hexafluoroisopropanol (HFIP) at 1 mg/mL, and the HFIP evaporated with a stream of N2. The defibrillated Aβ was dissolved in dimethyl sulfoxide (DMSO) at 1 mg/mL. Standards of both Aβ1‐40 and Aβ1‐42 were made in antigen capture buffer [(ACB) 20 mM NAH2PO4, 2 mM ethylenediaminetetraacetic acid (EDTA), 0.4 M NaCl, 0.5 g CHAPS, 1% BSA, pH 7.0] and loaded onto ELISA plates in duplicate and incubated overnight at 4°C. The plates were washed and probed with either Horseradish Peroxidase (HRP)‐conjugated anti‐Aβ 35‐40 (MM32‐13.1.1, for Aβ40) or anti‐Aβ 35‐42 (MM40‐21.3.4, for Aβ42) overnight at 4°C. 3,3′,5,5′‐Tetramethylbenzidine was used as the chromagen, and the reaction was stopped with the addition of 30% O‐phosphoric acid and read at 450 nm on a plate reader (Labsystems, Sunnyvale, CA, USA).
Cholesterol measurement
The experimental protocol used for lipid extraction and measurement of cholesterol in the brain has previously been described (43). Briefly, one‐half brains (n = 5/group/genotype) were homogenized in 1 mL of PBS. Three milliliters of methanol and 1.5 mL of chloroform were subsequently added to each sample, and the mix was incubated at Room Temperature for 15 minutes while shaking. The samples were then spun at 4000 rpm for 10 minutes. The supernatant was transferred into a new tube, and the pellet was reincubated with methanol and chloroform as described above. The supernatant from both centrifugations was combined and dried at 30°C in a speed vacuum. The dried pellet was then resuspended in 100 µL of 10 mL/L Triton X‐100, 6 mL/L Brij 96 and 0.1 M/L HCl in 0.9% NaCl. Cholesterol levels were measured using a cholesterol quantification kit from BioVision (Mountain View, CA, USA) and using the manufacturer's protocol.
Statistics
Data analysis was conducted either by t‐test or using one‐way analyses of variance (ANOVAs) with Bonferroni post‐test using GraphPad Prism version 3.00 for Windows (GraphPad Software, San Diego, CA, USA). All the data analyses were conducted in a blinded fashion.
RESULTS
To determine the effect of replacing mouse apoE with the human apoE4 gene on the onset and progression of tau pathology, we crossed homozygous 3xTg‐AD mice [PS1M146VKI+/+; APPswe+/+; tauP301L+/+(30)] with the human apoE4 knockin mice, where the human apoE4 gene is expressed under the control of the murine apoE regulatory sequences (23). The 3xTg‐AD mice developed an age‐ and region‐dependent accumulation of plaques and tangles, which is accompanied with an age‐associated cognitive decline 2, 29, 30, 33. For these studies, we used three different groups of mice: (i) 3xTg‐AD; (ii) 3xTg‐AD containing one mouse apoE allele and one human apoE4 allele (3xTg‐AD/ε4h); and (iii) 3xTg‐AD containing two human apoE4 alleles and no mouse apoE alleles (3xTg‐AD/ε4H; Table 1). Western blot analysis using antibodies specific against either mouse or human apoE yielded the anticipated dose‐dependent and species‐specific expression pattern (Figure 1). Considering that human apoE4 is driven by the endogenous mouse promoter, the cellular distribution of human apoE is similar to the mouse apoE as confirmed by confocal microscopy experiments. Sections form 3xTg‐AD/ε4h mice, which express both human and mouse apoE, where double labeled with a human‐specific apoE antibody and mouse‐specific apoE antibody. As expected, we found that the mouse and human apoE completely colocalize (Figure 1D). Notably, the expression of human apoE4 did not alter brain cholesterol levels (Figure 1E).
Table 1.
Genotype of the mice used in this study. Abbreviations: PS1‐KI = PS1M146V knockin; APP = amyloid precursor protein; apoE = apolipoprotein E; apoE4 = apolipoprotein E4.
| PS1‐KI | Human APP | Human tau | Mouse apoE | Human apoE4 | |
|---|---|---|---|---|---|
| 3xTg‐AD | +/+ | +/+ | +/+ | +/+ | −/− |
| 3xTg‐AD/ε4h | +/+ | +/+ | +/+ | +/− | +/− |
| 3xTg‐AD/ε4H | +/+ | +/+ | +/+ | −/− | +/+ |
Figure 1.
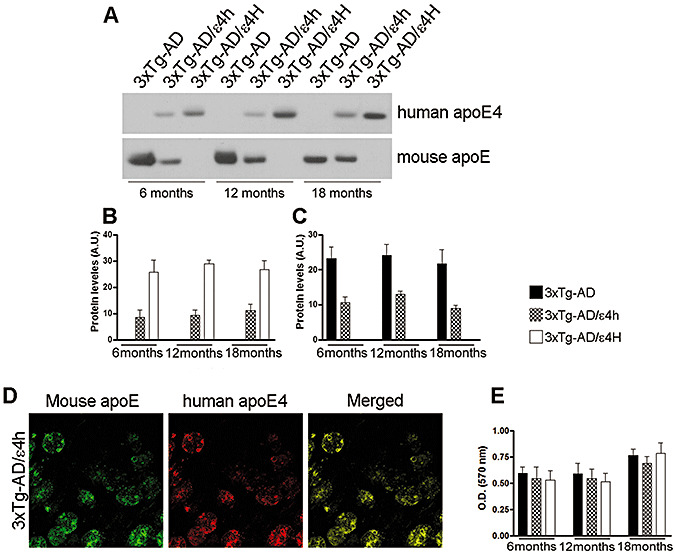
Dose‐dependent and species‐specific expression pattern of apolipoprotein E (apoE). To determine the expression profile of mouse and human apoE as a function of age and genotype, we measured the steady‐state levels of mouse apoE and human apolipoprotein E4 (apoE4) using species‐specific antibodies. A. Representative Western blot probed with a specific antibody against mouse apoE or human apoE4 of protein extracts from the brains of 3xTg‐AD, 3xTg‐AD/ε4h and 3xTg‐AD/ε4H mice at different time points. B,C. Quantitative analysis of the mouse and human apoE4 levels indicates a dose‐dependent and species‐specific expression of apoE. The graph represents the mean of the apoE levels measured in 10 mice per group. The error bar indicates standard error. D. Sections from 18‐month‐old 3xTg‐AD mice were stained with a human‐specific apoE antibody and a mouse‐specific apoE antibody and analyzed using confocal microscopy. The merged image shows a complete co‐localization of human and mouse apoE, consistent with the fact that the expression of both mouse and human apoE is controlled by the same endogenous promoter. E. Expression of the human apoE4 did not alter brain cholesterol levels (n = 5/group/genotype).
Expression of apoE4 leads to a reduction in parenchyma Aβ deposits and Aβ levels
To define the impact on the Aβ pathology after replacing the endogenous mouse apoE gene with one or two human apoE4 alleles in the 3xTg‐AD mice, we first determined if the steady‐state levels of APP and Aβ were altered. Using Western blot analysis, we measured the steady‐state levels of APP in the brains across different time points and found that the expression of the human apoE4 gene did not alter APP levels at any of the ages analyzed (Figure 2A–D).
Figure 2.

Reduction in Amyloid‐β 42 (Aβ42) levels following expression of human apolipoprotein E4 (apoE4). Panel A shows representative Western blots for amyloid precursor protein (APP) holoprotein. βActin was used as a loading control. B–D. Quantification analysis shows that the steady‐state levels of APP were not altered in any of the ages analyzed (n = 10 mice/group/time point). To determine how the replacement of mouse apolipoprotein E (apoE) with human apoE4 alters amyloid‐β (Aβ) levels in the brain, we performed Enzyme‐Linked ImmunoSorbent Assay (ELISA) measurements. E–G. Although soluble Aβ40 levels were similar among the three groups at any of the ages analyzed, soluble Aβ42 levels were significantly reduced in 6‐ and 12‐month‐old 3xTg‐AD/ε4h and in 6‐, 12‐ and 18‐month‐old 3xTg‐AD/ε4H mice compared with age‐matched 3xTg‐AD mice (n = 10 mice/group/time point). H–J. With the exception of 6‐month‐old mice, insoluble Aβ40 and Aβ42 levels were significantly decreased in the mice expressing human apoE4. * indicates a P‐value < 0.05; ** indicates a P‐value < 0.01; *** indicates a P‐value < 0.001.
To determine how Aβ levels change as a function of age, we performed ELISA measurements with protein extracts from the brains of 6‐, 12‐ and 18‐month‐old mice. Soluble Aβ40 levels did not differ among the three groups at any of the ages analyzed (Figure 2E–G). Although not statistically significant, soluble Aβ42 levels were lower in the brains of 6‐month‐old 3xTg‐AD/ε4h and 3xTg‐AD/ε4H compared with 3xTg‐AD mice (Figure 2E). As the mice age, soluble Aβ42 levels are significantly lower in 3xTg‐AD/ε4h (at 12 months) and in the 3xTg‐AD/ε4H (at 12 and 18 months) compared with the 3xTg‐AD mice expressing the mouse apoE gene (Figure 2F,G). The levels of insoluble Aβ40 and Aβ42 were similar among the three groups of mice at 6 months of age; however, a marked decrease was observed in the mice expressing one or two copies of the human apoE compared with the 3xTg‐AD mice at both 12 and 18 months of age (Figure 2H–J).
We next determined whether Aβ deposition was altered in the 3xTg‐AD mice expressing one or two human apoE4 alleles. Toward this end, we immunostained sections from the brains of 6‐, 12‐ and 18‐month‐old mice. These ages correspond to the time points in the 3xTg‐AD mice with different stages of Aβ pathology. At 6 months of age, the 3xTg‐AD mice showed intraneuronal Aβ deposition in several brain regions including the hippocampus (Figure 3A–C). Notably, the intraneuronal Aβ staining was markedly decreased in 6‐month‐old 3xTg‐AD/ε4h mice (Figure 3D–F), and little, if any, intraneuronal Aβ was evident in 6‐month‐old 3xTg‐AD/ε4H mice (Figure 3G–I). As the 3xTg‐AD mice age, the Aβ deposits become extracellular and are readily detectable in the hippocampus of 12‐month‐old mice (Figure 3J–L). In contrast, we found that 12‐month‐old 3xTg‐AD/ε4h mice lack extracellular Aβ staining, and only intraneuronal Aβ is apparent (Figure 3M–O). This is the first time point at which 3xTg‐AD/ε4H mice start to accumulate some intracellular Aβ in the CA1/subiculum region of the hippocampus (Figure 3R), which illustrates the delay in Aβ accumulation caused by completely replacing the mouse apoE gene with the human apoE4. In 18‐month‐old 3xTg‐AD mice, the Aβ pathology is widespread throughout the brain, and extensive extracellular plaques are detected (Figure 3S–U). In the 3xTg‐AD/ε4h mice, extracellular Aβ deposits start to be apparent in the hippocampus (Figure 3V–W), and in the 3xTg‐AD/ε4H mice, intraneuronal Aβ staining is now more evident in the hippocampus, although even at this advanced age, extracellular Aβ deposits are not detected (Figure 3Y–AA).
Figure 3.

Reduction in parenchymal amyloid‐β (Aβ) deposition in mice expression of human apolipoprotein E4 (apoE4). To define the impact of replacing the endogenous mouse apolipoprotein E (apoE) gene with one or two human apoE4 alleles in the 3xTg‐AD mice on the Aβ pathology, we immunostained sections from the brains of 6‐, 12‐ and 18‐month‐old mice (n = 10 mice/group/time point). A–I. Six‐month‐old 3xTg‐AD mice show a robust intracellular Aβ immunoreactivity throughout the hippocampus, and virtually every CA1‐pyramidal neuron shows intraneuronal Aβ immunoreactivity. In contrast, the intraneuronal Aβ immunoreactivity is reduced in mice expressing one copy of the human apoE4 allele and is not present in mice expressing two copies of the apoE4 gene. J–R. The Aβ pathology increases as a function of age, and extracellular Aβ deposits are readily detectable in the hippocampus of 12‐month‐old 3xTg‐AD mice. Although the Aβ pathology also increases as a function of age in mice expressing the human apoE4 gene, the degree of staining is always reduced compared with age‐matched 3xTg‐AD mice, in particular, no extracellular Aβ deposits are found in the brains of 12‐month‐old 3xTg‐AD/ε4h mice and only intraneuronal Aβ is detected. S–AA. The Aβ pathology is widespread throughout the brains of 18‐month‐old 3xTg‐AD mice with extensive extracellular plaques. In contrast, only at this age 3xTg‐AD/ε4h mice start to accumulate extracellular Aβ deposits in the CA1/subiculum region, whereas age‐matched 3xTg‐AD/ε4H start to accumulate intraneuronal Aβ staining, although even at this advanced age, extracellular Aβ deposits are not detected. The scale bar is 500 µm for A, D, G, J, M, P, S, V, Y, and 125 µm for B, C, E, F, H, I, K, L, N, O, Q, R, T, U, X, W, Z, AA.
These data are consistent with the marked decrease in insoluble Aβ levels measured by ELISA (Figure 2H–J) and with the work by Holtzman and colleagues showing that the introduction of human apoE4 into APP transgenic mice leads to a delay in parenchymal Aβ deposition 10, 11, 12, 13, 19, 20. They also reported that replacing mouse apoE with human apoE4 causes a robust increase in vascular Aβ12, 13. Similarly, we found that replacing the mouse apoE gene with the human apoE4 leads to an increase in vascular Aβ deposits as detected by thioflavin S staining (Figure 4). Semiquantitative analysis indicated that the number thioflavin S‐positive vascular deposits correlates positively with the apoE4 gene dosage (Figure 4D). As there is a marked buildup of Aβ in the walls of the cerebral blood vessels, we next determined if blood levels of Aβ40 and Aβ42 were altered. At all ages analyzed, the levels of Aβ40 were below detection (data not shown). Although the changes were not statistically significant, we found that the blood levels of Aβ42 were increased in the mice expressing human apoE4 in a dose‐dependent manner at 6 and 12 months of age (Figure 4E). This increase becomes significant between 18‐month‐old 3xTg‐AD and 3xTg‐AD/ε4H (Figure 4E).
Figure 4.

Replacing the mouse apolipoprotein E (apoE) gene with the human apolipoprotein E4 (apoE4) leads to an increase in vascular amyloid‐β (Aβ) deposits. To determine the effect of expressing human apoE4 on vascular Aβ deposits, we stained sections from 3xTg‐AD, 3xTg‐AD/ε4h and 3xTg‐AD/ε4H mice with thioflavin S (n = 10 mice/group/time point). A–C. Representative thioflavin S‐positive deposits from the brains of 3xTg‐AD (A), 3xTg‐AD/ε4h (B) and 3xTg‐AD/ε4H (C) showing Aβ deposits in the brain vasculature. D. Semiquantitative analysis of the stained section indicated that the number of thioflavin S‐positive vascular deposits correlates positively with the apoE4 gene dosage and increased as a function of age. Because we showed that there is an increase in vascular Aβ deposition in mice expressing human apoE4, we next measured Aβ levels in the blood of these mice. E. Although the changes were not statistically significant, we found that the blood levels of Amyloid‐β 42 (Aβ42) were increased in the mice expressing human apoE4 in a dose‐dependent manner at 6 and 12 months of age. This increase becomes significant between 18‐month‐old 3xTg‐AD and 3xTg‐AD/ε4H. Aβ40 was not detectable at any of the ages analyzed. * indicates a P‐value < 0.05; ** indicates a P‐value < 0.01; *** indicates a P‐value < 0.001.
Taken together, the results presented here indicate that there is an apoE4‐dependent delay in the progression of the intraneuronal and parenchyma Aβ deposition and a concomitant increase in vascular Aβ deposits. Toward this end, the parenchymal Aβ deposition in 18‐month‐old 3xTg‐AD/ε4h mice is comparable to that of 12‐month‐old 3xTg‐AD mice. Moreover, the Aβ burden in 18‐month‐old 3xTg‐AD/ε4H mice is less compared with that observed in 6‐month‐old 3xTg‐AD mice.
Marked decrease in tau pathology following expression of the human apoE4 gene
It has been suggested that there may be an interaction between different apoE genotypes and tau pathology 1, 9, 22, 28, 48. To determine whether the onset or distribution profile of the tau pathology was altered by replacing the mouse apoE gene with the human apoE4 alleles, we immunostained sections from 3xTg‐AD, 3xTg‐AD/ε4h and 3xTg‐AD/ε4H mice with a human‐specific anti‐tau antibody. We first noted that the localization pattern of tau was altered as a function of the apoE genotype. Whereas somatodendritic tau is readily detectable in the hippocampus of 6‐month‐old 3xTg‐AD mice, replacing the mouse apoE gene with different doses of the E4 allele had a marked impact on the subcellular distribution of tau accumulation (Figure 5A–C). Replacing a single mouse allele with the human E4 allele sufficed to significantly reduce somatodendritic tau localization in CA1 pyramidal neurons, whereas replacing both alleles virtually eliminated any tau localization in this compartment (Figure 5A–C). The human apoE4 gene delayed the onset of the tau pathology in these mice, an effect also dependent on the gene dosage. For example, age‐dependent increase in somotodendritic tau was also detected in the 3xTg‐AD/ε4h mice (Figure 5B, E, H); however, the number of CA1 pyramidal neurons and the intensity of the staining was always reduced compared with age‐matched 3xTg‐AD mice (Figure 5A, B, D, E, G, H). Notably, we did not find any somatodendritic tau accumulation in the hippocampi of 3xTg‐AD/ε4H mice at any of the ages analyzed (Figure 5C, F, I).
Figure 5.

Marked reduction in somatodendritic tau accumulation as a function of human apolipoprotein E4 (apoE4) expression. To determine if the distribution of tau accumulation was altered following expression of human apoE4, we stained brain sections from 3xTg‐AD, 3xTg‐AD/ε4h and 3xTg‐AD/ε4H mice (n = 10/group/time point) with the human‐specific anti‐tau antibody HT7. A–C. Somatodendritic tau deposits in the hippocampus are significantly reduced after replacing endogenous mouse apoE with human apoE4 in a dose‐dependent manner. D–I. Although tau accumulation increases as a function of age, replacing a single mouse allele with the human E4 allele sufficed to significantly reduce somatodendritic tau localization in CA1 pyramidal neurons, whereas replacing both alleles virtually eliminated any tau localization in this compartment. The scale bar is 250 µm.
In the 3xTg‐AD mice, tau undergoes age‐dependent changes in conformation and phosphorylation, mimicking events occurring in the AD brain 30, 35. To determine the impact of human apoE4 on tau phosphorylation, we measured the levels of tau phosphorylated at Ser202/205, using the anti‐phosphotau antibody AT8. Western blot analysis revealed a robust decrease in the level of AT8‐positive tau in the brains of the 3xTg‐AD/ε4h and ε4H mice vs. age‐matched 3xTg‐AD mice (Figure 6A,B). Thus, not only was there a delay in the somatodendritic localization of tau, but replacing the mouse apoE gene with an E4 allele also suppressed the phosphotau phenotype. To elucidate the mechanism underlying the reduction in tau phosphorylation, we used quantitative Western blot analysis to screen a panel of candidate tau kinases, using antibodies against the activated form of these enzymes. We found that the steady‐state levels of the active forms of GSK3α or CDK5 and its activators p25 and p35, were not significantly different among the groups analyzed (data not shown). GSK3β is another important kinase involved in tau phosphorylation, and it has been shown that phosphorylation at Ser9 inversely correlates with its activity. Using an antibody specific for GSK3β phosphorylated at Ser9, we found a significant increase in the levels of GSK3βSer9 in the brain of 3xTg‐AD/ε4h and ε4H mice compared with age‐matched 3xTg‐AD mice (Figure 6A,C). These data suggest that the decrease in tau phosphorylation in the mice expressing human apoE4 is mediated by the reduction of GSK3β activity and are consistent with data showing that apoE decreases the activity of tau kinases and tau phosphorylation in primary neurons (17).
Figure 6.

Age‐ and genotype‐dependent decrease in tau levels and phosphorylation. To determine if the phosphorylation state of tau was altered as a function of the apolipoprotein E (apoE) genotype, we measured the steady‐state levels of tau phosphorylated at Ser202/205 in the brains of 3xTg‐AD, 3xTg‐AD/ε4h and 3xTg‐AD/ε4H by Western blot analysis. Panel A shows representative Western blots for phosphorylated tau (AT8) and the inactive form of glycogen synthase kinase 3 beta (GSK3β). βActin was used as a loading control. B. Quantification analysis of the steady‐state levels of AT8 shows that there is a marked and significant reduction of tau phosphorylation at Ser202 in the mice expressing one or two copies of human apolipoprotein E4 (apoE4). To elucidate the mechanism underlying the reduction in tau phosphorylation following human apoE4 expression, we measured the levels of the inactive form of GSK3β, one of the major kinases involved in tau phosphorylation. D. Quantification analysis of the blots indicates that there is a decrease in GSK3β activity as a function of age and human apoE4 dosage. * indicates a P‐value < 0.05; ** indicates a P‐value < 0.01; *** indicates a P‐value < 0.001.
DISCUSSION
The inheritance of the apoE4 allele is the single most important risk factor for developing late‐onset AD (39), whereas the apolipoprotein E2 (apoE2) allele has a protective effect (37). In addition, the Aβ burden positively correlates with the apoE genotype (E4 > E3 > E2) 6, 37, 39, clearly indicating that diverse apoE isoforms differentially influence Aβ pathology. Toward this end, the data presented here indicate that mouse apoE facilitates parenchyma Aβ pathology to a greater extent than human apoE4. Conversely, we showed that replacing the endogenous apoE gene with the human apoE4 gene leads to a dose‐ and age‐dependent increase in Aβ accumulation in the walls of the brain vasculature. These data are consistent with the large body of evidence generated by Holtzman and colleagues showing in different transgenic models that the expression of human apoE4 shifts Aβ deposits from the parenchyma to the vasculature 4, 8, 10, 11, 12, 13, 18, 19, 20.
Recent evidence indicates that intracellular Aβ accumulation plays a pivotal role in the pathogenesis of AD (26). One of the mechanisms proposed for the intracellular Aβ accumulation is its reuptake mediated by apoE receptors, such as the low density lipoprotein receptor related protein (LRP) receptor. Here, we showed that replacing mouse apoE with the human apoE4 allele significantly reduced intracellular Aβ accumulation at all ages analyzed. Consistent with our data, it has been reported that mouse apoE and human apoE4 have different affinities for Aβ(25), and the Aβ/apoE complexes can differentially bind to the apoE receptors 40, 49, thereby influencing Aβ reuptake, and likely its intracellular accumulation.
We previously showed that clearing Aβ deposits from the brains of the 3xTg‐AD mice by Aβ immunization also reduces early tau pathology, suggesting that Aβ accumulation is upstream of tau pathology 31, 33, 34. However, our previous studies did not discriminate if the clearance of tau pathology was mediated by the clearance of parenchymal or vascular Aβ. Here, we showed that tau pathology is markedly reduced in the mice expressing human apoE4, with little or no somatodendritic tau accumulation present even in 18‐month‐old mice, despite the presence of robust vascular Aβ deposits. Previous reports have shown an increase in tau phosphorylation in transgenic mice overexpressing human apoeE4 3, 44. There is an apparent disagreement between these reports and our data. Notably, in both reports, the human apoE4 was overexpressed under the control of a foreign promoter, whereas for our experiments, we have used mice where human apoE4 is expressed at physiological levels under the endogenous promoter. These differences in the experimental paradigms can explain the apparent discordance in results. The data presented here indicate that under the experimental conditions used here, clearing Aβ from the parenchyma is sufficient to reduce tau pathology.
The mechanism underlying the clearance of tau pathology is likely dependent on the proteasome. Toward this end, we previously showed that Aβ can impair proteasome activity and its removal reestablishes proteasome function 31, 45. It has also been suggested that apoE may alter tau pathology either by a direct interaction with tau 3, 16, 21, 42 or via its receptors apolipoprotein E receptor 2 (ApoER2) and LRP, which mediate several intracellular signaling kinases including the tau kinases PI3K, GSK3β, CDK5 and MAPK(38). Consistent with these reports, here we showed a significant decrease in the activity of GSK3β in the mice expressing human apoE4. A previous report showed that while expressing apoE4 in combination with mutant APP and mutant presenilin‐1 increased the vascular deposition of Aβ (consistent with the data presented here), the cellular distribution of tau was not altered (46). Although this may seem inconsistent with our results, it should be noted that the 3xTg‐AD mice showed accumulation of phosphorylated and aggregated tau and neurofibrillary tangles, whereas none of the mice reported in Van Dooren et al showed robust tau pathology.
Taken together, the data presented here further support an upstream role of Aβ in triggering tau pathology and indicate that tau accumulation is highly dependent on the presence of intracellular and/or extracellular Aβ deposits, despite the fact that the tau pathology in the 3xTg‐AD mice is driven by its own transgene.
ACKNOWLEDGMENTS
This work was supported by funding from the National Institute on Aging (AG0212982) to F.M.L. and (AG029729A) to S.O.
REFERENCES
- 1. Bernardi L, Maletta RG, Tomaino C, Smirne N, Di Natale M, Perri M et al (2006) The effects of APOE and tau gene variability on risk of frontotemporal dementia. Neurobiol Aging 27:702–709. [DOI] [PubMed] [Google Scholar]
- 2. Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM (2005) Intraneuronal Abeta causes the onset of early Alzheimer's disease‐related cognitive deficits in transgenic mice. Neuron 45:675–688. [DOI] [PubMed] [Google Scholar]
- 3. Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q et al (2004) Neuron‐specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci 24:2527–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brendza RP, Bales KR, Paul SM, Holtzman DM (2002) Role of apoE/Abeta interactions in Alzheimer's disease: insights from transgenic mouse models. Mol Psychiatry 7:132–135. [DOI] [PubMed] [Google Scholar]
- 5. Chalmers K, Wilcock GK, Love S (2003) APOE epsilon 4 influences the pathological phenotype of Alzheimer's disease by favouring cerebrovascular over parenchymal accumulation of Abeta protein. Neuropathol Appl Neurobiol 29:231–238. [DOI] [PubMed] [Google Scholar]
- 6. Chapman J, Korczyn AD, Karussis DM, Michaelson DM (2001) The effects of APOE genotype on age at onset and progression of neurodegenerative diseases. Neurology 57:1482–1485. [DOI] [PubMed] [Google Scholar]
- 7. Chen K, Reiman EM, Alexander GE, Caselli RJ, Gerkin R, Bandy D et al (2007) Correlations between apolipoprotein E {epsilon}4 gene dose and whole brain atrophy rates. Am J Psychiatry 164:916–921. [DOI] [PubMed] [Google Scholar]
- 8. DeMattos RB, Cirrito JR, Parsadanian M, May PC, O'Dell MA, Taylor JW et al (2004) ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron 41:193–202. [DOI] [PubMed] [Google Scholar]
- 9. Engelborghs S, Dermaut B, Marien P, Symons A, Vloeberghs E, Maertens K et al (2006) Dose dependent effect of APOE epsilon4 on behavioral symptoms in frontal lobe dementia. Neurobiol Aging 27:285–292. [DOI] [PubMed] [Google Scholar]
- 10. Fagan AM, Younkin LH, Morris JC, Fryer JD, Cole TG, Younkin SG, Holtzman DM (2000) Differences in the Abeta40/Abeta42 ratio associated with cerebrospinal fluid lipoproteins as a function of apolipoprotein E genotype. Ann Neurol 48:201–210. [PubMed] [Google Scholar]
- 11. Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM (2002) Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis 9:305–318. [DOI] [PubMed] [Google Scholar]
- 12. Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, Holtzman DM (2003) Apolipoprotein E markedly facilitates age‐dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci 23:7889–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM (2005) Human apolipoprotein E4 alters the amyloid‐beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 25:2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grundke‐Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986) Abnormal phosphorylation of the microtubule‐associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83:4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 16. Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss‐Coray T et al (2003) Carboxyl‐terminal‐truncated apolipoprotein E4 causes Alzheimer's disease‐like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci U S A 100:10966–10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoe HS, Freeman J, Rebeck GW (2006) Apolipoprotein E decreases tau kinases and phospho‐tau levels in primary neurons. Mol Neurodegener 1:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holtzman DM (2001) Role of apoe/Abeta interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy. J Mol Neurosci 17:147–155. [DOI] [PubMed] [Google Scholar]
- 19. Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM et al (1999) Expression of human apolipoprotein E reduces amyloid‐beta deposition in a mouse model of Alzheimer's disease. J Clin Invest 103:R15–R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ et al (2000) Apolipoprotein E isoform‐dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 97:2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang Y, Liu XQ, Wyss‐Coray T, Brecht WJ, Sanan DA, Mahley RW (2001) Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle‐like intracellular inclusions in neurons. Proc Natl Acad Sci U S A 98:8838–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ingelson M, Fabre SF, Lilius L, Andersen C, Viitanen M, Almkvist O et al (2001) Increased risk for frontotemporal dementia through interaction between tau polymorphisms and apolipoprotein E epsilon4. Neuroreport 12:905–909. [DOI] [PubMed] [Google Scholar]
- 23. Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH et al (1999) Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J Clin Invest 103:1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kosik KS, Joachim CL, Selkoe DJ (1986) Microtubule‐associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 83:4044–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. LaDu MJ, Lukens JR, Reardon CA, Getz GS (1997) Association of human, rat, and rabbit apolipoprotein E with beta‐amyloid. J Neurosci Res 49:9–18. [PubMed] [Google Scholar]
- 26. LaFerla FM, Green KN, Oddo S (2007) Intracellular amyloid‐beta in Alzheimer's disease. Nat Rev Neurosci 8:499–509. [DOI] [PubMed] [Google Scholar]
- 27. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82:4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mehta SG, Watts GD, Adamson JL, Hutton M, Umberger G, Xiong S et al (2007) APOE is a potential modifier gene in an autosomal dominant form of frontotemporal dementia (IBMPFD). Genet Med 9:9–13. [DOI] [PubMed] [Google Scholar]
- 29. Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM (2003) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging 24:1063–1070. [DOI] [PubMed] [Google Scholar]
- 30. Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R et al (2003) Triple‐transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421. [DOI] [PubMed] [Google Scholar]
- 31. Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM (2004) Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43:321–332. [DOI] [PubMed] [Google Scholar]
- 32. Oddo S, Caccamo A, Green KN, Liang K, Tran L, Chen Y et al (2005) Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer's disease. Proc Natl Acad Sci U S A 102:3046–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM (2006) Temporal profile of amyloid‐beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem 281:1599–1604. [DOI] [PubMed] [Google Scholar]
- 34. Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM (2006) Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem 281:39413–39423. [DOI] [PubMed] [Google Scholar]
- 35. Oddo S, Caccamo A, Cheng D, Jouleh B, Torp R, Laferla FM (2007) Genetically augmenting tau levels does not modulate the onset or progression of Abeta pathology in transgenic mice. J Neurochem 4:1053–1063. [DOI] [PubMed] [Google Scholar]
- 36. Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, Garcia G et al (2003) Apolipoprotein Eepsilon4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann Neurol 54:163–169. [DOI] [PubMed] [Google Scholar]
- 37. Rebeck GW, Reiter JS, Strickland DK, Hyman BT (1993) Apolipoprotein E in sporadic Alzheimer's disease: allelic variation and receptor interactions. Neuron 11:575–580. [DOI] [PubMed] [Google Scholar]
- 38. Rebeck GW, Ladu MJ, Estus S, Bu G, Weeber EJ (2006) The generation and function of soluble apoE receptors in the CNS. Mol Neurodegener 1:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH et al (1993) Increased amyloid beta‐peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late‐onset Alzheimer disease. Proc Natl Acad Sci U S A 90:9649–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider WJ, Kovanen PT, Brown MS, Goldstein JL, Utermann G, Weber W et al (1981) Familial dysbetalipoproteinemia. Abnormal binding of mutant apoprotein E to low density lipoprotein receptors of human fibroblasts and membranes from liver and adrenal of rats, rabbits, and cows. J Clin Invest 68:1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81:741–766. [DOI] [PubMed] [Google Scholar]
- 42. Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R et al (1994) Isoform‐specific interactions of apolipoprotein E with microtubule‐associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A 91:11183–11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Svennerholm L, Bostrom K, Helander CG, Jungbjer B (1991) Membrane lipids in the aging human brain. J Neurochem 56:2051–2059. [DOI] [PubMed] [Google Scholar]
- 44. Tesseur I, Van Dorpe J, Spittaels K, Van den Haute C, Moechars D, Van Leuven F (2000) Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am J Pathol 156:951–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tseng BP, Green KN, Chan JL, Blurton‐Jones M, Laferla FM (2007) Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Dooren T, Muyllaert D, Borghgraef P, Cresens A, Devijver H, Van der Auwera I et al (2006) Neuronal or glial expression of human apolipoprotein e4 affects parenchymal and vascular amyloid pathology differentially in different brain regions of double‐ and triple‐transgenic mice. Am J Pathol 168:245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vassar R (2004) BACE1: the beta‐secretase enzyme in Alzheimer's disease. J Mol Neurosci 23:105–114. [DOI] [PubMed] [Google Scholar]
- 48. Verpillat P, Camuzat A, Hannequin D, Thomas‐Anterion C, Puel M, Belliard S et al (2002) Apolipoprotein E gene in frontotemporal dementia: an association study and meta‐analysis. Eur J Hum Genet 10:399–405. [DOI] [PubMed] [Google Scholar]
- 49. Weisgraber KH, Innerarity TL, Mahley RW (1982) Abnormal lipoprotein receptor‐binding activity of the human E apoprotein due to cysteine‐arginine interchange at a single site. J Biol Chem 257:2518–2521. [PubMed] [Google Scholar]