

Abstract
Amyotrophic lateral sclerosis (ALS) is a motor neuron disease characterized by progressive distal axonopathy that precedes actual motor neuron death. Triggers for neuromuscular junction degeneration remain to be determined, but the axon repulsion factor semaphorin 3A (Sema3A), which is derived from terminal Schwann cells, is a plausible candidate. This study examines the hypothesis that Sema3A signaling through its motor neuron neuropilin-1 (NRP1) receptor triggers distal axonopathy and muscle denervation in the SOD1G93A mouse model of ALS. Neuropilin-1 was found to be expressed in axonal terminals at the mouse neuromuscular junction in vivo and in NSC-34 motor neuron–like cells in vitro. In differentiated NSC-34 cells, an anti-NRP1A antibody that selectively blocks Sema3A binding to NRP1 prevented Sema3A-induced growth cone collapse. Furthermore, intraperitoneal injections of anti-NRP1A antibody administered twice weekly from age 40 days significantly delayed and even temporarily reversed motor functional decline while prolonging the life span of SOD1G93A mice. Histologic evaluation at 90 and 125 days revealed that anti-NRP1A antibody reduced neuromuscular junction denervation and attenuated pathologic alterations in ventral roots at late-stage disease. These data suggest that peripheral NRP1A signaling is involved in the pathobiology of this ALS model and that antagonizing Sema3A/NRP1 binding or downstream signals could have implications for the treatment of ALS.
Key Words: Amyotrophic lateral sclerosis, CRMP2, Neuromuscular junction, Neuropilin-1, NSC-34 motor neuron–like cells, Semaphorin 3A
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron disease that is, in part, a progressive distal axonopathy (1–4). In SOD1G93A transgenic mice, which model human familial ALS (fALS), one of the earliest neuronal disease alterations is neuromuscular junction (NMJ) disintegration, which is followed by axon degeneration that progresses to spinal motor neuron death (1, 4). In fALS mouse models and human sporadic ALS, changes suggesting a period of distal axon remodeling and muscle reinnervation occur before the death of motor neurons at end-stage disease (1). Triggers for axon retraction from motor end plates and subsequent drivers of axonal deterioration remain unknown. Because motor neuron disease in fALS mouse models linked to mutant superoxide dismutase 1 (SOD1) is non–cell-autonomous and depends on transgene expression in non-neuronal cells, glial factors may contribute to either the initiation or the progression of motor neuron degeneration (2, 5).
There is reason to suspect that aberrant expression of axon repulsion factors near the NMJ or activation of their downstream pathways may contribute to the distal axonopathy of ALS. Semaphorin 3A (Sema3A), an important axon guidance cue involved in developmental neural patterning, is upregulated in specific populations of terminal Schwann cells near fast-fatigable Type IIb/x muscle fibers that are particularly vulnerable to denervation in ALS (6). Semaphorin 3A signaling through its receptor neuropilin-1 (NRP1) and plexin A coreceptors triggers axonal retraction by destabilizing microtubules and microfilament networks via a mechanism involving the collapsin response mediator protein (CRMP) class of microtubule-associated proteins (7–11) (Fig. 1). Collapsin response mediator proteins have been independently implicated in ALS because 1) the cytoskeleton-stabilizing CRMP2 is antagonized by isoforms of CRMP4 that become induced in ALS motor neurons, probably in response to neuroinflammatory factors (12), and 2) treatment of SOD1G93A mice with the CRMP2-binding experimental therapeutic lanthionine ketimine ethyl ester slows disease progression (13).
FIGURE 1.
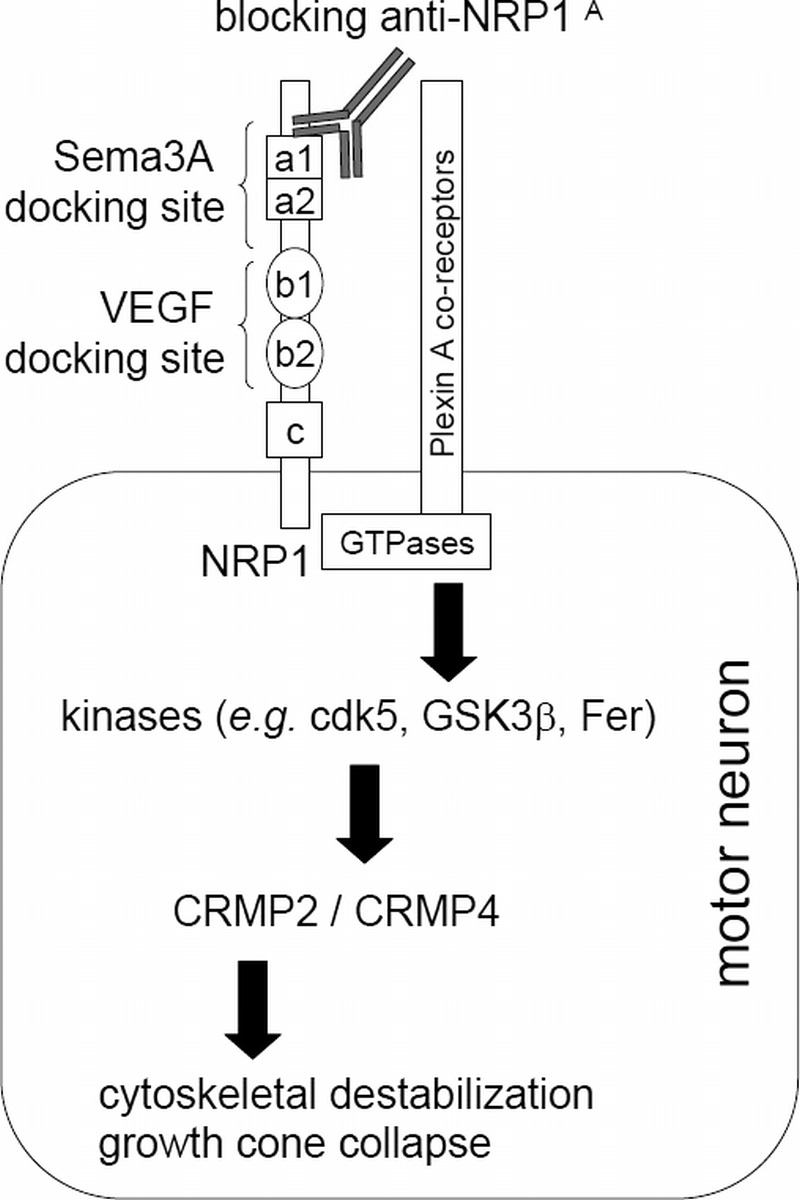
Schematic diagram of Sema3A signaling through CRMP2 and CRMP4 via the NRP1/plexin A receptor system and mechanism of anti-NRP1A antibody action. Neuropilin-1 can bind either Sema3A or VEGF at independent docking domains. Heterodimerization with plexin coreceptors triggers activation of small GTPases, leading to downstream events, particularly phosphorylation of the microtubule-associated protein CRMP2. Recent findings also implicate the CRMP4 isoform in some aspects of Sema3A signaling (11). CRMP2 phosphorylation leads to microtubule instability and indirectly to actin cytoskeletal rearrangements. In developing axons, such activation of the Sema3A/NRP1/CRMP2 axis promotes growth cone collapse and axon retraction away from inappropriate target cells. In the adult animal, Sema3A released from terminal Schwann cells could inhibit compensatory axon sprouting and help coordinate NMJ remodeling after injury. In the context of ALS, inappropriate or prolonged overstimulation of the system could contribute to distal axonopathy. We hypothesize that the ability of anti-NRP1A antibody to selectively bind to the CUB domains (a1 and a2) of NRP1 and to block Sema3A-induced neuron collapse without interfering with the binding of VEGF to the b1 and b2 domains of NRP1 may prevent axon retraction and protect the integrity of NMJs. Cdk5, cyclin-dependent kinase 5; Fer, tyrosine protein kinase Fer; GSK3β, glycogen synthase kinase 3β.
This study directly tests the hypothesis that ALS motor neuron retraction from vulnerable motor end plates occurs in response to local Sema3A signaling through NRP1 receptors. We antagonized this signaling with a high-affinity monoclonal antibody (anti-NRP1A antibody), which selectively blocks Sema3A binding sites on NRP1 (14, 15); the antibody slowed disease progression in SOD1G93A mice and even temporarily reversed motor functional deficits, most probably by alleviating motor unit damage when treatments were initiated near the time of onset of NMJ degeneration.
MATERIALS AND METHODS
Reagents
Semaphorin 3A–docking CUB domains (a1a2) of NRP1 antibody (anti-NRP1A antibody or YW64.3) and human anti-IgG2a antibody were kindly provided by Genentech Inc (South San Francisco, CA). Both therapeutic antibodies were obtained as sterile solution in PBS and stored at −80°C until use. The following special chemicals and antibodies were used in this study: recombinant mouse Sema3A Fc chimera (IgG2a) (R&D Systems Inc, Minneapolis, MN); isotype-matched control human IgG2a (Equitech-Bio Inc, Kerrville, TX); NRP1-blocking peptide (BioVision Inc, Milpitas, CA); anti-NRP1 antibody (Sigma Prestige antibody HPA030278; Sigma-Aldrich, St Louis, MO); monoclonal anti-neurofilament 160-kDa clone NN18 IgG antibody (NF 160; Millipore, Billerica, MA); synaptic vesicle-2 antibody (Iowa Developmental Hybridoma Bank, Iowa City, IA); anti–β-actin primary mouse monoclonal antibody (Sigma-Aldrich); anti–β-tubulin rabbit polyclonal antibody (pAb) (Abcam, Cambridge, United Kingdom); tetramethylrhodamine α-bungarotoxin (Molecular Probes/Invitrogen, Carlsbad, CA); and secondary fluorescently labeled Alexa Fluor 488 donkey anti-mouse IgG, Alexa Fluor 488 goat anti-rabbit IgG, and Alexa Fluor 594 goat anti-rabbit pAb antibodies (Molecular Probes/Invitrogen).
Cell Culture and Immunocytochemistry
NSC-34 cells (a gift from Dr Arthur Burghes of Ohio State University, Columbus, OH) were maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin (Sigma-Aldrich), and 2 mmol/L L-glutamine (Gibco/Invitrogen) at 37°C under a humidified atmosphere of 5% CO2. Cells were subcultured every 3 to 5 days and seeded at a density of approximately 50,000 cells/cm2 for experiments.
For immunocytochemistry, NSC-34 cells were grown on poly-D-lysine–coated coverslips, fixed in 4% paraformaldehyde for 30 minutes at 37°C, and permeabilized for 30 minutes in 0.1% Triton X-100 (Sigma-Aldrich) in PBS at room temperature. To visualize NRP1 expression, we incubated the cells overnight with anti-NRP1 primary rabbit antibody (1:20, Sigma Prestige antibody HPA030278) at 4°C. The secondary antibody was Alexa Fluor 488 goat anti-rabbit IgG antibody (1:250) applied at room temperature for 30 minutes. Blue fluorescent DAPI nucleic acid stain (Invitrogen) was used to visualize the cell nuclei.
A separate series of experiments using differentiated NSC-34 cells was performed to confirm the effects of anti-NRP1A antibody on Sema3A-induced growth cone collapse. To enhance cell differentiation, we replaced the maintenance medium with a differentiation medium consisting of Dulbecco’s modified Eagle medium/F-12 (Gibco/Invitrogen) supplemented with 1% fetal bovine serum, 1% minimum essential medium nonessential amino acids (Gibco/Invitrogen), and 1% penicillin/streptomycin. The medium was renewed every 2 to 3 days, and cells were grown under these conditions for 5 to 7 days. Because growth cone collapse is characterized by cytoskeletal reorganization manifested as changes in the relative distribution of actin filaments and microtubules, we used image analysis to quantify β-actin and β-tubulin fluorescence per growth cone. Differentiated NSC-34 cells were pretreated for 4 hours with either 100 μg/mL anti-NRP1A antibody or isotype-matched human 100 μg/mL IgG2a, and growth cone collapse was induced by 2 nmol/L Sema3A. After 20 hours, the cells were fixed in 4% paraformaldehyde for 30 minutes, permeabilized in 0.1% Triton X-100 in PBS for 30 minutes at room temperature, and immunostained with primary anti–β-actin mouse monoclonal antibody (1:100) and anti–β-tubulin rabbit pAb (1:200) at room temperature for 2 hours. The secondary antibodies used were Alexa Fluor 488 donkey anti-mouse IgG (1:100) and Alexa Fluor 594 goat anti-rabbit pAb (1:100) applied for 30 minutes at room temperature.
An Olympus IX 71 fluorescent microscope (Olympus America Inc, Center Valley, PA) was used for observation and image collection. Images were captured with a charge-coupled device camera (QImaging, Surrey, BC, Canada), recorded with Slidebook 4.2 digital microscopy software (Olympus America Inc), and later analyzed using MetaMorph for Olympus Basic Offline software. Four independent experiments, each including all treatments and controls, were used for growth cone collapse assessment. The images were acquired at constant settings: 20× magnification, 100-millisecond exposure time for red fluorescence emission, and 400-millisecond exposure time for green fluorescence emission. Data for each treatment were obtained from 5 to 10 fields per slide and up to 20 measured growth cones per field. The specific fluorescence of labeled actin (green) and tubulin (red) was evaluated at uniform threshold settings. Ratios between total green- and red-labeled areas along the 16-μm length at the tips of neurites were used as a measure of the relative abundance of actin and tubulin. Results were presented as percentages of actin and tubulin per growth cone.
Animals and Treatment Protocol
Mice used in the study were the F1 offspring of breeder pairs (stock no. 002726; strain designation B6SJL-TgN(SOD1-G93A)1-Gur) purchased from the Jackson Laboratory (Bar Harbor, ME (16). Male B6SJL-TgN(SOD1-G93A)1-Gur/J mice expressing a high copy number of human G93A mutant SOD1 were bred with B6SJLF1/J female mice to provide mixed litters of hemizygous transgenic and nontransgenic wild-type littermates. Pups were genotyped at age 10 days using a standard genotyping protocol provided by the Jackson Laboratory. At weaning, mice were litter-matched into groups with balanced numbers of male and female animals in each group. Results of the initial rotarod test performed at ages 38 to 40 days (before the first drug treatment) indicated that all SOD1G93A mice showed near-equivalent rotarod performance, but the time spent on the rotarod was always significantly shorter than the performance time of their nontransgenic littermates. This early loss of motor function was equivalent to that reported by Mead et al (17) for the (SJL×C57BL/6) SOD1G93A transgenic line, which allowed us to follow their protocol developed for early sensitive and rapid screening in the initial phases of muscle fiber denervation. Early readouts showing a large decline in motor function compared with normal littermates allowed a reduction in the numbers of mice per treatment group when testing the effects of therapeutics. In addition, 2-way analysis of variance with post hoc comparisons indicated no significant influence of sex on treatment outcome after 50 to 60 days of treatment.
Animals in the treatment groups received humanized monoclonal anti-NRP1A antibody specific for the Sema3A docking CUB domains (a1a2) of NRP1. To evaluate the effects of anti-NRP1A antibody treatment, we used 2 control groups of SOD1G93A mice: a group receiving isotype-matched but irrelevant control human IgG2a and another group receiving saline (vehicle control). In addition, nontransgenic littermates were studied as reference to healthy controls.
Both anti-NRP1A antibody and IgG2a were obtained solubilized in sterile saline and administered twice weekly by intraperitoneal injection of 20 mg/kg (130 nmol/kg). The vehicle control and nontransgenic groups received sterile saline injections. Dose of anti-NRP1A antibody, route of administration, and treatment regimen were chosen based on recommendations from Genentech Inc (unpublished pharmacokinetic data) and published experimental data showing functional blocking of NRP1A receptors in mouse tumor models (14, 15). Moreover, anti-NRP1A and anti-NRP1B antibodies have shown in vivo efficacy after twice-weekly intraperitoneal administration during 5-week treatments in models of other NRP1-dependent conditions such as chronic bladder inflammation (18).
The treatment was initiated at age 40 days, and the animals received 2 doses per week for the rest of their life span. In survival experiments, mice were either found dead in their cage or killed when they were no longer able to perform the rotarod task or no longer able to right themselves within 30 seconds of being placed on their sides. Recommendation to euthanize by criteria was made by animal care staff blinded to treatment group. For behavioral and survival experiments, mice were killed at end-stage disease by CO2 inhalation. The animals used for immunohistochemistry and histology were killed under deep anesthesia induced by intraperitoneal injections of ketamine (90 mg/kg) and xylazine (10 mg/kg), followed by transcardial perfusion with prewarmed (37°C) PBS (pH 7.4). The lumbosacral region of the vertebral column and the gastrocnemius muscles were removed and processed for histologic evaluation of ALS neuropathology. All procedures, including the maintenance of a breeding colony and the methods of euthanasia, were approved by the University of Toledo Institutional Animal Care and Use Committees (protocol no. 107662; National Institutes of Health Office of Laboratory Animal Welfare Animal Assurance no. A3414-01; Association for Assessment and Accreditation of Laboratory Animal Care accreditation no. 00557).
Rotarod Task
Motor function was assessed by rotarod performance (2). Mice were acclimated to a rotarod task at ages 37 to 39 days then divided into control and treatment groups for therapeutic intervention at age 40 or 90 days. Mice were litter-matched into groups and selected to ensure near equivalence of the mean initial rotarod performance before treatments. Each rotarod test consisted of 4 consecutive runs, the results of which were used to calculate the mean value. Mice were placed on a commercial horizontal rotarod (Columbus Instruments, Columbus, OH) set to rotate from rest at 1 rpm/10 seconds until the mouse fell from the rod to a safe surface located 34 cm below the rod. Rotarod tasks were conducted at 10-day intervals throughout the mouse life span.
Immunohistochemistry
Neuromuscular junction integrity was assessed as previously reported (1). Gastrocnemius muscles were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 30 minutes, rinsed in PBS, immersed in 20% sucrose in PBS, and stored overnight at 4°C. On the next day, the muscles were snap-frozen in cooled isopentane, sealed in plastic tubes, and stored at −80°C. The muscles were embedded in TFM tissue freezing medium (Triangle Biomedical Sciences Inc, Durham, NC) for cryosectioning. Longitudinal serial sections (30 µm thick) were taken approximately 90 µm apart and thaw-mounted 3 per slide on Colorfrost Plus Microscopic Slides (Fisher Scientific, Waltham, MA). The slides were kept at −30°C and used for immunohistochemistry within 48 hours of sectioning. The frozen slides were placed in a humidified chamber and allowed 5 minutes to adapt to room temperature; the next steps were carried out in a humidified chamber at room temperature.
Neuromuscular junctions were visualized by labeling nicotinic acetylcholine receptors (AChRs) with tetramethylrhodamine α-bungarotoxin (Molecular Probes/Invitrogen). Stock solution of fluorescent α-bungarotoxin (1 mg/mL in water) was diluted 1:40 in PBS, placed onto tissue sections, and allowed to stay for 30 minutes. Sections were then washed 3 times with PBS, fixed in ice-cold methanol for 5 to 10 minutes, and washed 3 times with PBS. Nonspecific binding sites were blocked by incubation in 2% bovine serum albumin in PBS for 30 minutes. Blocking buffer was substituted with bovine serum albumin in PBS containing monoclonal anti-neurofilament 160-kDa clone NN18 IgG antibody (1:200) and synaptic vesicle protein-2 antibody (1:30). Samples were incubated with primary antibodies; the slides were then washed 3 times with PBS and incubated with a secondary fluorescently labeled antibody (1:100) for 30 minutes. The slides were washed 3 times with PBS; the sections were sealed with ProLong Gold (Molecular Probes/Invitrogen), coverslipped, and allowed to solidify for 24 hours. Sections were examined using an Olympus IX 71 fluorescent microscope (×40 air lens). Images were captured with a charge-coupled device camera (QImaging), recorded with Slidebook 4.2 digital microscopy software (Olympus America Inc), and later analyzed using MetaMorph for Olympus Basic Offline software. Images were taken to allow the detection of all end plates present in a muscle section. Each visible end plate (labeled red by tetramethylrhodamine α-bungarotoxin bound selectively to AChRs) was counted as a single neuromuscular NMJ. The number of NMJs per section and their structural integrity were evaluated by a researcher blinded to animal age and treatment group. Neuromuscular junctions were classified as normally innervated or denervated based on the overlap between axon terminals (labeled green by the fluorescence of neurofilaments and presynaptic vesicles) and postsynaptic AChRs and on the morphology of the end plate. Completely denervated end plates showed full uncoupling between nerve and muscle and were visualized as red clusters of AChRs with distorted end-plate morphology. Partially denervated NMJs were seen as a green fluorescent nerve fiber approaching the end plate but only partially overlapping with the red fluorescence; for purposes of quantification, both completely and partially denervated end plates were counted as denervated NMJs. Neuropilin-1 labeling of motor neurons was accomplished using a commercial anti-NRP1 (1:20) primary rabbit IgG and a fluorophore-conjugated secondary antibody, Alexa Fluor 488 goat anti-rabbit IgG (1:250), because attempts to directly conjugate fluorophores to anti-NRP1A antibody (therapeutic agent) resulted in a loss of antibody affinity for NRP1 (data not shown).
Ventral Root Histology
Ventral root histology was assessed similarly to published methods (1). The dissected vertebral columns were immersed in 5% glutaraldehyde (pH 7.4) immediately after isolation and fixed for 48 to 72 hours. The lumbar ventral roots at L4 were isolated using a stereomicroscope, stored overnight in 0.1 mol/L phosphate buffer at 4°C, and processed as described by Fischer et al (1). Tissue was treated with 1% osmium tetroxide for 90 minutes, dehydrated through graded (30%–100%) ethanol and acetone, and embedded in Spurr Low Viscosity Embedding Media (Polysciences Inc, Warrington, PA). Cross sections (80 nm) were stained with methylene blue azure II, rinsed, dried, and coverslipped. Ventral root sections were imaged at 20× magnification using an Olympus BH-2 microscope (Olympus America Inc), and images were captured using an attached camera (Evolution MP; Media Cybernetics, Rockville MD). Each whole-section image was divided into image segments of constant area (735 × 865 pixels) using MetaMorph for Olympus Basic Offline Software (Olympus America Inc). The number of axons and their caliber were evaluated visually and marked manually by an observer blinded to treatment. For each ventral root, 7 to 9 representative nonoverlapping images were used to identify the diameter of axons by imposing a 4-µm measuring bar over the image of each of the axonal cross sections. Axons with a diameter of 4 µm or larger were categorized as large-diameter axons (i.e. axons of large-caliber α-motor neurons); the rest of the axons were classified as small-diameter axons (i.e. axons of small-caliber γ-motor neurons). The number of large-caliber axons was evaluated as percentage of the total number of axons per cross section.
Statistics
While collecting the data, the researchers were blinded to treatment groups. All data were graphically presented as mean ± SE, unless otherwise specified. In the case of single mean comparisons, data were analyzed by 2-tailed unpaired t-tests or Mann-Whitney U tests appropriate for data distributions. In the case of multiple comparisons, data were analyzed by 1-way or 2-way analysis of variance with post hoc Bonferroni multiple comparisons using GraphPad Prism Software (GraphPad Prism, San Diego, CA). Distribution of categorical variables was assessed using Fisher exact test.
RESULTS
NRP1 Is Expressed in NSC-34 Motor Neuron–Like Cells and in Mouse Motor Neuron Axons In Vivo
In recent years, oncologists have come to appreciate the biologic importance and therapeutic relevance of NRP1 in cancer biology; signaling through the NRP1 receptor has been implicated in tumor cell metastasis and tumor neovascularization (14, 15, 19). Neuropilin-1 expression and function in normal or diseased motor neurons have been much less studied. Initial experiments were undertaken in NSC-34 cells—a hybridoma cell line obtained by the fusion of mouse embryonic motor neurons and neuroblastoma cells—to confirm NRP1 expression in motor neurons (20). Antibody against NRP1 labeled the cytosol and/or plasma membranes of NSC-34 cells, whereas omission of the primary antibody indicated specificity of immunolabeling (Figs. 2A-C). The specific labeling of the NRP1 receptor was also confirmed by the lack of immunolabeling in the presence of excessive concentrations of NRP1-blocking peptide (Fig. 2D).
FIGURE 2.
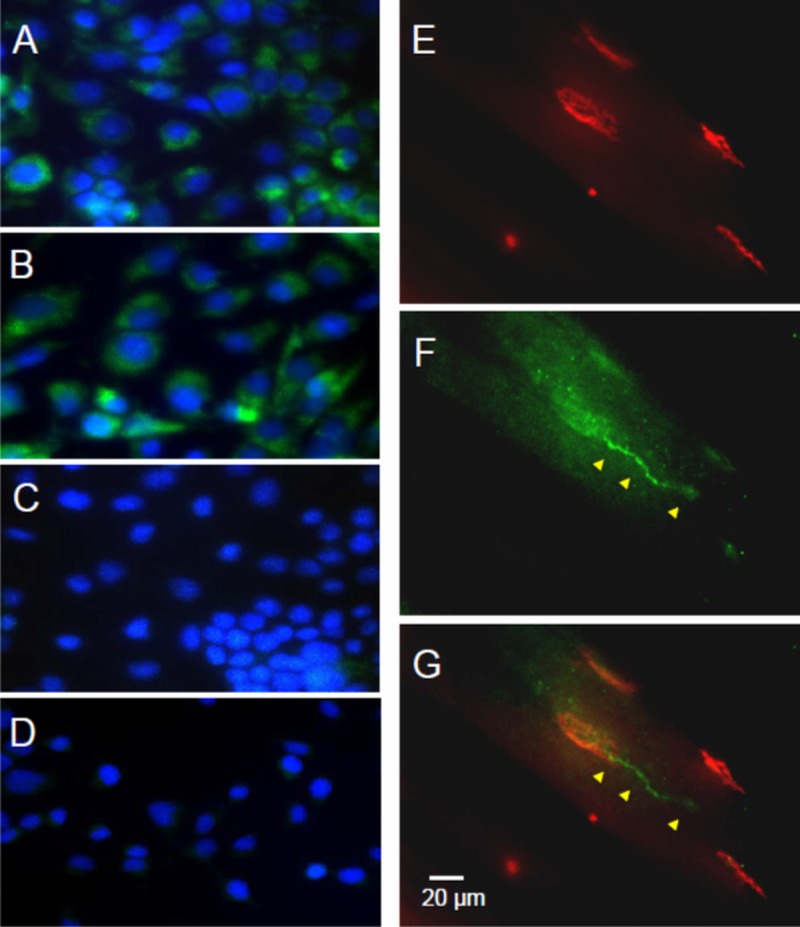
Immunolabeling indicates NRP1 expression in motor neuron–derived NSC-34 cells in culture and at the terminal part of motor neuron axons forming NMJs in mouse gastrocnemius muscle. (A, B) Neuropilin-1 immunolabeling (green) was observed in the cytosol and/or membrane of undifferentiated NSC-34 cells. Nuclei are labeled with DAPI (blue). (C, D) Omission of the primary antibody (C) or inclusion of 10 mg/mL blocking peptide (D) confirmed the specificity of immunocytochemical labeling in NSC-34 cells. (E–G) Immunohistochemistry of NMJs in mouse gastrocnemius muscle using the same anti-NRP1 probe revealed NRP1 immunolabeling (F; green) in axons (arrowheads) and terminal synaptic endings that superimpose upon α-bungarotoxin–labeled motor end plates (E, red); (G) Merged image.
After validation of the antibody as a labeling tool, immunohistochemistry was used to assess whether NRP1 is expressed in distal motor axons or near the NMJ. Anti-NRP1–labeled motor axons innervating end plates were labeled in cryosections of mouse gastrocnemius muscle (Figs. 2F, G). No readily discernible differences in the quantity or distribution of NRP1-binding sites on motor neurons were observed between nontransgenic and SOD1G93A mice (data not shown). Anti-NRP1 labeling of motor neurons was present along the whole length of the axons innervating the end plates. Moreover, the density of NRP1 binding was high enough to follow the labeling pattern normally observed when the neurofilaments and synapses of motor axons are deliberately labeled for morphologic analysis.
Sema3A-Blocking Antibody Against NRP1 Prevents Sema3A-Induced Growth Cone Collapse in NSC-34 Cells
Cell culture experiments were undertaken to validate the concept that anti-NRP1A antibody can block Sema3A effects on the distal part of motor neuron axons. In axon growth cones, actin filaments are distributed throughout the lamellipodium and filopodia and are often clustered around lamellipodium edges. In contrast, tubulin is present only in the central region of the growth cone, where it forms an extensive network of microtubules. Growth cone collapse is characterized by an altered relative distribution of actin filaments and microtubules in the growth cone, mainly caused by a major loss of actin filaments at the tip of the axon. Thus, the relative abundance of actin and tubulin at the distal end of axons can be used as a quantitative indicator of growth cone collapse. Treatment of differentiated NSC-34 cells with 2 nmol/L recombinant Sema3A for 20 hours induced a dramatic growth cone collapse compared to untreated cells (Figs. 3A, B, E). Pretreatment of the cells with 100 μg/mL anti-NRP1A antibody in the culture medium almost completely prevented the Sema3A effect (Figs. 3C, E). In contrast, pretreatment with 100 μg/mL of the isotype-matched control human IgG2a failed to prevent Sema3A-induced growth cone collapse (Figs. 3D, E).
FIGURE 3.
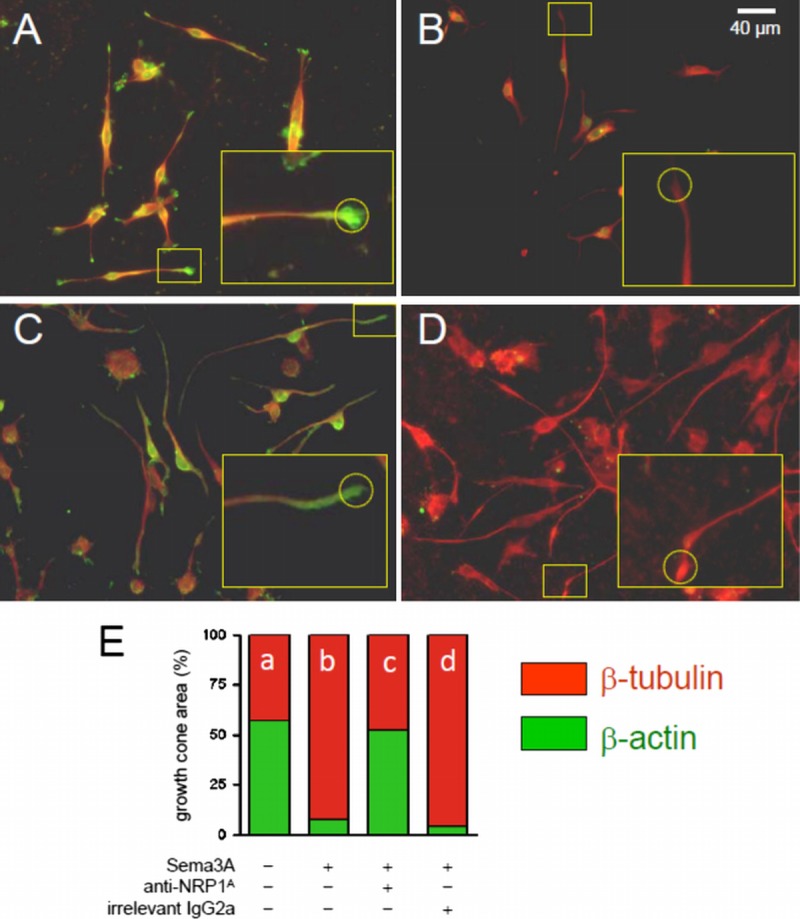
Anti-NRP1 antibody (NRP1A) blocks Sema3A-induced collapse of growth cones in differentiating NSC-34 motor neuron–like cells. (A–D) Growth cone areas were defined for morphometric measurements (20× magnification) by a standard circle area (d = 16 μm) superimposed at the tip of neurites (insets). Areas occupied by fluorescently labeled β-actin (green) and β-tubulin (red) were measured in untreated NSC-34 cells (A); cells treated with 2 nmol/L Sema3A for 20 hours (B); cells pretreated with 100 μg/mL anti-NRP1A antibody then treated with Sema3A in the presence of anti-NRP1A antibody (C); and cells pretreated with 100 μg/mL irrelevant IgG2a then treated with Sema3A in the presence of IgG2a (D). Typical immunolabeling of growth cones is shown in expanded inset boxes. (E) Bar graph summarizing the area of green-labeled β-actin and red-labeled β-tubulin within the growth cones is presented as a percentage of total growth cone area for each treatment (4 independent experiments, 5–10 fields per slide, and up to 20 measured growth cones per field). Data are presented as mean ± SE. Differences between Sema3A effect in the absence of anti-NRP1A antibody (bar b) and Sema3A effect in the presence of anti-NRP1A antibody (bar c) were significant at p < 0.001 (Fisher exact test).
Anti-NRP1A Antibody Improves Outcomes in SOD1G93A Mice
Previous studies in SOD1G93A mice have reported that motor end-plate denervation occurs at a relatively early age, with significant denervation observed at 47 days (1). At the same age, numerous Sema3A-expressing terminal Schwann cells were found in the gastrocnemius muscle of SOD1G93A mice but were only occasionally observed in age-matched nontransgenic controls (6). To investigate whether Sema3A drives initial motor end-plate denervation, our experiments used a protocol for anti-NRP1A antibody treatment beginning at age 40 days and continuing until end-stage disease criteria were reached and the animal was killed. Male and female animals were equally represented in each group. In this paradigm, anti-NRP1A antibody significantly extended mouse life span by a median of 11 days (mean, 24 days) (Figs. 4A, B). Dose and isotype-matched irrelevant IgG had no effect (Fig. 4A).
FIGURE 4.
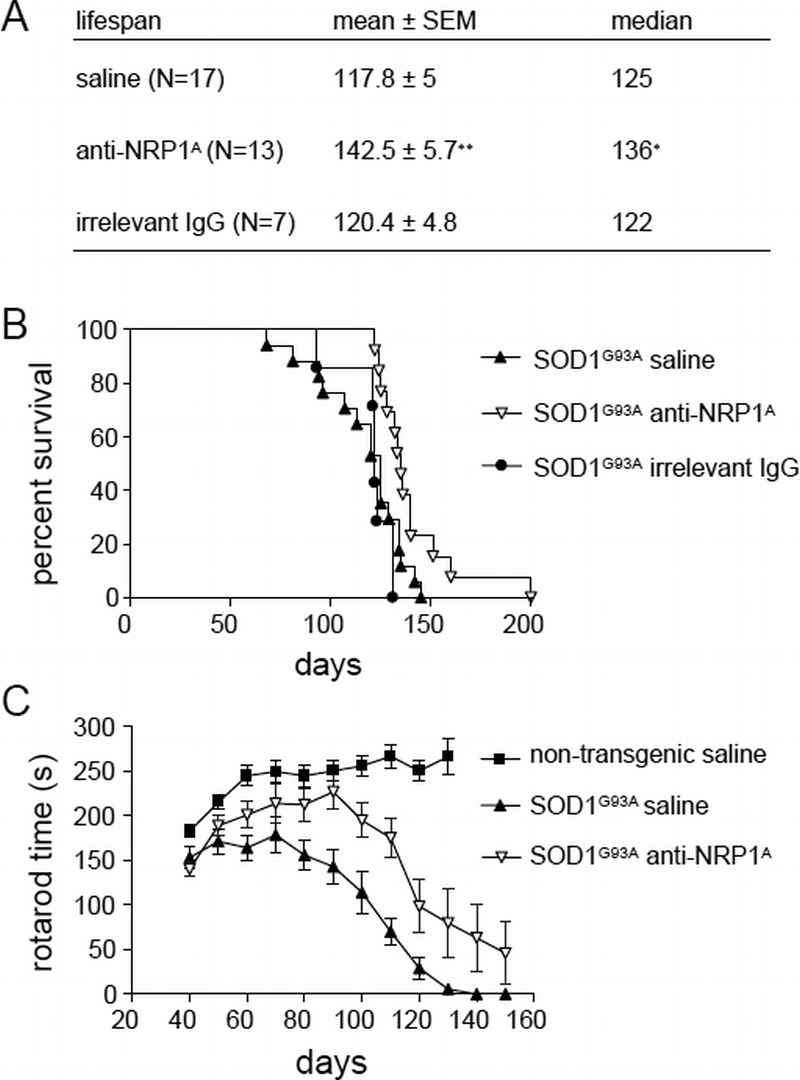
Peripheral administration of anti-NRP1 antibody (anti-NRP1A antibody) beginning at age 40 days improves outcomes in the SOD1G93A mouse model of ALS. Statistical analysis of data for the groups of SOD1G93A mice receiving saline vehicle, anti-NRP1A antibody, or irrelevant IgG (20 mg/kg/dose twice weekly from 40 days throughout the remaining life span). (A) Tabulated survival statistics. * p = 0.0047 by Mann-Whitney U test; ** p = 0.0032 by Student t-test. (B) Survival curves for logrank analysis (χ2 = 8.14 and p = 0.017 for anti-NRP1A antibody–treated vs vehicle-treated SOD1G93A mice). (C) Rotarod motor function assessed in serial tests of nontransgenic or SOD1G93A mice receiving saline vehicle or anti-NRP1A antibody from 40 days throughout the remaining life span. Repeated-measures analysis of variance of SOD1G93A groups (age effect: F10,310 = 51.73, p < 0.0001; anti-NRP1A antibody treatment effect: F1,310 = 10.25, p = 0.0032; age × treatment interaction: F10,310 = 3.68, p = 0.001). Numbers of male versus female mice in each group: saline-treated, 8 male mice versus 9 female mice (54% male); anti-NRP1A antibody–treated, 7 male mice versus 6 female mice (47% male).
For motor function, SOD1G93A mice displayed slight (approximately 10%) but measurable and significant motor weakness on a rotarod task at age 40 days relative to age-matched nontransgenic littermates (Fig. 4C). Nontransgenic littermate mouse motor function improved steadily between ages 40 and 60 days (Fig. 4C). In contrast, the motor performance of vehicle (saline)–treated SOD1G93A mice did not improve but instead decreased in accelerated fashion after age 60 days (Fig. 4C). In comparison with vehicle-treated animals, SOD1G93A mice receiving anti-NRP1A antibody displayed age-dependent improvement in motor function after as few as 2 to 3 doses of the antibody (Fig. 4C). This improvement continued up to age 90 days, at which time anti-NRP1A antibody–treated SOD1G93A mice were not statistically distinguishable in rotarod performance from age-matched nontransgenic littermates (Fig. 4C). At ages older than 90 days, however, the motor function of anti-NRP1A antibody–treated SOD1G93A mice began to decline in parallel with the later-stage performance of vehicle-treated transgenic mice (Fig. 4C).
There have been separate experiments where anti-NRP1A antibody treatment was begun at 90 days—an age when untreated animals show significant motor deficits, advanced NMJ deterioration, and initiation of a loss of large α-motoneuron axons in the spinal cord ventral horns (1). In this paradigm, anti-NRP1A antibody had no significant effect on SOD1G93A mouse life span or motor functional decline (Fig. 5).
FIGURE 5.
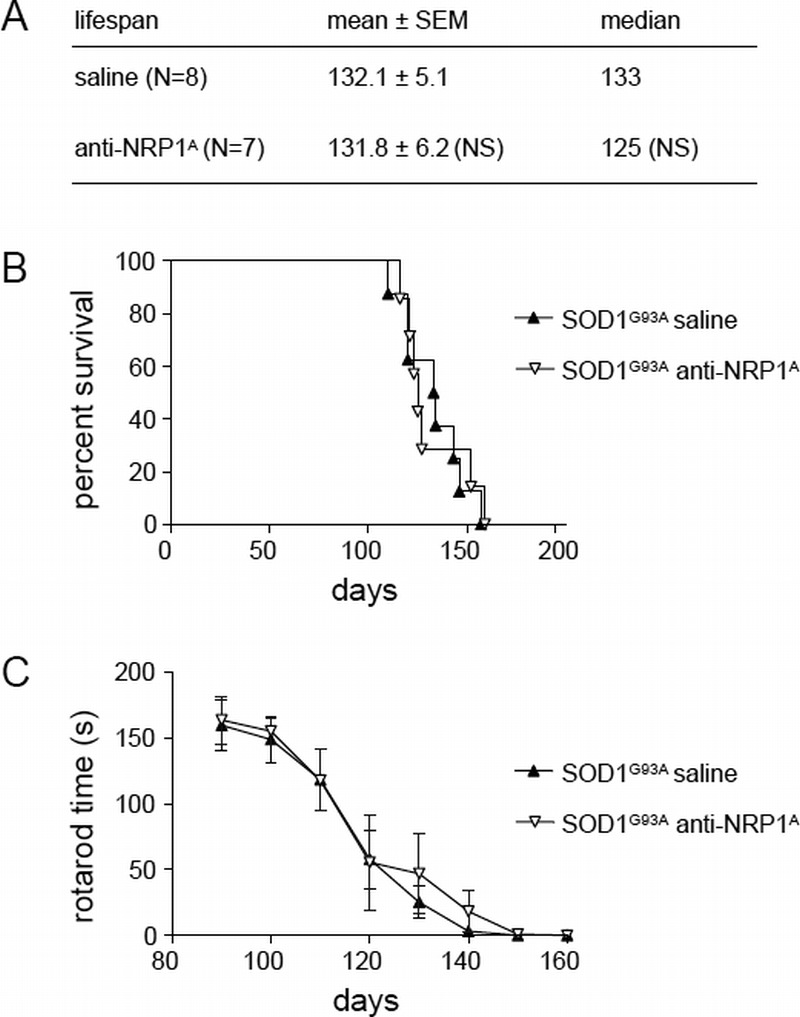
Anti-NRP1 antibody (NRP1A) fails to affect motor functional decline or life span in SOD1G93A mice when treatment is initiated at age 90 days. (A) Tabulated survival statistics are shown for SOD1G93A mice receiving anti-NRP1A antibody (20 mg/kg/dose twice weekly) or saline vehicle beginning at age 90 days. (B) Survival curves are shown for the same groups of mice. (C) Rotarod motor functional decline is shown for the same groups of mice. NS, not significant.
Anti-NRP1A Antibody Reduces NMJ Denervation in SOD1G93A Mice
The histology of NMJs was examined at 90 and 125 days as a function of mouse genotype and anti-NRP1A antibody treatment initiated at age 40 days. The morphologic integrity of NMJs was assessed in muscle sections labeled with antibodies against neurofilaments and synaptic vesicle-2 (green) and bungarotoxin (red) according to the definition of intact versus denervated motor end plates used in this protocol (Fig. 6A). SOD1G93A mice displayed a substantial increase in compromised NMJ at 90 days compared with age-matched nontransgenic littermates (47.6% in vehicle-treated SOD1G93A mice vs 82.1% in nontransgenic littermates; Fig. 6B). SOD1G93A mice receiving anti-NRP1A antibody treatment since age 40 days showed a significantly greater percentage of innervated motor end plates than vehicle-treated SOD1G93A mice (Fig. 6B). By age 125 days, the percentage of intact NMJs in surviving SOD1G93A mice continued to decline in the absence of anti-NRP1A antibody treatment, whereas mice receiving anti-NRP1A antibody largely retained NMJ integrity (Fig. 6B).
FIGURE 6.
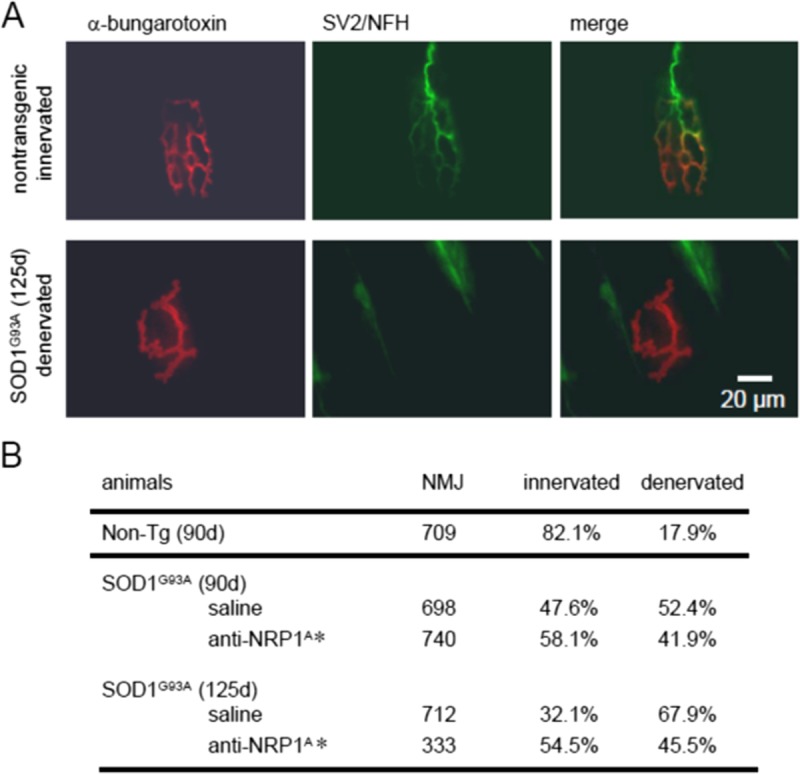
Anti-NRP1 antibody (NRP1A) treatment, begun at age 40 days, diminishes middle late-stage NMJ denervation in SOD1G93A mice. (A) Fluorescence micrographs of individual NMJs labeled with rhodamine-conjugated α-bungarotoxin for nicotinic AChRs at the motor end plate (red) plus anti-neurofilament and SV2 proteins to visualize axons and synaptic terminals (green). Note the loss of fine structure in motor end plates in the SOD1G93A specimen. (B) Quantification of intact (innervated end-plate AChRs) and detached (denervated AChRs) NMJs in untreated Non-Tg animals and SOD1G93A mice treated with anti-NRP1A antibody or vehicle. The effect of treatment was evaluated at ages 90 and 125 days. * p < 0.0001 for treatment effect on the percentage of innervation (Fisher exact test). NFH, neurofilament heavy chain; Non-Tg, nontransgenic; SV2, synaptic vesicle-2.
Anti-NRP1A Antibody Attenuates Ventral Root Pathology in SOD1G93A Mice
Ventral root pathology was also assessed as a function of mouse genotype and anti-NRP1A antibody treatment. As anticipated and widely reported, the loss of large-caliber ventral root axons in SOD1G93A mice started at about age 90 days and worsened with time (Fig. 7). By 125 days, there was a significant reduction in the percentage of large-caliber axons in the ventral roots of saline-treated SOD1G93A mice versus nontransgenic littermates (Fig. 7). Compared with saline-treated controls, mice receiving anti-NRP1A antibody maintained a greater percentage of large-caliber ventral root axons from ages 90 to 125 days (Fig. 7). Despite the protective effect, a profound loss of motor function (as assessed by rotarod performance) and animal mortality persisted at this late disease stage.
FIGURE 7.
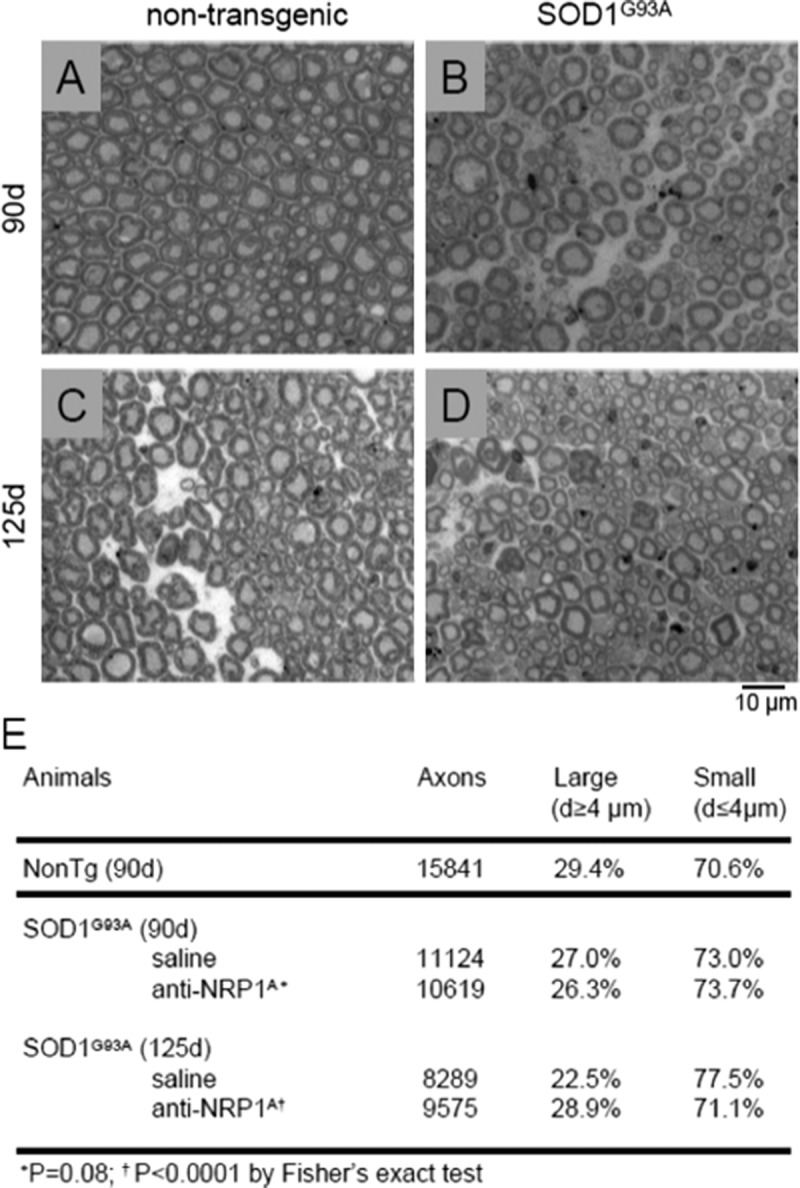
Anti-NRP1 antibody (NRP1A) treatment initiated at age 40 days attenuates late-stage axonal degeneration in the L4 ventral roots from SOD1G93A mice. (A–D) Representative light microscopy micrographs (40× magnification) showing cross sections at age 90 or 125 days in SOD1G93A mice and Non-Tg littermates. Note the presence of degenerating large-caliber axons in SOD1G93A mice at age 125 days. (E) Effect of anti-NRP1A antibody treatment on the percentage of large-caliber axons evaluated at ages 90 and 125 days. * p < 0.0001 for treatment effect (Fisher exact test). Non-Tg, nontransgenic.
DISCUSSION
In this study, we report that peripheral administration of a humanized monoclonal antibody against the Sema3A-binding domain of the NRP1 receptor (anti-NRP1A antibody) delayed the appearance of motor functional deficits and increased the life span of SOD1G93A mice, a widely used model of fALS. The efficacy of anti-NRP1A antibody depended on disease stage. Anti-NRP1A antibody showed a significant protective effect when the treatment was initiated at an early disease stage but had no significant efficacy when applied at a late disease stage characterized by advanced neuromuscular pathology. Nevertheless, the effect of early anti-NRP1A antibody treatment was confirmed histologically at middle late-stage disease by a reduced fraction of denervated motor end plates and reduced ventral root histopathology at ages 90 and 125 days. Taken together, these findings suggest that the Sema3A/NRP1 axis is a trigger—but probably not the sole trigger—of distal axonopathy in fALS and that peripheral NRP1 receptors present a novel target located outside the CNS for pharmacotherapeutic intervention.
Fischer et al (1) previously noted that NMJ pathology occurs early in SOD1G93A mice—before evident ventral root pathology or loss of motor neuron somas. The same study reported a similar NMJ pathology found postmortem in an ALS patient who died unexpectedly at the early stages of the disease; the autopsy revealed muscle fiber–type grouping indicating cyclic motor end-plate denervation and reinnervation without apparent changes in motor neurons (1). In recent years, there has been speculation about possible factors that could trigger pathology near the NMJ in early ALS. The finding that motor neuron disease in SOD1-linked cases of fALS is largely non–cell-autonomous (5) has lent favor to hypotheses involving glia-derived factors. Axon repulsion factors have been implicated because the natural function of such molecules is to trigger asymmetric cytoskeletal deterioration and reconstruction in growth cones of developing axons (21). The physiologic and/or pathologic function of these factors has been much less well studied in the adult nervous system. Among such guidance cues, the axon repulsion cue Sema3A is a prime suspect because De Winter et al (6) reported in situ hybridization evidence for Sema3A upregulation in specific populations of terminal Schwann cells near fast-fatigable muscle fibers that are most prone to denervation in early ALS. Also, Hernandez et al (22) recently reported finding circulating antibodies against semaphorins in patients with motor neuron disease. Semaphorin 3A normally acts as a growth cone retraction signal in the developing CNS by signaling through NRP1 receptors to promote CRMP2 phosphorylation and downstream cytoskeletal depolymerization (8, 19) (Fig. 1). Therefore, aberrant engagement of this pathway in a compromised NMJ might trigger the distal axonopathy observed in this disease. To date, however, there have been little empirical data published to implicate Sema3A signaling directly in the distal axonopathy of ALS.
Signaling through the NRP1 receptor and plexin coreceptors has been implicated in cancer metastasis and neovascularization. Indeed, humanized blocking antibodies against vascular endothelial growth factor (VEGF) or Sema3A docking sites of NRP1 are now in human clinical trials for solid tumors (23). In particular, the anti-NRP1A antibody YW64.3 has been shown to block Sema3A binding to NRP1 while having virtually no effect on VEGF binding to the same receptor (14, 15). These studies also rule out the action of anti-NRP1A antibody on neuropilin-2 because of the low affinity of the antibody for the receptor. The report that intraperitoneal administration of VEGF-blocking anti-NRP1B antibody, either alone or in combination with the Sema3A-blocking anti-NRP1A antibody, blocks tumor growth in mouse xenograft models (14, 15) guided us in selecting the treatment paradigm for targeting Sema3A binding to NRP1 in our experiments using SOD1G93A mice. We used a higher dose (20 mg/kg) to overcome the significant antigenic sinks for anti-NRP1 antibody that must be saturated before effective local concentrations are achieved (24). Because the presumptive targets in the ALS model are the Sema3A receptors at the NMJs (which lie outside the CNS), penetration by the antibody through the blood-brain barrier was not deemed to be a concern.
As predicted, anti-NRP1A antibody delayed the deterioration of motor function in the SOD1G93A mouse, whereas the irrelevant isotype-matched IgG had no effect, thus confirming the specific nature of the protection afforded by anti-NRP1A antibody. Neuromuscular junction pathology in SOD1G93A mice occurs very early and progresses rapidly with an estimated decrease in motor end-plate innervation from 95% at age 30 days to approximately 40% at age 50 days and only 20% at age 100 days (1). In general, our estimate of NMJ innervation in untreated SOD1G93A mice agrees with these findings, except that our methods resulted in a higher number of noninnervated motor end plates, which could reflect a systematic difference in tissue processing and histopathologic evaluation. Our decision to begin treatment at 40 days was based on interpolation of these data on NMJ pathology to select a feasible prevention/treatment dosing regimen to test our hypothesis.
Anti-NRP1A antibody treatment delayed the appearance of motor deficits and histopathologic deterioration of NMJs, which is predictive of its ability to interfere with early ALS changes that take place in the periphery; however, such therapy is not designed to prevent or abolish alterations in the CNS that take place later in the disease. Nevertheless, the delayed deterioration of NMJs was accompanied by a positive effect of anti-NRP1A antibody on ventral root histopathology, promoting the retention of large-caliber motor axons at later-stage disease (125 days). We did not observe a significant effect of anti-NRP1A antibody on ventral root deterioration at age 90 days despite the observed effect of the monoclonal antibody on motor end-plate innervation at 90 days (Fig. 6). The relatively larger effect of anti-NRP1A antibody on later-stage NMJ pathology, compared with ventral root pathology, might reflect an effect of the Sema3A-blocking antibody (removing Sema3A inhibition of compensatory axon sprouting at the distal motor axon) so that remaining axons at 125 days could serve more muscle fibers. Intriguingly, anti-NRP1A antibody–treated SOD1G93A mice surviving up to 125 days displayed similar degrees of NMJ disintegration and ventral root pathology as correspondingly treated 90-day-old mice (Figs. 6, 7). Nevertheless, paralysis of anti-NRP1A antibody–treated SOD1G93A mice at later disease stages appeared to progress despite the rescue of NMJ and ventral root histopathologic progression. This might suggest late-stage onset in motor neurons of somatic death programs whose triggers do not depend on changes taking place at the NMJ and are not mitigated by the anti-NRP1A antibody. Nevertheless, the ability of anti-NRP1A antibody to protect peripheral NMJs could prove valuable when used as combined therapy with centrally acting neuroprotectants such as riluzole.
Semaphorin 3A is known to disrupt the polymerization of both actin and tubulin, reducing the presence of β-actin relative to tubulin within the growth cone and the entire cell (25). With respect to the rearrangement of the cytoskeleton during Sema3A-mediated growth cone collapse, our results showed a strong protective effect of anti-NRP1A antibody on neurites of differentiated NSC-34 cells. Because other positive and negative guidance factors are expressed by non-neuronal cells, Sema3A is not the only axon guidance factor that might be implicated in ALS. A very recent study (26) identified the axon retraction factor ephrin receptor EphA4 as a modifier gene in a zebra fish model of ALS. The same EphA4 was implicated in SOD1G93A mice and humans because antagonism of EphA4 improves outcomes in mice and its expression in human ALS patients correlates significantly with age at onset and survival (26). Thus, further research is warranted to better understand the role of axon guidance cues in motor neuron disease and pharmacologic strategies for modulating their actions.
Finally, the Sema3A/NRP1/CRMP axis may prove important in neurodiseases other than ALS. For example, components of this axis have been implicated in Alzheimer disease, where hyperphosphorylated CRMP2 collects in neurofibrillary tangles, thus possibly depleting neurons of functional CRMP2 (9, 27, 28). Moreover, at least 1 study found evidence for intraneuronal Sema3A accumulation (in complex with plexins and CRMP2) in vulnerable fields of Alzheimer disease–afflicted hippocampi (29). Thus, further research is justified to explore the possible pathogenic roles of axon repulsion factors in general, and of Sema3A in particular, in neurodegenerative diseases affecting both lower and upper motor neurons.
ACKNOWLEDGEMENTS
NSC-34 cells used in this study were a gift from Dr Arthur Burghes (Ohio State University). We thank Genentech Inc for the generous gift of anti-NRP1A antibody used in this work and Dr Ryan Watts (Genentech Inc) for helpful advice.
Footnotes
This work was supported by the Muscular Dystrophy Association (Grant No. MDA-217526) and by a University of Toledo Biomedical Innovation Award.
REFERENCES
- 1. Fischer LR, Culver DG, Tennant P, et al. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp Neurol 2004; 185: 232– 40 [DOI] [PubMed] [Google Scholar]
- 2. Hensley K, Mhatre M, Mou S, et al. On the relation of oxidative stress to neuroinflammation: Lessons learned from the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid Redox Signal 2006; 8: 2075– 87 [DOI] [PubMed] [Google Scholar]
- 3. Bento-Abreu A, Van Damme P, Van Den Bosch L, et al. The neurobiology of amyotrophic lateral sclerosis. Eur J Neurosci 2010; 31: 2247– 65 [DOI] [PubMed] [Google Scholar]
- 4. Bilsland LG, Sahai E, Kelly G, et al. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci U S A 2010; 107: 20523– 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clement AM, Nguyen MD, Roberts EA, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003; 302: 113– 17 [DOI] [PubMed] [Google Scholar]
- 6. De Winter F, Vo T, Stam FJ, et al. The expression of the chemorepellent semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol Cell Neurosci 2006; 32: 102– 15 [DOI] [PubMed] [Google Scholar]
- 7. Pasterkamp RJ, Kolodkin AL. Semaphorin junction: Making tracks toward neural connectivity. Curr Opin Neurobiol 2003; 13: 79– 89 [DOI] [PubMed] [Google Scholar]
- 8. Deo RC, Schmidt EF, Elhabazi A, et al. Structural bases for CRMP function in plexin-dependent semaphorin 3A signaling. EMBO J 2004; 23: 9– 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uchida Y, Ohshima T, Sasaki Y, et al. Semaphorin3A signaling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 2005; 10: 165– 79 [DOI] [PubMed] [Google Scholar]
- 10. Lin PC, Chan PM, Hall C, et al. Collapsin response mediator proteins (CRMPs) are a new class of microtubule-associated protein (MAP) that selectively interacts with assembled microtubules via a Taxol-sensitive binding interaction. J Biol Chem 2011; 286: 41466– 78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Niisato E, Nagai J, Yamashita N, et al. CRMP4 suppresses apical dendrite bifurcation of CA1 pyramidal neurons in the mouse hippocampus. Dev Neurobiol 2012; 72: 1447– 57 [DOI] [PubMed] [Google Scholar]
- 12. Duplan L, Bernard N, Casseron W, et al. Collapsin response mediator protein 4a (CRMP4a) is upregulated in motoneurons of mutant SOD1 mice and can trigger motoneuron axonal degeneration and cell death. J Neurosci 2010; 30: 785– 96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hensley K, Venkova K, Christov A. Emerging biological importance of central nervous system lanthionines. Molecules 2010; 15: 5581– 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liang W-C, Dennis MS, Stawicki Y, et al. Function blocking antibodies to neuropilin-1 generated from a designed human synthetic antibody phage library. J Mol Biol 2007; 366: 815– 29 [DOI] [PubMed] [Google Scholar]
- 15. Pan Q, Chanthery Y, Liang W-C, et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell 2007; 11: 53– 67 [DOI] [PubMed] [Google Scholar]
- 16. Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu Zn superoxide dismutase mutation. Science 1994; 264: 1772– 75 [DOI] [PubMed] [Google Scholar]
- 17. Mead RJ, Bennett EJ, Kennerley AJ, et al. Optimised and rapid pre-clinical screening in the SOD1-G93A transgenic mouse model of amyotrophic lateral sclerosis (ALS). PLoS One 2011; 6: e23244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saban MR, Sferra TJ, Davis CA, et al. Neuropilin-VEGF signaling pathway acts as a key modulator of vascular, lymphatic, and inflammatory cell responses of the bladder to intravesicular BCG treatment. Am J Ren Physiol 2010; 299: F1245– 56 [DOI] [PubMed] [Google Scholar]
- 19. Grandclement C, Borg C. Neuropilins: A new target for cancer therapy. Cancers 2011; 3: 1899– 928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cashman NR, Durham HD, Blusztajn JK, et al. Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn 1992; 194: 209– 21 [DOI] [PubMed] [Google Scholar]
- 21. Schmidt ERE, Pasterkamp RJ, van den Berg L. Axon guidance proteins: Novel therapeutic targets for ALS? Prog Neurobiol 2009; 88: 286– 301 [DOI] [PubMed] [Google Scholar]
- 22. Hernandez S, Texido L, Caldero J, et al. Increased intramuscular nerve branching and inhibition of programmed cell death of chick motoneurons by immunoglobulins from patients with motoneuron disease. J Neuroimmunol 2010; 229: 157– 68 [DOI] [PubMed] [Google Scholar]
- 23. Jubb AM, Strickland LA, Liu SD, et al. Neuropilin-1 expression in cancer and development. J Pathol 2012; 226: 50– 60 [DOI] [PubMed] [Google Scholar]
- 24. Bumbaca D, Xiang H, Boswell CA, et al. Maximizing tumor exposure to anti–neuropilin-1 antibody requires saturation of non-tumor antigenic sinks in mice. Br J Pharmacol 2012; 166: 368– 77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalil K, Li L, Hutchins BI. Signaling mechanisms in cortical axon growth, guidance, and branching. Front Neuroanat 2011; 5: 1– 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Van Hoecke A, Schoonaert L, Lemmens R, et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med 2012; 18: 1418– 22 [DOI] [PubMed] [Google Scholar]
- 27. Cole AR, Noble W, van Aalten L, et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J Neurochem 2007; 103: 1132– 44 [DOI] [PubMed] [Google Scholar]
- 28. Takata K, Kitamura Y, Nakata Y, et al. Involvement of WAVE1 accumulation in Aβ/APP pathology–dependent tangle modification in Alzheimer’s disease. Am J Pathol 2009; 175: 17– 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Good PF, Alapat D, Hsu A, et al. A role for semaphoring 3A signaling in the degeneration of hippocampal neurons during Alzheimer’s disease. J Neurochem 2004; 91: 716– 36 [DOI] [PubMed] [Google Scholar]