

Abstract
Background
The ability to establish genetic risk models is critical for early identification and optimal treatment of breast cancer. For such a model to gain clinical utility, more variants must be identified beyond those discovered in previous genome wide association studies (GWAS). This is especially true for women at high risk because of family history, but without BRCA1/2 mutations.
Methods
This study incorporates three datasets in a GWAS analysis of women with Ashkenazi Jewish (AJ) homogeneous ancestry. Two independent discovery cohorts were comprised of 239 and 238 AJ women with invasive breast cancer or preinvasive ductal carcinoma in situ and strong family histories of breast cancer, but lacking the three BRCA1/2 founder mutations, along with 294 and 230 AJ controls, respectively. An independent, third cohort of 203 AJ cases with familial breast cancer history and 263 healthy controls of AJ women was used for validation.
Results
A total of 19 SNPs were identified as associated with familial breast cancer risk in AJ women. Among these SNPs, 13 were identified from a panel of 109 discovery SNPs, including an FGFR2 haplotype. Additionally, 6 previously identified breast cancer GWAS SNPs were confirmed in this population. Seven of the 19 markers were significant in a multivariate predictive model of familial breast cancer in AJ women, 3 novel SNPs [rs17663555(5q13.2), rs566164(6q21), and rs11075884(16q22.2)], the FGFR2 haplotype, and 3 previously published SNPs [rs13387042(2q35), rs2046210(ESR1), and rs3112612(TOX3)], yielding moderate predictive power with an area under the curve (AUC) of the ROC (receiver-operator characteristic curve) of 0.74.
Conclusion
Population-specific genetic variants in addition to variants shared with populations of European ancestry may improve breast cancer risk prediction among AJ women from high-risk families without founder BRCA1/2 mutations.
Keywords: Ashkenazi Jewish, breast cancer, genome-wide association study, SNP, risk model, AUC
Breast cancer continues to be the most common gender-specific malignancy and a leading cause of death in the United States, accounting for nearly one-third of all new cancers in females (Jemal et al. 2010; Siegel et al. 2011). Breast cancer ranks as the most common cause of cancer deaths among women between the ages of 20 and 59 years, highlighting the need for accurate risk assessment for women in this age group.
Family history is an important risk factor for breast cancer. The risk of developing breast cancer for a woman with a first-degree affected relative is increased two-fold (Easton et al. 2007). The risk is even greater for women with multiple cases in family members. Breast cancer risk may be attributable to mutations in high-penetrance genes such as BRCA1, BRCA2, p53 and PTEN, as well as moderate or low penetrance genes (e.g., CHEK2, ATM, HRAS1, BRIP1, and PALB2), but these mutations account for a relatively small proportion of the heritable risk in these breast cancer families (Easton 1999; Walsh et al. 2006; Walsh et al. 2010).
To date genome-wide association studies (GWAS) have used high-density genotyping successfully primarily in European-American populations to identify SNPs associated with breast cancer risk in several genes including FGFR2, TNRC9, MAP3K1, LSP1, CASP8, SLC4A7, NEK10 and COX11 and the 8q and 2q35 chromosomal regions (Ahmed et al. 2009; Cox et al. 2007; Easton et al. 2007; Rahman et al. 2007; Stacey et al. 2007). However, each of these SNPs is a low penetrance allele that is not adequate to predict individual risk even in combination. Thus, identification of additional risk variants is necessary for prediction of individual risk. Additionally, whereas these SNPs are well-represented in European-American groups, their allele frequencies and the attendant risks vary in other populations. This is a general phenomenon. Variability of risk allele frequency and effect size has been observed among major ethnic groups (European, African and Asian) for a panel of complex disease SNPs that had reached genome-wide significance (p≤5×10-8) in at least one of the groups (Ntzani et al. 2011). Other studies point to the importance of controlling for ancestry /ethnicity in designing genetic association studies (Haiman and Stram 2010).
The current study utilizes the Ashkenazi Jewish (AJ) population, the largest genetic isolate in the United States, comprising 2% of the total population (Stacey et al. 2007). The study of this group reduces the major confounding effect of population stratification and holds the promise of identifying founder mutations and less common mutations not easily identifiable in the general population. Accordingly, GWAS have successfully utilized this homogeneous group to identify disease-associated alleles including a recent study by a member of our group that identified 5 new risk alleles for Crohn disease, known to have up to 4-fold higher prevalence in the AJ population (Kenny EE 2012). Similarly, another group identified a new region at chromosome 6q22.33 that is associated with breast cancer in AJ women, and this dataset was used as the starting point for the current study (Gold et al. 2008).
The current study pays special attention to AJ women that are classified as high-risk for breast cancer based on family history but do not carry any of the 3 founder BRCA1/2 mutations, and, therefore, have no strategy for surveillance/prevention (Rubinstein 2004). Whereas there are many advantages to using genetically isolated populations, recruiting large cohorts of these distinct groups can be challenging, and this is further compounded by the need for high-risk women with no known BRCA1/2 mutations. Another potential difficulty is that other known/published risk variants besides BRCA1/2 mutations may not be replicable in these AJ women either. To accommodate for the drop in statistical power that results from the use of smaller cohorts this study employed a three-step design, in which each independent dataset included AJ women with familial breast cancer and an average risk control population to identify genetic variants associated with risk of breast cancer in these AJ women. We identified 7 new risk SNPs, one 6-SNP FGFR2 haplotype and confirm 6 known breast cancer SNPs in the AJ population. These SNPs were then evaluated in a multivariate predictive model and showed moderate discriminatory accuracy for separating familial breast cancer cases from controls among AJ women.
Methods
Patient datasets
The Discovery Phase included 1008 AJ females from 2 datasets. The first study group was comprised of a publicly available dataset provided by Memorial-Sloan Kettering Cancer Center (MSKCC) (Gold et al. 2008). In the second dataset, cases were women with invasive breast cancer or preinvasive ductal carcinoma in situ (DCIS) recruited as part of the Breast Cancer Family Registry (BCFR) [27], and unaffected controls without family histories through the New York Cancer Project (NYCP) (John et al. 2004; Mitchell et al. 2004). The BCFR cases were recruited at Columbia University in New York, NY (111 patients), Ontario Cancer Center in Toronto, ON (101 patients), the Cancer Prevention Institute of California in Fremont, CA (16 patients), and the Fox Chase Cancer Center in Philadelphia, PA (10 patients). Both the MSKCC and BCFR datasets were comprised of AJ women with breast cancer from high-risk families (at least 3 affected relatives in a single lineage for MSKCC and at least 1 first- or second-degree affected relative for the BCFR) who do not carry any of the three AJ BRCA1/2 founder mutations (BRCA1-5382insC, BRCA1-185delAG, and BRCA2-6174delT). Average age of diagnosis was 51.0±9.4 for MSKCC and 47.5±10.0 for BCFR. Both study groups included unaffected AJ controls (age-matched to the cases in the BCFR dataset: 47.9±9.8) with no family history of breast cancer. All subjects recruited for this study were considered AJ if they reported that all four grandparents were of Eastern European Jewish ancestry. 239 cases were compared to 294 controls from MSKCC in step one of the Discovery Phase followed by 238 cases and 230 controls from BCFR in step two (Figure 1).
Figure 1. Flow diagram of data analysis.
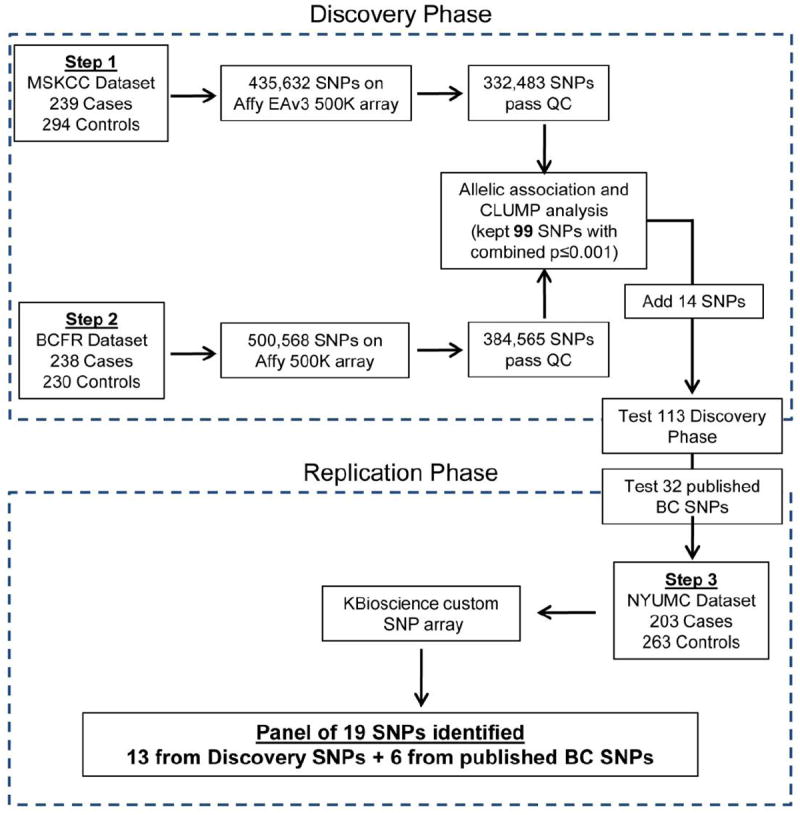
For the Replication Phase, 203 cases and 263 controls were recruited at New York University Medical Center (NYUMC). Cases were selected based on having 3 or more affected relatives in a single lineage (including the proband), and negative for the three BRCA1/2 founder mutations. Average age of diagnosis was 52.2±9.5. Controls were determined to lack personal and family histories of breast cancer and were at least 60 years old at the time of enrollment (68.5±7.0). DNA was extracted from NYUMC samples using the Puregene method (Qiagen).
Genotyping
MSKCC samples were genotyped on early-access Affymetrix 500K (EAv3) SNP arrays as described previously (Gold et al. 2008). BCFR samples were genotyped on commercially available Affymetrix 500K SNP arrays according to manufacturer instructions. There is an overlap of 435,632 SNPs between the early access and commercial platforms.
For the replication study, custom SNP assays were designed by KBioscience (Herts, England) using KASPar chemistry (http://www.kbioscience.co.uk/reagents/KASP/KASP.html) for testing 147 SNPs. Included in this list were 113 SNPs from the Discovery Phase; 4 assays failed leaving 109 SNPs for analysis. The other 34 are known breast cancer SNPs based on associations published at the time this study was designed. This list includes SNPs associated with breast cancer specifically in the AJ population or SNPs with genome-wide significant associations (p≤5×10-8) with breast cancer in other populations thereby having the greatest chance of replicating in the AJ population (Easton et al. 2007; Fletcher et al. 2011; Hunter et al. 2007; Li et al. 2011; Long et al. 2010; Stacey et al. 2007; Thomas et al. 2009; Turnbull et al. 2010; Zheng et al. 2009). One of these assays failed, leaving 33 known breast cancer SNPs for analysis.
Data Analysis
All genetic analyses were performed using PLINK (Purcell et al. 2007). 533 MSKCC and 468 BCFR samples were used in the final analysis for the Discovery Phase. IBD/IBS analysis (PLINK genome command) was used to eliminate subjects who were identical or closely related to others within or between the two datasets (IBD ≥ 0.46, IBS ≥0.85; IBD/IBS>0.99 for identical subjects). Principle components analysis (PCA; R software) was used to confirm AJ ancestry and remove 6 outlier subjects showing different or more admixed ancestry in the combined MSKCC and BCFR datasets versus reference HapMap3 populations. The utility of PCA for determining AJ ancestry has been previously demonstrated (Need et al. 2009). In the current study, the subjects who were removed were ≥5 standard deviations from the mean for at least 1 of the first 10 principle components (data not shown). All patient samples had at least 90% genotype calls so none were removed based on this.
SNPs that had genotyping rates <95% or minor allele frequencies <1% were excluded. For the remaining SNPs, chi-square association analyses were performed on minor allele counts (0, 1, 2) and genotype distributions between cases and controls. In addition to standard association analyses the CLUMP command was used to identify regions of SNPs in moderate linkage disequilibrium (LD). SNPs within a region spanning 1000kb and an LD threshold of r2=0.5 were considered, if an index SNP had p<0.01 and CLUMP SNPs had p<0.05.
Subjects and SNPs for the Replication Phase were subject to the same QC filtering as that performed for the Discovery Phase. Chi-square association analyses for both minor allele count (0, 1, or 2) and genotype distribution were performed. Haplotype analysis was also performed using the blocks function in PLINK. Pooled p-values and odds ratios (ORs) were obtained by combining the 3 datasets for chi-square association analysis. We also calculated and reported the Fisher meta p-value from p-values of three independent cohorts. Common measures of heterogeneity for meta analysis including Cochran’s Q statistic and I2 statistic (Higgins et al. 2003) were calculated and reported for each of the SNPs in Tables 1-3.
Table 1.
Novel candidate SNPs for familial AJ breast cancer.
| Pooled | Meta | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| SNP | Chr | MA | Dataset | MAFA | MAFU | Allelic p-value | OR | Genotypic p-value | Allelic P-value | OR | Genotypic P-value | Fisher Meta P | I2/Q |
| rs566164 | 6q21 | A | MSKCC | 0.32 | 0.24 | 4.37×10-3 | 1.49 | 1.55×10-2 | 6.67×10-6 | 1.45 | 6.31×10-5 | 2.00×10-4 | 0%/0.16 |
| BCFR | 0.36 | 0.29 | 1.50×10-2 | 1.41 | 5.36×10-2 | ||||||||
| NYUMC | 0.32 | 0.25 | 2.40×10-2 | 1.40 | 1.01×10-1 | ||||||||
|
| |||||||||||||
| rs16882214 | 6p22.3 | G* | MSKCC | 0.09 | 0.19 | 1.75×10-6 | 0.40 | 1.00×10-5 | 7.25×10-6 | 0.70 | 2.13×10-5 | 1.53×10-6 | 89%/18 |
| BCFR | 0.12 | 0.17 | 2.30×10-2 | 0.65 | 6.30×10-2 | ||||||||
| NYUMC | 0.15 | 0.12 | 1.96×10-1 | 1.28 | 3.50×10-2 | ||||||||
|
| |||||||||||||
| rs16956185 | 18p11.22 | A | MSKCC | 0.12 | 0.08 | 3.91×10-2 | 1.52 | 9.30×10-2 | 3.14×10-5 | 1.70 | 1.73×10-4 | 1.00×10-4 | 0%/1.2 |
| BCFR | 0.11 | 0.05 | 1.22×10-3 | 2.25 | 8.70×10-3 | ||||||||
| NYUMC | 0.13 | 0.08 | 2.66×10-2 | 1.62 | 1.21×10-2 | ||||||||
|
| |||||||||||||
| rs5965136 | Xq12 | A | MSKCC | 0.03 | 0.01 | 1.46×10-2 | 3.39 | 6.40×10-3 | 4.83×10-5 | 4.78 | 2.03×10-5 | 5.00×10-4 | 0%/1.0 |
| BCFR | 0.02 | 0.002 | 7.40×10-3 | 9.89 | 3.70×10-2 | ||||||||
| NYUMC | 0.012 | 0.002 | 4.91×10-2 | 6.53 | 1.19×10-1 | ||||||||
|
| |||||||||||||
| rs17663555 | 5q13.2 | G* | MSKCC | 0.61 | 0.48 | 2.09×10-3 | 1.66 | 9.85×10-3 | 5.74×10-5 | 1.38 | 3.82×10-4 | 5.00×10-4 | 0%/1.4 |
| BCFR | 0.58 | 0.52 | 1.53×10-2 | 1.26 | 5.82×10-3 | ||||||||
| NYUMC | 0.56 | 0.47 | 1.85×10-1 | 1.45 | 7.15×10-2 | ||||||||
|
| |||||||||||||
| rs12906542 | 15q24.3 | A | MSKCC | 0.04 | 0.07 | 3.65×10-2 | 0.54 | 3.22×10-2 | 6.70×10-6 | 0.50 | 7.12×10-6 | 7.34×10-7 | 80%/10 |
| BCFR | 0.007 | 0.07 | 9.90×10-7 | 0.09 | 2.20×10-7 | ||||||||
| NYUMC | 0.11 | 0.15 | 9.67×10-2 | 0.72 | 2.70×10-2 | ||||||||
|
| |||||||||||||
| rs11075884 | 16q22.2 | G | MSKCC | 0.22 | 0.28 | 2.43×10-2 | 0.72 | 2.49×10-2 | 9.15×10-3 | 0.80 | 1.31×10-4 | 7.00×10-4 | 80%/10 |
| BCFR | 0.20 | 0.29 | 1.24×10-3 | 0.61 | 3.78×10-3 | ||||||||
| NYUMC | 0.29 | 0.25 | 2.71×10-1 | 1.18 | 4.92×10-4 | ||||||||
G/C loci: minor allele = G on positive strand.
MA=minor allele; MAF=minor allele frequency for Cases (“A”) and Controls (“U”); p-values based on χ2 test on allele counts or genotype distribution between cases and controls. All 3 datasets were combined for pooled P-values and ORs and Fisher Meta-analysis. Cochran Q and I2 statistics for heterogeneity are reported (“I2/Q”).
Table 3.
Confirmation of known breast cancer variants.
| SNP | Chr | Gene | MA | Dataset | MAFA | MAFU | P-value | OR | Pooled P-value | Pooled OR | Fisher Meta P | I2/Q |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs13387042 | 2q35 | Unknown | G | NYUMC | 0.34 | 0.48 | 3.31×10-2 | 0.57 | N/A | N/A | N/A | |
|
| ||||||||||||
| rs889312 | 5q | MAP3K1 | C | NYUMC | 0.38 | 0.30 | 1.38×10-2 | 1.41 | N/A | N/A | N/A | |
|
| ||||||||||||
| rs2046210 | 6q25.1 | ESR1 | A | MSKCC | 0.37 | 0.35 | 0.360 | 1.13 | 1.61×10-2 | 1.21 | 7.71×10-2 | 0%/1.1 |
| BCFR | 0.39 | 0.36 | 0.299 | 1.15 | ||||||||
| NYUMC | 0.39 | 0.32 | 3.13×10-2 | 1.35 | ||||||||
|
| ||||||||||||
| rs3803662 | 16q | TOX3 | A | MSKCC | 0.46 | 0.38 | 1.76×10-2 | 1.35 | 7.19×10-5 | 1.35 | 6.00×10-4 | 0%/0.34 |
| BCFR | 0.42 | 0.36 | 5.39×10-2 | 1.30 | ||||||||
| NYUMC | 0.44 | 0.36 | 7.72×10-3 | 1.43 | ||||||||
|
| ||||||||||||
| rs4784227 | 16q | TOX3 | T | MSKCC | 0.38 | 0.40 | 0.629 | 0.94 | 2.74×10-2 | 1.19 | 9.80×10-3 | 65%/5.7 |
| BCFR | 0.34 | 0.28 | 2.90×10-2 | 1.36 | ||||||||
| NYUMC | 0.36 | 0.29 | 1.20×10-2 | 1.43 | ||||||||
|
| ||||||||||||
| rs3112612 | 16q | TOX3 | G | NYUMC | 0.40 | 0.51 | 1.55×10-3 | 0.65 | N/A | N/A | N/A | |
MA=minor allele; MAF=minor allele frequency for Cases (“A”) and Controls (“U”); p-values based on χ2 test on allele counts between cases and controls. All 3 datasets were combined for pooled P-values and Ors and Fisher Meta-analysis. Cochran Q and I2 statistics for heterogeneity are reported (“I2/Q”).
The NYUMC replication dataset was also used to evaluate utility in the multivariate models in terms of the discriminatory accuracy of the identified 19 SNP panel. Number of risk alleles for each SNP was used as an independent predictor in binary logistic regression analysis of the GLM function using SPSS v.19 with phenotype (cases versus controls) as the dependent variable. For each individual patient, each SNP was assigned a score (0, 1, or 2) based on the number of risk alleles. For FGFR2 a gene score was based on the total number of risk alleles across all 6 FGFR2 SNPs (0-12) for each person was used. Two redundant TOX3 SNPs were removed, only leaving the SNP (rs3112612) in the multivariate model. Five other SNPs were removed based on p>0.05 (rs16882214, rs12906542, rs16956185, rs5965136, and rs889312) in the multivariate logistic model. A receiver operating characteristic (ROC) curve was constructed using the final panel of 7 markers, and area under the ROC curve (AUC) was calculated to measure the power of the panel to distinguish breast cancer occurrence from controls. The predictive model was cross-validated using the ROCR package in the R platform. The NYUMC cohort was randomly split 50:50 to generate two new sub-cohorts. One-thousand iterations were performed, and at each iteration the model was fit using the first sub-cohort and tested using the second sub-cohort. A ROC curve was generated and AUC values calculated for the second cohort at each iteration. An average cross-validation AUC with empirical 95% confidence interval values are reported.
Results
Seven new candidate SNPs are identified in association with familial breast cancer in AJ women
To identify familial breast cancer risk variants we designed a GWAS workflow that utilized two separate datasets for the Discovery Phase and a third dataset for the Replication Phase (Figure 1). Quality control (QC) filtering (see Methods) resulted in 332,483 SNPs for Step 1 and 384,565 SNPs for Step 2 of the Discovery Phase. Overlapping top-ranked SNPs from both steps revealed 84 SNPs that were potentially associated with breast cancer in both the MSKCC and BCFR datasets (with Fisher combined p≤0.001). An additional 2 SNPs that were highly significant (p<10-7) in the BCFR dataset but untested in the MSKCC dataset were also carried forward.
Additionally, trying to take advantage of the relative genetic homogeneity of the AJ population, we identified shared ancestral haploblocks to find risk variants. A method for long-range haplotyping to investigate changes in linkage disequilibium (LD) patterns between breast cancer patients and controls (utilizing the CLUMP function in PLINK) was used to identify less significant SNPs that might otherwise be masked by the more significant risk variants (Wang et al. 2010). We have identified 15 SNPs in 7 regions that overlap between the MSKCC and BCFR datasets using this method (data not shown)
In Step 3 (the Replication Phase) the 101 SNPs from standard association analysis plus long-range haplotyping were tested in a third AJ dataset of high-risk familial breast cancer cases versus hyper-controls (AJ women with no family history and at least 60 years old). Allelic (Amitage trend test) and genotypic chi-square tests revealed 7 new candidate SNPs with pooled p-values across the phases, totaling 680 cases and 787 controls, ranging from 9.15×10-3 – 6.67×10-6 (allelic χ2) and 3.82×10-3 – 7.12×10-6 (genotypic χ2) (Table 1).
SNPs previously reported to have significant associations in the MSKCC GWAS were re-examined in the BCFR and NYUMC datasets (Table 2a). Since a slightly different subset of the MSKCC subjects was used (removing subjects closely related to each other or to subjects in the BCFR dataset), the p-values reported here vary somewhat from those published. The FGFR2 SNP, rs1078806, was determined to be significant in the NYUMC Replication cohort (Table 2a) as well as the BCFR Discovery dataset and similar effects, indicated by the odds ratios, were observed among all three datasets. rs7203563 (A2BP1) was significant in the BCFR dataset (p=2.43×10-2) and rs3012642 (PHKA1/HDAC8) nearly reached significance in this dataset (p=5.37×10-2), but neither SNP was significant in the NYUMC dataset. Similar odds ratios were also observed for the four ECHDC1/RNF146 SNPs in the BCFR and NYUMC datasets as that previously reported for the MSKCC dataset.
Table 2.
Previously identified variants for MSKCC dataset (a) and FGFR2 haplotype (b).
| a)
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Gene | MA | Dataset | MAFA | MAFU | P-value | OR | Pooled P-value | Pooled OR | Fisher Meta P | I2/Q |
| rs2180341 | 6q22.33 | ECHDC1; RNF146 | C | MSKCC | 0.28 | 0.19 | 6.86×10-4 | 1.64 | 4.77×10-3 | 1.28 | 6.40×10-3 | 56%/4.6 |
| BCFR | 0.25 | 0.22 | 2.51×10-1 | 1.20 | ||||||||
| NYUMC | 0.25 | 0.24 | 7.48×10-1 | 1.05 | ||||||||
|
| ||||||||||||
| rs6569479 | 6q22.33 | ECHDC1; RNF146 | A | MSKCC | 0.27 | 0.19 | 5.81×10-3 | 1.50 | 1.06×10-2 | 1.25 | 2.94×10-2 | 15%/2.4 |
| BCFR | 0.25 | 0.22 | 2.70×10-1 | 1.19 | ||||||||
| NYUMC | 0.26 | 0.24 | 5.74×10-1 | 1.09 | ||||||||
|
| ||||||||||||
| rs6569480 | 6q22.33 | ECHDC1; RNF146 | A | MSKCC | 0.27 | 0.19 | 1.77×10-3 | 1.58 | 5.62×10-3 | 1.27 | 1.19×10-2 | 39%/3.3 |
| BCFR | 0.25 | 0.22 | 2.70×10-1 | 1.19 | ||||||||
| NYUMC | 0.26 | 0.24 | 5.82×10-1 | 1.09 | ||||||||
|
| ||||||||||||
| rs7776136 | 6q22.33 | ECHDC1; RNF146 | A | MSKCC | 0.27 | 0.19 | 2.58×10-3 | 1.56 | 7.85×10-3 | 1.26 | 1.68×10-2 | 38%/3.2 |
| BCFR | 0.25 | 0.22 | 2.54×10-1 | 1.19 | ||||||||
| NYUMC | 0.26 | 0.24 | 6.60×10-1 | 1.07 | ||||||||
|
| ||||||||||||
| rs1078806 | 10q | FGFR2 | C | MSKCC | 0.44 | 0.39 | 7.66×10-2 | 1.25 | 1.70×10-6 | 1.43 | 9.64×10-6 | 25%/2.7 |
| BCFR | 0.46 | 0.38 | 7.72×10-3 | 1.43 | ||||||||
| NYUMC | 0.48 | 0.35 | 1.05×10-4 | 1.69 | ||||||||
|
| ||||||||||||
| rs7203563 | 16p13.3 | A2BP1 | C | MSKCC | 0.12 | 0.08 | 2.26×10-2 | 1.60 | 4.10×10-2 | 1.30 | 5.50×10-3 | 75%/8.0 |
| BCFR | 0.11 | 0.07 | 2.43×10-2 | 1.70 | ||||||||
| NYUMC | 0.07 | 0.09 | 1.93×10-1 | 0.72 | ||||||||
|
| ||||||||||||
| rs3012642 | Xq13.1 | PHKA1; HDAC8 | G | MSKCC | 0.06 | 0.03 | 2.44×10-2 | 2.01 | 8.91×10-3 | 1.64 | 3.84×10-2 | 18%/2.4 |
| BCFR | 0.06 | 0.03 | 5.37×10-2 | 1.86 | ||||||||
| NYUMC | 0.03 | 0.03 | 9.85×10-1 | 0.99 | ||||||||
|
b)
| ||||||||||||
| SNP | Chromosome | Gene | Minor Allele | Dataset | MAF (cases) | MAF (controls) | P-value | OR | Pooled P-value | Pooled OR | Meta P-value | |
|
| ||||||||||||
| rs11200014 | 10q | FGFR2 | A | NYUMC | 0.48 | 0.35 | 1.23×10-4 | 1.68 | N/A | N/A | N/A | |
|
| ||||||||||||
| rs2981579 | 10q | FGFR2 | A | NYUMC | 0.48 | 0.36 | 1.67×10-4 | 1.66 | N/A | N/A | N/A | |
|
| ||||||||||||
| rs1078806 | 10q | FGFR2 | G | MSKCC | 0.44 | 0.39 | 7.66×10-2 | 1.25 | 1.70×10-6 | 1.43 | 9.64×10-6 | |
| BCFR | 0.46 | 0.38 | 7.72×10-3 | 1.43 | ||||||||
| NYUMC | 0.48 | 0.35 | 1.05×10-4 | 1.69 | ||||||||
|
| ||||||||||||
| rs1219648 | 10q | FGFR2 | G | NYUMC | 0.48 | 0.36 | 2.67×10-4 | 1.63 | N/A | N/A | N/A | |
|
| ||||||||||||
| rs2420946 | 10q | FGFR2 | T | NYUMC | 0.48 | 0.37 | 9.57×10-3 | 1.56 | N/A | N/A | N/A | |
|
| ||||||||||||
| rs2981582 | 10q | FGFR2 | A | NYUMC | 0.48 | 0.37 | 9.57×10-3 | 1.56 | N/A | N/A | N/A | |
MA=minor allele; MAF=minor allele frequency for Cases (“A”) and Controls (“U”); p-values based on χ2 test on allele counts between cases and controls. All 3 datasets were combined for pooled P-values and Ors and Fisher Meta-analysis. Cochran Q and I2 statistics for heterogeneity are reported (“I2/Q”).
We chose 5 additional SNPs for haplotype analysis of FGFR2, based on Discovery Phase results plus published data. Analysis of a 6-SNP FGFR2 haplotype (rs11200014, rs2981579, rs1078806, rs1219648, rs2420946, rs2981582) revealed a significant minor (risk) allele haplotype (AAGGTA) in the cases (47.9%) compared to controls (36.7%, p=2.18×10-4) in the NYUMC Replication cohort (Table 2b). This result agrees with findings of a previous study of AJ women that identified a haplotype of four FGFR2 SNPs [rs11200014 (A), rs2981579 (A), rs1219648 (G), and rs2420946 (T)] significantly associated with breast cancer (p=5.90×10-3; OR=1.25) (Raskin et al. 2008). Our findings are similar, adding two additional SNPs [rs1078806 (G) and rs2981582 (A)] for a longer haplotype in association with breast cancer in AJ women.
In total, 113 SNPS from Discovery cohorts were carried forward to the Replication phase, the top 84 from standard association analysis, 2 highly-significant SNPs in the BCFR cohort that were absent from the platform used for MSKCC, 15 from LD Clumping, 7 from the original MSKCC analysis, and 5 additional SNPs for haplotype analysis. Custom SNP assays for 4 of the top 84 SNPs failed, leaving 109 SNPs tested in the NYUMC Replication cohort. Significant associations (P<0.05) were indicated in 13 of these 109 SNPs (11.9%) in the Replication phase.
Six previously identified SNPs were potentially associated with AJ familial breast cancer risk
In addition to the 113 SNPs identified in the Discovery Phase, we tested 34 SNPs previously reported to be associated with breast cancer risk, either in the AJ population or having reached genome-wide significance in other populations (p≤5×10-8), many of which were not present on the Affymetrix 500K array used in MSKCC and BCFR. One of the custom SNP assays failed. Six of the remaining 33 assays for known breast cancer SNPs (18.2%) were significant in the NYUMC dataset (Table 3, p<0.05). Three of these 6 known SNPs were on the 500K arrays and the data for the MSKCC and BCFR datasets are presented here as well.
SNPs moderately predict familial breast cancer risk in AJ women
We investigated the 19 SNPs (7 new candidate SNPs, the 6-SNP FGFR2 haplotype and the 6 previously published BC SNPs that were confirmed (p<0.05) in our population), for their ability to discriminate between AJ familial breast cancer cases and AJ controls. Scores for individual SNPs (0, 1, or 2 risk alleles) plus a gene score for FGFR2 (0-12 risk alleles) were used in stepwise logistic regression analysis comparing cases versus controls from the NYUMC replication dataset. SNPs with p>0.05 were eliminated one-by-one, resulting in a final list of 6 SNPs plus the FGFR2 gene score that significantly contributed to this risk model (Figure 2). This list included 3 new SNPs [rs17663555(5q13.2), rs566164(6q21), and rs11075884(16q22.2)] and 3 known SNPs [rs13387042 (2q35), rs2046210 (ESR1), and rs3112612 (TOX3)]. The table in Figure 2 shows the effect of each genotype (0, 1 or 2 risk alleles) or FGFR2 gene score as a covariate in the model. The predictive accuracy of the model was reflected in an AUC of 0.74 [95%CI: 0.69, 0.79] for the ROC curve. Cross-validation analysis generated an average AUC of 0.67 [95%CI: 0.62, 0.72].
Figure 2. Predictive model for breast cancer in AJ women.
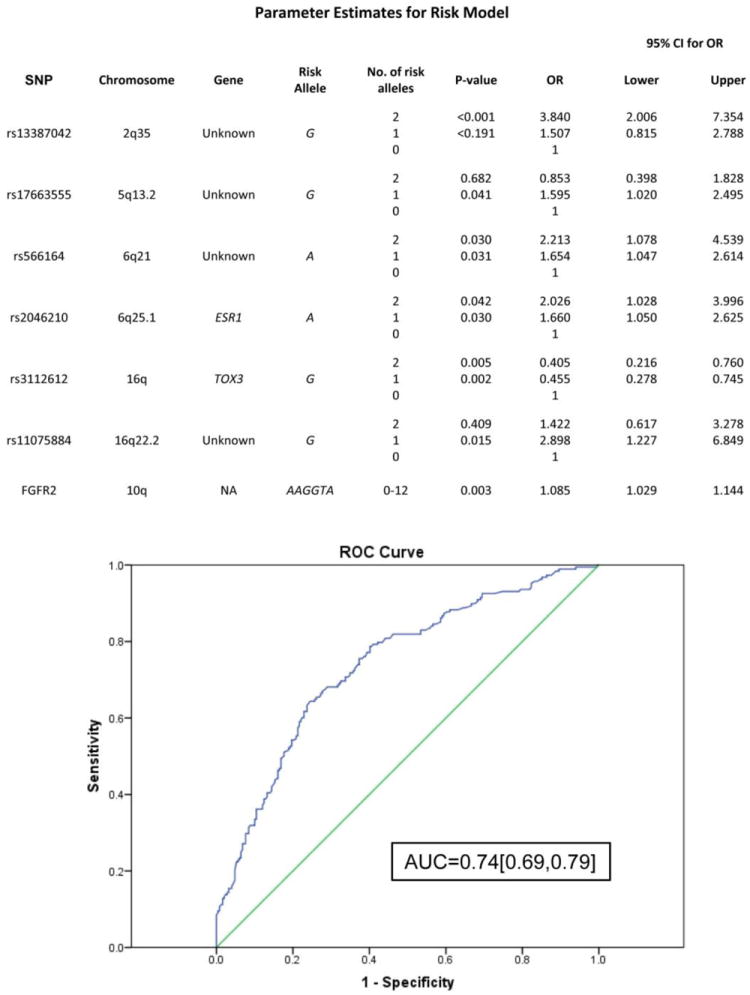
Seven markers were used to construct a multivariate logistic model for predicting risk in the NYUMC dataset. P-values and Odds Ratios (ORs) are listed for each SNP for 1 or 2 copies of the risk allele compared to 0 copies of the risk allele, or FGFR2 gene score (0-12 risk alleles). Predictability of the 7-marker panel is evaluated with AUC of the ROC curve shown below.
Discussion
The notion of ‘common disease-common variant’ has come under scrutiny in part because of the inability to identify enough variants to explain most of the heritability of complex diseases, such as breast cancer (Cazier and Tomlinson 2010; McClellan and King 2010). GWAS have identified several reproducible SNPs, but these high-frequency (MAF>5%), low-penetrance (<1.5 fold increase in risk) alleles account only for a small percentage of familial breast cancer risk (Harlid et al. 2012). At the same time studies have revealed a strong impact of population structure on common-versus- rare allelic variation and the need for more focus on ancestry in GWAS (Gravel et al. 2011; Guthery et al. 2007). In one study of the 4 major U.S. populations (African, Asian, European, and Hispanic), more than half of the common SNPs (MAF ≥10%) were shared among the 4 populations, but only one-third of these SNPs were common in all 4 populations (Guthery et al. 2007). More than half of all of the tested SNPs were private, occurring only in 1 of the 4 populations. Also, synonymous variants were more commonly shared among the populations than non-synonymous or nonsense mutations, indicating the presence of more recent, deleterious mutations in distinct populations.
Several GWAS have likewise identified disease-associated variants in the AJ population that were not previously identified in other populations. In addition to Crohn disease, AJ population studies have aided in the discovery of variants associated with Parkinson disease, bipolar disorder, and schizophrenia (Fallin et al. 2004; Liu et al. 2011; Shifman et al. 2006). Similar to the Crohn study, one group confirmed common variants associated with Parkinson disease in people of European origin, but also identified 6 new variants. While some of these disease-variants have and will replicate in non-AJ populations, it is clear that using subjects with homogeneous ancestry will, at the very least, facilitate the discovery of SNPs that are present at lower frequencies in members of that heterogeneous group.
The majority of the breast cancer SNPs identified to date, particularly those that have reached genome-wide significance, have been discovered using study populations of European ancestry. Likewise, commercial genotyping platforms have been designed with particular attention to more common variants, so odds are in favor of identifying only common risk alleles or common alleles that tag the actual causal variant. The AJ population is a genetically isolated one that harbors rare and private variants that are present at a much higher frequency in this group than the general population, such as the BRCA1/2 founder mutations. European ancestry is a major contributor to AJ genetics, and our data indicated 6 known breast cancer SNPs that seemingly survived admixture in this population (and more than these 6 are likely to be significant if our sample sizes are larger). But studying this population has allowed us to identify 7 additional variants that have not yet been linked to breast cancer, and it is expected that using denser coverage and larger sample sizes will reveal still more risk alleles.
Among the known breast cancer SNPs tested in the Replication Phase, 7 were originally identified in the MSKCC dataset (Gold et al. 2008). Statistical significance was not reached for 4 of these SNPs in the BCFR dataset and for 6 of the SNPs in the NYUMC dataset, though similar odds ratios were consistently observed across all three cohorts. Thus, it is likely that all of these SNPs would have achieved significance had the sample sizes been larger. Minor differences in study design may also have contributed to the observed differences in allele frequencies among cases and controls. Similar strategies were used for recruiting the cases at MSKCC, BCFR and NYUMC, selecting women with ≥3 affecteds in a single lineage at MSKCC and NYUMC and women with an average 2.86 cases of breast cancer per family through BCFR, but control recruitment differed for the NYUMC dataset compared to the Discovery Phase groups. Women aged 60 years or older were selected to exclude the chance of an early breast cancer diagnosis. The MSKCC controls were not selected based on age, but controls used in the BCFR dataset were age-matched to the cases. Similarly, phase 2 of the original MSKCC study [10] did select age-matched controls but did not select cases based on family history, and phase 3 utilized sporadic breast cancer cases. Two SNPs (rs7203563 and rs3012642) were not significant in phase 3 of the original MSKCC study, but it is unclear if this is a result of the use of sporadic cases. The remaining 5 SNPs were validated in both of the MSKCC phase 2 and 3 datasets. In the current study it is most likely that sample size impacted the results observed in the BCFR and NYUMC datasets because the observed odds ratios are all similar to the MSKCC group. Indeed, in the 3 phases of the original MSKCC study p-value ranges vary with the size of the respective datasets: phase 1 [548 subjects, p-values=8.9×10-4 – 4.5×10-2], phase 2 [1929 subjects, p-values=9.8×10-5 – 3.3×10-3], and phase 3 [430 subjects, p-values= 1.8×10-2 - 0.57]. Pooled p-values were examined for the 3 datasets in our study, for a total of 1467 subjects, and the range was 1.70×10-6 - 4.10×10-2. Fisher meta p-value from p-values of the three independent cohorts was calculated and reported together with common measures of heterogeneity for meta analysis including Cochran’s Q statistic and I2 statistic. Also, complete clinical data may reveal phenotypic subtype(s) with which some SNPs are more strongly associated.
To our knowledge, breast cancer risk models that do not incorporate BRCA1 or BRCA2 mutations have not been previously attempted for the AJ population. Cases in our study groups are without BRCA1/2 mutations. To assess usefulness of the identified markers in predicting AJ familial breast cancer cases versus AJ controls, we constructed a preliminary model using the NYUMC group starting with14 markers: 7 new candidate SNPs, a gene score for FGFR2 and 6 previously identified SNPs. We arrived at a 7-marker panel in a stepwise manner, eliminating 7 of the 14 SNPs that were not significant in the multivariate model (p>0.05). More details on the construction of the multivariate logistic model can be found in the Data Analysis section. In short, the model based on the 7 markers achieved an AUC of 0.74 (95% CI: 0.69, 0.79). However, it is important to note that since the NYUMC cohort was used to evaluate discovery SNPs and to construct the risk model, the model is subject to some degree of over-fitting or overestimation of the predictability. For this reason, cross-validation of the model was performed revealing an AUC of 0.67 (95%CI: 0.62, 0.72).
It is clear that many more SNPs of similar effect size will be required to predict familial breast cancer risk more accurately in AJ women. However, we provide evidence here of progress towards that goal, achieving moderate discriminatory accuracy with a panel of just 7 markers. It might also be valuable to test the Breast Cancer Risk Assessment Tool BCRAT data, based on age at menarche, age at first live birth, number of first-degree relatives with breast cancer, and number of previous benign breast biopsies, combined with this SNP panel. These clinical covariates have previously been shown with modest predictability for breast cancer risk [23]. Unfortunately, these covariates were not measured in this current study. Unlike the previous study, though, the NYUMC dataset was recruited to include controls without family histories of breast cancer. It will be necessary to recruit high-risk controls and cases to test the current model and build a potentially better predictive model.
Finally, 6 of the 7 candidate SNPs newly identified in this study reside in chromosomal regions previously known are linked to cancer. Deletions in 6q21 and 16q22.2 and gains in Xq12 have each been associated with breast cancer (Kuukasjarvi et al. 1997; Negrini et al. 1994; Nordgard et al. 2008). Likewise, amplification of 6p22.3 has been linked to multiple cancers including bladder and hereditary prostate cancer (Janer et al. 2003; Veltman et al. 2003). 18p11.22 has been linked to lung cancer and 5q13.2 deletions to chemoresistant ovarian tumors, colorectal cancer, and chondrosarcomas (Ahn et al. 2012; Dyrso et al. 2011; Hameed et al. 2009; Kim et al. 2007). However, we were unable to directly determine the specific biological relevance for these SNPs. GWAS SNP arrays were designed to detect common alleles with the assumption that they themselves are causal alleles or that they tag those that are. It is possible that the low-penetrance risk SNPs identified here are actually tagging causal high-penetrance mutations undetectable at this point due to low frequency and/or incomplete coverage of variants. Complete analysis using whole genome or exome sequencing might make the hunt for the alleles with the largest effect on their respective diseases feasible.
One of the new SNPs [rs5965136(Xq12)], however, was rare in this population and exhibited a large effect (pooled OR for the risk allele= 4.78). Therefore, another possible explanation is that the genes in proximity to these SNPs are not fully annotated, or that the SNPs affect genes further away. As more eQTL data becomes available, the function of these SNPs may be revealed.
We have addressed the need for better utilizing ancestry in GWAS to identify new candidate SNPs for predicting breast cancer risk in a specific population. One limitation to this study is the relatively small sample size resulting in limited power to achieve genome-wide significance or replicate known variants. This limitation might be the main cause for the insignificance of the 27 previously identified breast cancer SNPs in the NYUMC cohort, many of which had reached genome-wide significance in other studies. As mentioned earlier, some of the SNPs found to be associated with breast cancer risk in different populations may not be replicable in the AJ population without BRCA1/2 mutations even with a larger sample size. Nonetheless, our goal for examining these previously published SNPs in the NYUMC cohort was to identify potential candidates that might be useful in the context of building multivariate risk prediction models. Another limitation was the incomplete coverage of variants on the SNP arrays available at the onset of the study, impeding the detection of rare and/or causal variants. Similarly, there was incomplete coverage of SNPs in the MSKCC dataset compared to the BCFR dataset due to the use of early-access versus commercially available Affymetrix 500K arrays. As a result, about 13% of the SNPs genotyped in the BCFR cohort were not genotyped in the MSKCC cohort. Since 111 SNPs were identified from the 435,632 genotyped SNPs (0.025%) we expect that successful imputation of the remaining SNPs in the MSKCC cohort would have resulted in an approximately additional 17 SNPs for further testing in the NYUMC cohort, 1 to 2 of which may have been of interest. As high-density genotyping data from next-generation sequencing for the AJ population becomes available, imputation for missing and untyped SNPs, as well as genotyping of rare and/or casual variants, will become more achievable. Finally, because we were already limited by sample size we did not divide the cohorts further by stage or histopathological subtype. Rather this study was designed to identify common predispositional risk factors for AJ women across subtypes of breast cancer.
The major strength of this studylies in the use of three independent, genetically homogeneous populations for identification of new potential risk alleles, and strong, consistent effects were observed for several SNPs across all three independent datasets. Seven new candidate SNPs were identified, 3 of which contributed to a preliminary risk model. While moderate predictive power was accomplished, complete coverage of genetic variants (e.g., next-generation sequencing) combined with clinical and demographic data in a larger cohort will undoubtedly result in a more powerful model for predicting individual risk for this and other homogeneous populations.
Acknowledgments
The authors gratefully acknowledge the generous contribution of the patients who participated in this study. We also wish to thank Gord Glendon, Teresa Selander, Nayana Weerasooriya and members and participants in the Ontario Familial Breast Cancer Registry for their contributions to the study. We would like to thank Peter Pressman from the Weill Cornell Medical College and Ina Ratner of the Maimonides Medical Center for their help with patient recruitment. Finally we would like to thank Dr. Bert Gold from the National Cancer Institute–Frederick for his help with obtaining data for the MSKCC dataset.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics Statement
This study was performed under the NYU School of Medicine Institutional Review Board-approved protocol, “Genetic Modifiers of Breast and Ovarian Cancer Risk” (07-333).
References
- Ahmed S, Thomas G, Ghoussaini M, Healey CS, Humphreys MK, Platte R, Morrison J, Maranian M, Pooley KA, Luben R, Eccles D, Evans DG, Fletcher O, Johnson N, dos Santos Silva I, Peto J, Stratton MR, Rahman N, Jacobs K, Prentice R, Anderson GL, Rajkovic A, Curb JD, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Diver WR, Bojesen S, Nordestgaard BG, Flyger H, Dork T, Schurmann P, Hillemanns P, Karstens JH, Bogdanova NV, Antonenkova NN, Zalutsky IV, Bermisheva M, Fedorova S, Khusnutdinova E, Kang D, Yoo KY, Noh DY, Ahn SH, Devilee P, van Asperen CJ, Tollenaar RA, Seynaeve C, Garcia-Closas M, Lissowska J, Brinton L, Peplonska B, Nevanlinna H, Heikkinen T, Aittomaki K, Blomqvist C, Hopper JL, Southey MC, Smith L, Spurdle AB, Schmidt MK, Broeks A, van Hien RR, Cornelissen S, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Schmutzler RK, Burwinkel B, Bartram CR, Meindl A, Brauch H, Justenhoven C, Hamann U, Chang-Claude J, Hein R, Wang-Gohrke S, Lindblom A, Margolin S, Mannermaa A, Kosma VM, Kataja V, Olson JE, Wang X, Fredericksen Z, Giles GG, Severi G, Baglietto L, English DR, Hankinson SE, Cox DG, Kraft P, Vatten LJ, Hveem K, Kumle M, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat Genet. 2009;41:585–90. doi: 10.1038/ng.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn MJ, Won HH, Lee J, Lee ST, Sun JM, Park YH, Ahn JS, Kwon OJ, Kim H, Shim YM, Kim J, Kim K, Kim YH, Park JY, Kim JW, Park K. The 18p11.22 locus is associated with never smoker non-small cell lung cancer susceptibility in Korean populations. Hum Genet. 2012;131:365–72. doi: 10.1007/s00439-011-1080-z. [DOI] [PubMed] [Google Scholar]
- Cazier JB, Tomlinson I. General lessons from large-scale studies to identify human cancer predisposition genes. J Pathol. 2010;220:255–62. doi: 10.1002/path.2650. [DOI] [PubMed] [Google Scholar]
- Cox A, Dunning AM, Garcia-Closas M, Balasubramanian S, Reed MW, Pooley KA, Scollen S, Baynes C, Ponder BA, Chanock S, Lissowska J, Brinton L, Peplonska B, Southey MC, Hopper JL, McCredie MR, Giles GG, Fletcher O, Johnson N, dos Santos Silva I, Gibson L, Bojesen SE, Nordestgaard BG, Axelsson CK, Torres D, Hamann U, Justenhoven C, Brauch H, Chang-Claude J, Kropp S, Risch A, Wang-Gohrke S, Schurmann P, Bogdanova N, Dork T, Fagerholm R, Aaltonen K, Blomqvist C, Nevanlinna H, Seal S, Renwick A, Stratton MR, Rahman N, Sangrajrang S, Hughes D, Odefrey F, Brennan P, Spurdle AB, Chenevix-Trench G, Beesley J, Mannermaa A, Hartikainen J, Kataja V, Kosma VM, Couch FJ, Olson JE, Goode EL, Broeks A, Schmidt MK, Hogervorst FB, Van’t Veer LJ, Kang D, Yoo KY, Noh DY, Ahn SH, Wedren S, Hall P, Low YL, Liu J, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Sigurdson AJ, Stredrick DL, Alexander BH, Struewing JP, Pharoah PD, Easton DF. A common coding variant in CASP8 is associated with breast cancer risk. Nat Genet. 2007;39:352–8. doi: 10.1038/ng1981. [DOI] [PubMed] [Google Scholar]
- Dyrso T, Li J, Wang K, Lindebjerg J, Kolvraa S, Bolund L, Jakobsen A, Bruun-Petersen G, Li S, Cruger DG. Identification of chromosome aberrations in sporadic microsatellite stable and unstable colorectal cancers using array comparative genomic hybridization. Cancer Genet. 2011;204:84–95. doi: 10.1016/j.cancergencyto.2010.08.019. [DOI] [PubMed] [Google Scholar]
- Easton DF. How many more breast cancer predisposition genes are there? Breast Cancer Res. 1999;1:14–7. doi: 10.1186/bcr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–93. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin MD, Lasseter VK, Wolyniec PS, McGrath JA, Nestadt G, Valle D, Liang KY, Pulver AE. Genomewide linkage scan for bipolar-disorder susceptibility loci among Ashkenazi Jewish families. Am J Hum Genet. 2004;75:204–19. doi: 10.1086/422474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher O, Johnson N, Orr N, Hosking FJ, Gibson LJ, Walker K, Zelenika D, Gut I, Heath S, Palles C, Coupland B, Broderick P, Schoemaker M, Jones M, Williamson J, Chilcott-Burns S, Tomczyk K, Simpson G, Jacobs KB, Chanock SJ, Hunter DJ, Tomlinson IP, Swerdlow A, Ashworth A, Ross G, dos Santos Silva I, Lathrop M, Houlston RS, Peto J. Novel breast cancer susceptibility locus at 9q31.2: results of a genome-wide association study. J Natl Cancer Inst. 2011;103:425–35. doi: 10.1093/jnci/djq563. [DOI] [PubMed] [Google Scholar]
- Gold B, Kirchhoff T, Stefanov S, Lautenberger J, Viale A, Garber J, Friedman E, Narod S, Olshen AB, Gregersen P, Kosarin K, Olsh A, Bergeron J, Ellis NA, Klein RJ, Clark AG, Norton L, Dean M, Boyd J, Offit K. Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci U S A. 2008;105:4340–5. doi: 10.1073/pnas.0800441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S, Henn BM, Gutenkunst RN, Indap AR, Marth GT, Clark AG, Yu F, Gibbs RA, Bustamante CD. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci U S A. 2011;108:11983–8. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthery SL, Salisbury BA, Pungliya MS, Stephens JC, Bamshad M. The structure of common genetic variation in United States populations. Am J Hum Genet. 2007;81:1221–31. doi: 10.1086/522239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiman CA, Stram DO. Exploring genetic susceptibility to cancer in diverse populations. Curr Opin Genet Dev. 2010;20:330–5. doi: 10.1016/j.gde.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hameed M, Ulger C, Yasar D, Limaye N, Kurvathi R, Streck D, Benevenia J, Patterson F, Dermody JJ, Toruner GA. Genome profiling of chondrosarcoma using oligonucleotide array-based comparative genomic hybridization. Cancer Genet Cytogenet. 2009;192:56–9. doi: 10.1016/j.cancergencyto.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Harlid S, Ivarsson MI, Butt S, Grzybowska E, Eyfjord JE, Lenner P, Forsti A, Hemminki K, Manjer J, Dillner J, Carlson J. Combined effect of low-penetrant SNPs on breast cancer risk. Br J Cancer. 2012;106:389–96. doi: 10.1038/bjc.2011.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–60. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–4. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janer M, Friedrichsen DM, Stanford JL, Badzioch MD, Kolb S, Deutsch K, Peters MA, Goode EL, Welti R, DeFrance HB, Iwasaki L, Li S, Hood L, Ostrander EA, Jarvik GP. Genomic scan of 254 hereditary prostate cancer families. Prostate. 2003;57:309–19. doi: 10.1002/pros.10305. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- John EM, Hopper JL, Beck JC, Knight JA, Neuhausen SL, Senie RT, Ziogas A, Andrulis IL, Anton-Culver H, Boyd N, Buys SS, Daly MB, O’Malley FP, Santella RM, Southey MC, Venne VL, Venter DJ, West DW, Whittemore AS, Seminara D. The Breast Cancer Family Registry: an infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res. 2004;6:R375–89. doi: 10.1186/bcr801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny EE, Pe I, Karban A, Ozelius L, Mitchell AA, Ng SM, Erazo M, Ostrer H, Abraham C, Abreu MT, Atzmon G, Barzilai N, Brant S, Burns ER, Chowers Y, Clark LN, Darvasi A, Doheny D, Duerr RH, Eliakim R, Giladi N, Gregersen PK, Hakonarson H, Jones MR, McGovern DPB, Mulle J, Orr-Urtreger A, Proctor DD, Pulver A, Rotter JI, Silverberg MS, Ullman T, Warren ST, Waterman M, Zhang W, Bergman A, Mayer L, Katz S, Desnick RJ, Cho JH, Peter I. A Genome-Wide Scan of Ashkenazi Jewish Crohn’s Disease Suggests Novel Susceptibility Loci. PLoS Genetics. 2012 doi: 10.1371/journal.pgen.1002559. accepted/in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Kim JW, Kim YT, Kim JH, Kim S, Yoon BS, Nam EJ, Kim HY. Analysis of chromosomal changes in serous ovarian carcinoma using high-resolution array comparative genomic hybridization: Potential predictive markers of chemoresistant disease. Genes Chromosomes Cancer. 2007;46:1–9. doi: 10.1002/gcc.20384. [DOI] [PubMed] [Google Scholar]
- Kuukasjarvi T, Karhu R, Tanner M, Kahkonen M, Schaffer A, Nupponen N, Pennanen S, Kallioniemi A, Kallioniemi OP, Isola J. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997;57:1597–604. [PubMed] [Google Scholar]
- Li J, Humphreys K, Heikkinen T, Aittomaki K, Blomqvist C, Pharoah PD, Dunning AM, Ahmed S, Hooning MJ, Martens JW, van den Ouweland AM, Alfredsson L, Palotie A, Peltonen-Palotie L, Irwanto A, Low HQ, Teoh GH, Thalamuthu A, Easton DF, Nevanlinna H, Liu J, Czene K, Hall P. A combined analysis of genome-wide association studies in breast cancer. Breast Cancer Res Treat. 2011;126:717–27. doi: 10.1007/s10549-010-1172-9. [DOI] [PubMed] [Google Scholar]
- Liu X, Cheng R, Verbitsky M, Kisselev S, Browne A, Mejia-Sanatana H, Louis ED, Cote LJ, Andrews H, Waters C, Ford B, Frucht S, Fahn S, Marder K, Clark LN, Lee JH. Genome-wide association study identifies candidate genes for Parkinson’s disease in an Ashkenazi Jewish population. BMC Med Genet. 2011;12:104. doi: 10.1186/1471-2350-12-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Cai Q, Shu XO, Qu S, Li C, Zheng Y, Gu K, Wang W, Xiang YB, Cheng J, Chen K, Zhang L, Zheng H, Shen CY, Huang CS, Hou MF, Shen H, Hu Z, Wang F, Deming SL, Kelley MC, Shrubsole MJ, Khoo US, Chan KY, Chan SY, Haiman CA, Henderson BE, Le Marchand L, Iwasaki M, Kasuga Y, Tsugane S, Matsuo K, Tajima K, Iwata H, Huang B, Shi J, Li G, Wen W, Gao YT, Lu W, Zheng W. Identification of a functional genetic variant at 16q12.1 for breast cancer risk: results from the Asia Breast Cancer Consortium. PLoS Genet. 2010;6:e1001002. doi: 10.1371/journal.pgen.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–7. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- Mitchell MK, Gregersen PK, Johnson S, Parsons R, Vlahov D. The New York Cancer Project: rationale, organization, design, and baseline characteristics. J Urban Health. 2004;81:301–10. doi: 10.1093/jurban/jth116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Need AC, Kasperaviciute D, Cirulli ET, Goldstein DB. A genome-wide genetic signature of Jewish ancestry perfectly separates individuals with and without full Jewish ancestry in a large random sample of European Americans. Genome Biol. 2009;10:R7. doi: 10.1186/gb-2009-10-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini M, Sabbioni S, Possati L, Rattan S, Corallini A, Barbanti-Brodano G, Croce CM. Suppression of tumorigenicity of breast cancer cells by microcell-mediated chromosome transfer: studies on chromosomes 6 and 11. Cancer Res. 1994;54:1331–6. [PubMed] [Google Scholar]
- Nordgard SH, Johansen FE, Alnaes GI, Bucher E, Syvanen AC, Naume B, Borresen-Dale AL, Kristensen VN. Genome-wide analysis identifies 16q deletion associated with survival, molecular subtypes, mRNA expression, and germline haplotypes in breast cancer patients. Genes Chromosomes Cancer. 2008;47:680–96. doi: 10.1002/gcc.20569. [DOI] [PubMed] [Google Scholar]
- Ntzani EE, Liberopoulos G, Manolio TA, Ioannidis JP. Consistency of genome-wide associations across major ancestral groups. Hum Genet. 2011 doi: 10.1007/s00439-011-1124-4. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–7. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin L, Pinchev M, Arad C, Lejbkowicz F, Tamir A, Rennert HS, Rennert G, Gruber SB. FGFR2 is a breast cancer susceptibility gene in Jewish and Arab Israeli populations. Cancer Epidemiol Biomarkers Prev. 2008;17:1060–5. doi: 10.1158/1055-9965.EPI-08-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein WS. Hereditary breast cancer in Jews. Fam Cancer. 2004;3:249–57. doi: 10.1007/s10689-004-9550-2. [DOI] [PubMed] [Google Scholar]
- Shifman S, Levit A, Chen ML, Chen CH, Bronstein M, Weizman A, Yakir B, Navon R, Darvasi A. A complete genetic association scan of the 22q11 deletion region and functional evidence reveal an association between DGCR2 and schizophrenia. Hum Genet. 2006;120:160–70. doi: 10.1007/s00439-006-0195-0. [DOI] [PubMed] [Google Scholar]
- Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–36. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- Stacey SN, Manolescu A, Sulem P, Rafnar T, Gudmundsson J, Gudjonsson SA, Masson G, Jakobsdottir M, Thorlacius S, Helgason A, Aben KK, Strobbe LJ, Albers-Akkers MT, Swinkels DW, Henderson BE, Kolonel LN, Le Marchand L, Millastre E, Andres R, Godino J, Garcia-Prats MD, Polo E, Tres A, Mouy M, Saemundsdottir J, Backman VM, Gudmundsson L, Kristjansson K, Bergthorsson JT, Kostic J, Frigge ML, Geller F, Gudbjartsson D, Sigurdsson H, Jonsdottir T, Hrafnkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Jonsson T, von Holst S, Werelius B, Margolin S, Lindblom A, Mayordomo JI, Haiman CA, Kiemeney LA, Johannsson OT, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007;39:865–9. doi: 10.1038/ng2064. [DOI] [PubMed] [Google Scholar]
- Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, Hankinson SE, Hutchinson A, Wang Z, Yu K, Chatterjee N, Garcia-Closas M, Gonzalez-Bosquet J, Prokunina-Olsson L, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Diver R, Prentice R, Jackson R, Kooperberg C, Chlebowski R, Lissowska J, Peplonska B, Brinton LA, Sigurdson A, Doody M, Bhatti P, Alexander BH, Buring J, Lee IM, Vatten LJ, Hveem K, Kumle M, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Chanock SJ, Hunter DJ. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1) Nat Genet. 2009;41:579–84. doi: 10.1038/ng.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, Seal S, Ghoussaini M, Hines S, Healey CS, Hughes D, Warren-Perry M, Tapper W, Eccles D, Evans DG, Hooning M, Schutte M, van den Ouweland A, Houlston R, Ross G, Langford C, Pharoah PD, Stratton MR, Dunning AM, Rahman N, Easton DF. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42:504–7. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman JA, Fridlyand J, Pejavar S, Olshen AB, Korkola JE, DeVries S, Carroll P, Kuo WL, Pinkel D, Albertson D, Cordon-Cardo C, Jain AN, Waldman FM. Array-based comparative genomic hybridization for genome-wide screening of DNA copy number in bladder tumors. Cancer Res. 2003;63:2872–80. [PubMed] [Google Scholar]
- Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, Higgins J, Roach KC, Mandell J, Lee MK, Ciernikova S, Foretova L, Soucek P, King MC. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA. 2006;295:1379–88. doi: 10.1001/jama.295.12.1379. [DOI] [PubMed] [Google Scholar]
- Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, Nord AS, Mandell JB, Swisher EM, King MC. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci U S A. 2010;107:12629–33. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Dickson SP, Stolle CA, Krantz ID, Goldstein DB, Hakonarson H. Interpretation of association signals and identification of causal variants from genome-wide association studies. Am J Hum Genet. 2010;86:730–42. doi: 10.1016/j.ajhg.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Long J, Gao YT, Li C, Zheng Y, Xiang YB, Wen W, Levy S, Deming SL, Haines JL, Gu K, Fair AM, Cai Q, Lu W, Shu XO. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet. 2009;41:324–8. doi: 10.1038/ng.318. [DOI] [PMC free article] [PubMed] [Google Scholar]