

Summary
Two transcriptional regulators, the FadR activator and the FabR repressor control biosynthesis of unsaturated fatty acids in Escherichia coli. FabR represses expression of the two genes, fabA and fabB, required for unsaturated fatty acid synthesis and has been reported to require the presence of an unsaturated thioester (of either acyl carrier protein or CoA) in order to bind the fabA and fabB promoters in vitro. We report in vivo experiments in which unsaturated fatty acid synthesis was blocked in the absence of exogenous unsaturated fatty acids in a ΔfadR strain and found that the rates of transcription of fabA and fabB were unaffected by the lack of unsaturated thioesters. To examine the discrepancy between our in vivo results and the prior in vitro results we obtained active, natively folded forms of the E. coli and V. cholerae FabRs by use of a vitro transcription-translation system. We report that FabR binds the intact promoter regions of both fabA and fabB in the absence of unsaturated acyl thioesters, but FabR binds the two promoters differently. Native FabR binds the fabA promoter region provided that the canonical FabR binding site is extended by inclusion of flanking sequences that overlap the neighboring FadR binding site. In contrast, although binding to the fabB operator also requires flanking sequences, a nonspecific sequence can suffice. However, unsaturated thioesters do allow FabR to binding to the minimal FabR operator sites of both promoters which otherwise are not bound. Thus unsaturated thioester ligands are not essential for FabR/target DNA interaction, but act to enhance binding. The gel mobility shift data plus in vivo expression data indicate that despite their remarkably similar arrangements of promoter elements, FadR predominately regulates fabA expression whereas FabR is the dominant regulator of fabB expression. We also report that E. coli fabR expression is not autoregulated. Complementation, RT-qPCR and fatty acid composition analyses demonstrated that V. cholerae FabR is a functional repressor of unsaturated fatty acid synthesis. However, in contrast to E. coli, gel mobility shift assays indicated that neither E. coli nor V. cholerae FabR bind the V. cholerae fabB promoter, although both efficiently bind the V. cholerae fabA promoter. This asymmetry was shown to be due the lack of a FabR-binding site within the V. cholerae fabB promoter region.
Introduction
Fatty acid moieties make up the essential hydrophobic component of bacterial membrane phospholipids and can also be used as carbon sources (Fujita et al., 2007; Zhang and Rock, 2009). Unsaturated fatty acids (UFAs), which play key roles in the physical properties of bacterial membrane phospholipids, are synthesized by either of two mechanisms. In the anaerobic pathway the double bond is introduced at the C10 level by isomerization of a standard biosynthetic intermediate, the trans-2-decenoyl thioester of acyl carrier protein (ACP) whereas the aerobic pathway proceeds by oxygen-requiring desaturation of fully elongated acyl chains. Some bacteria (e.g., Pseudomonas aeruginosa) have both pathways (Zhu et al., 2006).
E. coli, the paradigm of bacterial fatty acid metabolism, has only the anaerobic pathway which requires the products of two genes, fabA and fabB (Cronan et al., 1972; de Mendoza et al., 1983). FabA introduces the double bond into the growing acyl chain by catalyzing dehydration of β-hydroxydecanoyl-ACP and the isomerization of the trans-2 dehydration product to cis-3-decanoyl-ACP (Magnuson et al., 1993). FabB, a 3-ketoacyl-ACP synthase, is required for the elongation of cis-3-decanoyl-ACP to cis-5-dodecanoyl-ACP (Feng and Cronan, 2009a; Magnuson et al., 1993) which enters the standard fatty acid synthesis cycle and becomes elongated to the sixteen and eighteen carbon UFAs of the membrane phospholipids. Coordinated expression of fabA and fabB is therefore required for a proper ratio of UFA to saturated fatty acid (SFA) which determines the stability and fluidity of the membrane bilayer (Clark et al., 1983; DiRusso et al., 1993; Feng and Cronan, 2009a; Magnuson et al., 1993; Wang and Cronan, 2004). Two regulatory proteins, FadR and FabR, are known to regulate transcription of fabA and fabB (Fig. 1). FadR is the more thoroughly studied and functions as a global regulator of both UFA synthesis and the β-oxidative utilization of fatty acids as carbon sources (Cronan and Subrahmanyam, 1998; Henry and Cronan, 1991, 1992). FadR, which belongs to the GntR family of transcription factors, acts as an activator of fabA and fabB where it acts as a repressor of the β-oxidation (fad) regulon. Upon binding long chain fatty acyl-CoA thioesters, FadR dissociates from its binding DNA sites resulting in decreased transcription of fabA and fabB and increased transcription of the fad regulon genes (Cronan, 1997; Henry and Cronan, 1992). FabR binding to the fabA and fabB promoter regions was discovered by McCue and coworkers (McCue et al., 2001). These workers first identified sequences upstream of E. coli genes that were conserved in other γ-proteobacteria and then sought a protein that would bind the closely related sequences found upstream of E. coli fabA, fabB and yqfA, a gene encoding a membrane protein of the hemolysin 3 family. These workers isolated proteins from crude extracts of E. coli using affinity chromatography on columns of immobilized double stranded oligonucleotides containing the sequences of interest. Proteomic analyses of the isolated proteins resulted in identification of FabR. The in vivo function of FabR was subsequently demonstrated by construction of a fabR deletion strain in which transcription of both fabA and fabB were increased relative to the wild parental strain thereby indicating that FabR acted as a repressor (Zhang et al., 2002). Recently Zhu et al. (Zhu et al., 2009) reported that presence of a CoA or ACP thioester of a long chain UFA was required for binding of FabR to the fabA and fabB promoter regions. This report seems to disagree with the original report because McCue and coworkers (McCue et al., 2001) performed their chromatographic separations on E. coli crude extracts that had been concentrated by ammonium sulfate precipitation, a treatment that should effectively remove both CoA and ACP thioesters. Moreover, the FabR preparations of Zhu and coworkers (Zhu et al., 2009) consisted of a heterologous mixture of protein species that had been refolded following denaturing extraction from inclusion bodies. This raised the possibility that the UFA CoA or ACP thioesters might have aided folding of incompletely folded proteins and thereby indirectly facilitated DNA binding. Finally, these workers (Zhu et al., 2009) tagged the FabR N-terminus which raised a possible caveat because the predicted DNA binding domain is located adjacent to the N-terminus. For these reasons we have used a different approach to study DNA binding by FabR. First, we used a specific UFA synthesis inhibitor to block synthesis of UFA in vivo and found that this treatment had no effect on transcription of fabA and fabB. Second, we synthesized FabR in vitro using a coupled transcription-translation system derived from components individually purified from E. coli (Shimizu et al., 2001) to avoid the insoluble protein aggregates that invariably resulted upon overproduction of FabR. This approach not only avoided the solubility problems, but also allowed the use of an untagged protein and precluded the possibility of binding of endogenous ligands by FabR. We report that FabR binds to both the fabA and fabB promoter regions in the absence of unsaturated fatty acid thioesters provided that the DNA sequences required for FabR binding are longer than the canonical binding sites. However, FabR binding to the two E. coli promoters differed and this, plus in vivo expression data, indicated that although FabR plays a major role in regulation of fabB expression, it plays only a minor role in fabA expression. We have also compared the in vitro results obtained with E. coli FabR with those of Vibrio cholerae FabR and its cognate promoters. Finally, FabR was found not to regulate yqfA expression and to not be subject to autoregulation.
Fig. 1.

Results
The dependence of FabR regulation on UFA synthesis in vivo
To test the effects of blocking UFA synthesis in vivo on transcription of fabA and fabB we synthesized the specific FabA inhibitor, 3-decynoyl-N-acetylcysteamine (DNAC) (Bloch, 1971, 1986). FabA catalyzes two reactions within the same active site and the second of these reactions is responsible for the irreversible inactivation of the enzyme by DNAC. The enzyme first dehydrates 3-hydroxydecanoyl-ACP to trans-2-decenoyl-ACP and then isomerizes the trans-2 double bond to form cis-3-decenoyl-ACP. The cis-3 intermediate is then passed through several elongation cycles to form the cis UFAs found in the cellular phospholipids. DNAC is a substrate analogue of cis-3-decenoyl-ACP in which the double bond is replaced with a triple bond and ACP is replaced with N-acetylcysteamine, the distal third of the ACP 4′-phosphopanthetheine prosthetic group. Upon binding DNAC FabA isomerizes the triple bond to form 2,3-decadienoyl-NAC, an extremely reactive allene, which reacts with the active site histidine residue to form a covalent adduct. Since this was the first example of a mechanism activated (“suicide”) inhibitor, the reaction has been characterized in great detail (Annand et al., 1993; Leesong et al., 1996; Schwab and Klassen, 1984). Kass (Kass, 1968) first showed that DNAC rapidly blocked UFA synthesis in vivo and that growth inhibition by DNAC is reversed by UFA supplementation.
In our experiments we used ΔfadR strains lacking the FadR activator protein in order to limit regulation to FabR. We added DNAC (100 μM final concentration) to growing cultures of fadR null mutant strains and took samples of the cultures to assay the rate of UFA synthesis and transcription of fabA and fabB (Fig. 2). The rate of UFA synthesis was determined by [1-14C]acetate labeling followed by separation of the UFA and SFA species by argentation thin layer chromatography of their methyl esters whereas transcription was assayed by qRT-PCR. As expected from prior reports (Kass, 1968; Nunn and Cronan, 1974) DNAC inhibition of UFA synthesis was very rapid and became essentially quantitative 10 min after addition of the inhibitor. The inhibition was specific in that SFA synthesis continued (Fig. 2A & B) and the effects of 100 μM DNAC on growth were reversed by addition of oleic acid (data not shown).
Fig. 2. Effects of DNAC Treatment on UFA Synthesis and Transcription of fabA and fabB.

A & B. Argentation thin-layer chromatographic analyses of [I-14C]-acetate labeled fatty acids from the ΔfadR strain (FYJ57). A log-phase culture was treated with 100 μM DNAC and 10 ml samples were removed at the times indicated labeled for 10 min with [I-14C]-sodium acetate. The methyl esters of fatty acids were obtained as we previously described (Feng and Cronan, 2009a; Wang and Cronan, 2003). The thin-layer chromatographic assays were performed in triplicate and visualized using a phosphorimager (FLA-3000, Fujifilm). A representative autoradiogram is given in panel A. SFA, saturated fatty acids; Δ11 C18:1, cis-vaccenic acid; Δ9 C16:1, palmitoleic acid. C. qPCR analyses of fabA and fabB expression levels, following addition of DNAC. Log-phase cultures grown in LB liquid medium were treated with 100 μM DNAC for the intervals given in minutes at which time samples were taken for RNA preparation. The expression levels were measured in triplicate. “ – ” denotes an untreated sample of the culture
In the model of Zhu and coworkers (Zhu et al., 2009) FabR bound to an unsaturated acyl-ACP was the form of FabR responsible for repression of fabB expression in the absence of exogenous UFAs. If so, depletion of the pool of unsaturated acyl-ACP species should result in dissociation of FabR from the fabB operator resulting in increased fabB expression. However, inhibition of UFA synthesis had the opposite result, fabB transcription showed a modest decrease rather than the increase expected (Fig. 2C). Moreover, the effects on transcription of fabA and fabB were very similar although the effects of FabR on expression of fabA and fabB were reported to differ (Zhang et al., 2002; Zhu et al., 2009) (Fig. 2D).
To examine the reported differential expression of fabA and fabB and to test the in vivo effects of exogenous unsaturated fatty acids on FabR-mediated regulation we constructed lacZ transcriptional fusions to the E. coli fabA and fabB genes. The fusions were constructed by inserting the lacZ cassette immediately downstream of the termination codon of the target gene such that the fabA and fabB genes retained function (both are transcribed as single genes which precluded polar effects). Assay of the β-galactosidase levels of these strains showed that deletion of both fabR and fadR eliminated all regulation of fabA and fabB expression observed in the wild type strain in the presence of the exogenous UFA, oleate (compare Figs. 3A and B) as previously reported (Zhu et al., 2009). Moreover, in the ΔfadR strain (which lacked FadR, but retained FabR) addition of oleate had no detectable effect on fabA expression whereas oleate addition decreased fabB expression about 2-fold (Fig. 3D). The lacZ transcriptional fusion data indicate that the oleate-responsive regulator for fabA expression is FadR whereas FabR plays the dominant role in oleate regulation of fabB expression as reported by Zhu and coworkers (Zhu et al., 2009). Taken together the DNAC plus qRT-PCR and lacZ transcriptional fusion data indicated that only fabB expression was subject to significant FabR regulation and only exogenous UFAs (imported as acyl-CoAs) affected the severity of the regulation.
Fig. 3. Effect of oleate on fabA and fabB expression.

A–D. Effects of oleate supplementation on E. coli fabA and fabB expression levels in the wild type, ΔfabR, ΔfadR and ΔfadR ΔfabR strains, respectively. The fabA-lacZ transcription fusion strains used to measure fabA expression were JT180 (wild type denoted WT), FYJ30 (ΔfadR), FYJ31 (ΔfabR), and FYJ90 (ΔfadR ΔfabR), respectively. The fabB-lacZ transcription fusion strains used to measure fabB expression were JT181 (wild type), FYJ32 (ΔfadR), FYJ33 (ΔfabR), and FYJ91 (ΔfadR ΔfabR). 5 mM oleate was added to RB medium. The cultures were grown to mid-log phase and assayed for β-galactosidase activity. The data shown are from three independent experiments.
The FabR proteins of E. coli and V. cholerae
Our in vivo data plus the prior in vitro data outlined in the Introduction argued that FabR binds the fabA and fabB promoters in the absence of an unsaturated thioester ligand. To test this possibility we required a source of natively folded FadR. However, despite many attempts to obtain soluble E. coli FabR by production of hexahistidine-tagged versions (both N-terminal and C-terminal) in E. coli by use of a large variety of expression conditions only insoluble inclusion bodies resulted. Although in some cases a small fraction of the protein appeared soluble by western blotting with a tag-specific antibody and showed specific DNA binding activity (data not shown), we were unable to purify the protein. Similarly disappointing results were obtained with glutathione S-transferase fusion proteins (data not shown). In hopes of finding a more tractable FadR we found that several diverse γ-proteobacteria encode a protein having strong sequence similarity to E. coli FabR (Fig. 4). Indeed the putative DNA binding domains of these proteins showed remarkably strong conservation suggesting that they may bind to very similar operator sequences. We chose one of these, V. cholerae FabR, to test if it might provide a soluble protein that could serve as a surrogate for the E. coli protein. Unfortunately, upon expression in E. coli V. cholerae FabR showed the same frustrating properties as the E. coli protein. However, since V. cholerae FabR is interesting in its own right (see below), we have studied this protein further and although in some figures (for space considerations) V. cholerae FabR data are sometimes given together with data on the E. coli protein, these results will be presented in a separate section.
Fig. 4. Multiple sequence alignments of FabR homologues.

The conserved putative FadR DNA binding motif, TetR_N, is underlined. White letters with black background denotes identical residues, black letters with grey background denotes similar residues, and grey letters on a white background denote different residues. Vibrio cholerae is indicated in bold. The bacterial genome sequences are E, coli K-12 MG1655 (NC_000913); Salmonella enterica serovar Typhimurium LT2 (NC_003197); Shigella flexneri 2a str. 301 (NC_004337); Yersinia pestis Angola (NC_010159); Vibrio cholerae O395 (NC_009457); Pasteurella multocida subsp. multocida str. Pm70 (NC_002663); Haemophilus influenzae Rd KW20 (NC_000907); Pseudomonas aeruginosa PAO1 (NC_002516) and Xanthomonas campestris pv. campestris str. 8004 (NC_007086).
Given our inability to obtain a soluble FabR and our wish to avoid refolding a denatured protein we turned to the reconstituted in vitro cell-free transcription/translation system of Shimizu and coworkers (Shimizu et al., 2001). Use of this system in which all components are well defined avoided the solubility problems and the possibility of the presence of regulatory ligands (e.g. acyl-thioesters). Note that our attempts to purify the FabR proteins produced in vitro were unsuccessful due to loss of the protein presumably due to aggregation on the chromatographic columns. Thus, although FabR was the dominant protein observed by gel electrophoresis (Fig. 5), the preparations also contained the proteins of the expression system. However, no binding to the fabA and fabB DNA probes was observed unless the system had been programmed with a FabR-encoding plasmid (data not shown). Production and identification of the E. coli and V. cholerae FabR proteins was demonstrated by liquid chromatography-mass spectral analysis of the tryptic peptides derived from the proteins (Fig. 5) and by [35S]-methionine labeling (Fig. 6) whereas gel mobility shift assays tested function of the proteins (Fig. 7 and below). E. coli FabR was synthesized in both native and N-terminal hexahistidine-tagged forms to test if the tag interfered with DNA binding. The DNA binding properties of the two forms were indistinguishable (Fig. 7).
Fig. 5. Expression and identification of in vitro synthesized FabR proteins by analysis of tryptic peptides and antibody reactivity.

A. and B. Expression of E. coli FabR or V. cholerae FabR. Arrows denote the candidate protein bands.
C. Western blotting of the expressed FabR proteins. The first antibody was an anti-hexahistidine mouse monoclonal antibody.
D. and E. Liquid chromatography quadrupole time-of-flight electmass spectrometry identification of the peptides of FabR proteins of E. coli and V. cholerae, respectively. The peptides that matched the database sequences are given in bold type and underlined.
Fig. 6. Size exclusion chromatography of in vitro synthesized FabR proteins.

A. Size exclusion chromatographic profile and of in vitro synthesized E. coli FabR. The inset is an autoradiogram of a SDS-PAGE analysis of the labeled protein.
B. Size exclusion chromatographic profile and of in vitro synthesized V. cholerae FabR. A Superdex 75 column (Pharmacia) was the matrix. The elution peaks of the molecular weight standards are given at the top of the panels. The inset is an autoradiogram of a SDS-PAGE analysis of the labeled protein.
Fig. 7. The presence of an N-terminal His-tag on FabR has no effect on DNA binding.
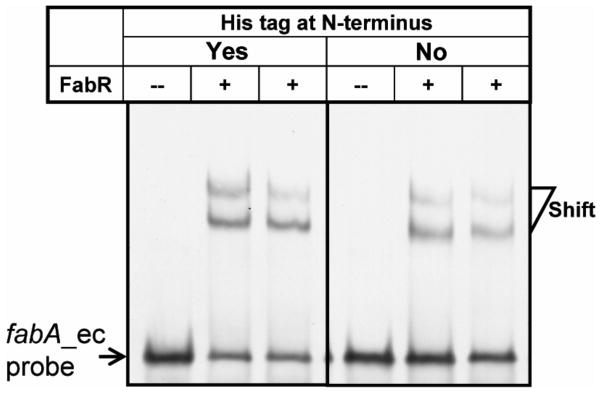
The gel mobility shift assays were conducted using 6% native PAGE. An N-terminal hexahistidine tagged FabR and the native protein obtained by in vitro synthesis were incubated with about 0.1 pmol of a 56 bp fabA_ec probe containing the FabR binding site. The shifted DIG-labeled DNA bands are indicated. A plus sign denotes addition of FabR (~0.6 pmol) whereas the minus sign denotes a lane lacking FabR.
To our surprise and contrary to the report of Zhu and coworkers (Zhu et al., 2009) DNA binding was readily observed in the absence of added unsaturated acyl-thioesters (Fig. 7). To determine the solution forms of the E. coli and V. cholerae FabR proteins the [35S]methionine labeled proteins were synthesized in vitro and subjected to size exclusion chromatography. The two FabR proteins behaved similarly in that both gave three peaks indicating monomeric, dimeric and polymeric forms (Fig. 6). Since the FabR binding site is a degenerate palindrome and bacterial transcription factors generally bind to the target DNA sites as symmetric structures (Collado-Vides et al., 1991; McCue et al., 2001) (e.g., dimer or tetramer), it seemed likely that the functional forms of both FabRs were the dimeric species.
Binding of FabR to its cognate promoters does not require unsaturated acyl-CoA or acyl-ACP thioesters
As mentioned above Zhu et al. (Zhu et al., 2009) reported that either an unsaturated acyl-ACP or acyl-CoA species are required for FabR-binding to its operator sites. As detailed in the introduction, this conclusion seemed to conflict with the original report of McCue and coworkers (McCue et al., 2001) and directly conflicted with our observations (Fig. 7). Based on the work of Zhu and coworkers (Zhu et al., 2009) we titrated binding of the fabB probe with FabR over a range of concentrations. This was done in the absence of acyl-CoA and in the presence of either oleoyl-CoA or palmitoyl-CoA. As seen above FabR shifted the 57 bp fabB probe in the absence of acyl-CoA. Moreover, the presence of oleoyl-CoA did not shift the binding curve to lower FabR concentrations, although the super-shift seen in the absence of acyl-CoA was no longer present. In contrast, the presence of palmitoyl-CoA shifted the binding curve to higher FabR concentrations (Fig. 8). Since Zhu and coworkers (Zhu et al., 2009) had failed to report the sequence or identity of their probe1, we designed probes from the fabA and fabB promoter sequences (Fig. 9A–C) to explore possible effects of differing lengths of the binding partner on FabR binding. Four different probes were made where B denotes the E. coli FabR binding sequence and D denotes the FadR binding sequence. These were: the B+D probes (56 bp for E. coli fabA, 57 bp for E. coli fabB and 54 bp for V. cholerae fabA), the D1 probes which contained only the FadR-binding palindrome and probes B1 and B2 which contained only the canonical FabR binding site with the B2 probe [39 bp, which resembled the original probe used (McCue et al., 2001)], being slightly longer than the B1 probe of 30 bp (Fig. 9A–C). In the absence of a long chain acyl-CoA or acyl-ACP, FabR binding shifted the B+D probes (which acted as positive controls) whereas the D1 probes showed no shifted bands (negative controls) (Fig. 9A–C). Although the shorter B1 version of the FabR-specific probes contained the full-length FabR-binding site, FabR protein failed to bind this probe whereas the slightly longer B2 probes were bound indicating that the additional DNA sequences adjacent to the FabR-binding site allowed binding (Fig. 9C). Surprisingly, upon the addition of either palmitoleoyl-CoA (C16:1-CoA) or oleoyl-CoA (18:1-CoA), the shorter E. coli fabA and fabB B1 probes were bound by FabR (Fig. 9C). Therefore, the presence of an unsaturated acyl-CoA ligand had the same effect as an increase in the length of the DNA probe. This raised the question of whether the extra bp of the B2 probe provided specific or nonspecific augmentation of FabR binding relative to the B1 probe. Since the additional bp of the B2 probes overlapped the adjacent FadR-binding sites, it seemed possible that binding might indicate interplay in binding of FabR and FadR. We therefore made probes (called B2A) for the E. coli fabA and fabB promoters in which the probes were of the same length as B2, but the FadR binding site sequence was replaced with a random sequence. FabR bound both B2A probes in the absence of an unsaturated ligand. However, the E. coli fabB B2A and B2 probes were bound to similar extents whereas the binding to the E. coli fabA B2A probe was significantly weaker than to the B2 probe. These data were the first indication that binding of FabR to the fabA promoter was less robust than its interaction with the fabB promoter.
Fig. 8. Determination of minimal effective concentration of E. coli FabR protein for in vitro gel shift assays.

A & B. Binding of the fabBec probe by various concentrations of E. coli FabR in the absence of acyl-CoAs.
C & D. Binding of the fabBec probe by various concentration E. coli FabR in the presence of C18:1-CoA.
E & F. Binding of the fabBec probe by various concentrations of E. coli FabR in the presence of C16:0-CoA. The minus sign denotes no addition of protein. The FabR concentrations in the right hand eight lanes of each panel were (left to right) 0.00006, 0.0006, 0.0012, 0.006, 0.06, 0.12, 0.3 and 0.6 pmol. The protein samples were incubated with 0.1 pmol of DIG-labeled probe in a total volume of 20 μl. Representative gels are shown The data were derived from at least three independent gel shift assays.
Fig. 9. Effects of probe length on FabR binding to its target DNA sequences.

A–C. The probe length dependence of FabR binding to the fabAec, fabBec, and fabAvc promoter regions were tested. The sequences of the FadR/FabR binding sites are given. The FadR-binding site and FabR-binding site are abbreviated “D” and “B”, respectively. Probes B1 and B2 are two versions of FabR binding sites having different lengths of the upstream and adjacent FadR binding site. The four E. coli fabA probes B+D, B2, B1 and D) were, respectively, 56, 39, 30 and 30 bp in length (Table 2) and the four E. coli fabB probes (B+D, B2, B1 and D, in Panel B) were 57, 39, 30 and 30 bp in length, respectively (Table 2). The, V. cholerae fabA probes (B+D, B2, B1, and D were 54, 37, 30 and 30 bp in length, respectively (Table 2). The B2A probes were modified versions of the B2 probes in which the 9 bp that overlapped the FadR binding site were replaced with the random nucleotide sequence, 5′-TACAGAGAA-3′. The gel shift assays were carried out using 7% native PAGE. The binding reactions (20 μl total) contained about 0.1 pmol of probe and about 0.6 pmol of FabR.
The effects of long chain acyl thioesters on FabR binding to the E. coli fabA and fabB B+D probes were also tested. Neither holo-ACP nor decanoyl-ACP had any apparent effects on binding of FabR_ec to the promoter regions of fabAec or fabBec (Fig. 10A & B). In agreement with the observations of Zhu and coworkers (Zhu et al., 2009), addition of oleoyl-ACP (Fig. 10A & B) or palmitoleoyl-CoA (or oleoyl-CoA, Fig. 10C & D) gave increased binding of FabR_ec to its targets although in our hands binding was also seen in the absence of these ligands (the super-shifted bands sometimes seen in our assays may be attributable to the minor polymeric forms of FabR observed by the size exclusion chromatography (Fig. 7). We also observed that the inhibition of FabR-DNA binding by the saturated acyl-CoA species, palmitoyl- and stearoyl-CoA, was offset by addition of an equimolar concentration of the monounsaturated acyl-CoA of the same chain length (Fig. 10E & F).
Fig. 10. Effects of acyl-thioesters on FabR-operator interactions.

A. E. coli fabA and fabB promoter regions.
B. Holo-, Holo-ACP; Dec-, decanoyl-ACP (C10:0-ACP); Pal-, palmitoyl-ACP (C16:0-ACP); Ole-. oleoyl-ACP (C18:1-ACP).
C & D. Effect of long chain unsaturated -acyl CoAs on the binding of FabR to fabA and fabB probes, respectively.
E & F. Long chain unsaturated acyl-CoA species allow binding of FabR protein to the minimal FabR-binding site of fabA and fabB, respectively. Designations C16:0, palmitoyl-CoA; C16:1, palmitoleoyl-CoA; C18:0, stearoyl-CoA; C18:1, oleoyl-CoA. In the f binding reaction mixtures (20 μl total), the components were added as follows: probe, ~0.1 pmol; FabR, ~0.6 pmol; acyl-ACP (or holo-ACP), ~50 pmol and acyl-CoA, ~50 pmol.
G & H. Competitive effects of unsaturated and saturated acyl-CoA thioesters on FabR binding to the fabB promoter. G shows 16 carbon acyl chains whereas H shows 18 carbon acyl chains. The minus and plus signs denote no addition of protein, and addition of protein (0.3 pmol). In these experiments, 0.1 pmol of DIG-labeled fabBec probe was added. For the concentration of acyl-CoA, “0” denotes no addition of acyl-CoA , and a value of 1 corresponds to 50 pmol of acyl-CoA. Representative gels are shown.
FabR plays no detectable role in yqfA expression
Based on the results of phylogenetic footprinting, McCue et al. (McCue et al., 2001) suggested that E. coli yqfA, encoding a membrane protein of unknown function was a third FabR regulated gene. A putative FabR-binding palindrome upstream of the yqfA coding sequence was highly similar to those of fabA and fabB (McCue et al., 2001) (Fig. 11A). To examine FabR binding to this region two probes covering the putative binding site (43 bp and 32 bp) (Fig. 11B) were used in gel shift assays. As seen above for fabA (Fig. 11C), the longer yqfA probe bound FabR protein (Fig. 11C), although the binding was weak compared to the fabA sequence assayed in parallel whereas the shorter probe was shifted by FabR only upon addition of palmitoleoyl-CoA or oleoyl-CoA (Fig. 11D). However, this binding was without physiological consequence in that yqfA-lacZ fusion activity in vivo was identical in wild type and ΔfabR strains under both aerobic and anaerobic conditions (Fig. 11E) and in the presence or absence of oleate (Fig. 11F).
Fig. 11. FabR plays no regulatory role in expression of E. coli yqfA.

A. Sequence alignment of the putative FabR-binding site of E. coli yqfA with those of fabA and fabB.
B. Design of DNA probes carrying the predicted FabR-binding site in the promoter region of yqfA gene.
C. Gel shift analyses of binding activity of FabR to the fabAec probe and the yqfA probe 1.
D. Long chain unsaturated acyl-CoA species allowed the binding of FabR to the shorter yqfA probe 2. In the binding system (20 μl total), the probe and FabR protein were added to ~0.1 pmol and ~0.6 pmol, respectively.
E. Expression of yqfA monitored by use of a chromosomal yqfA-lacZYA fusion. β-Galactosidase activity was measured in cells grown under either aerobic or anaerobic conditions. The strains carrying the yqfA-lacZ transcription fusion were FYJ110 (WT) and FYJ111 (ΔfabR::Cm), respectively. Anaerobic conditions were obtained as previously described (Feng and Cronan, 2010).
F. Effects of oleate supplementation on yqfA expression. Cultures were grown in RB medium with or without 5 mM oleate and cultures in mid-log phase were collected for β-galactosidase assays. The strains carrying the yqfA-lacZ transcription fusion were FYJ110 (WT) and FYJ111 (ΔfabR::Cm), respectively.
FabR does not regulate V. cholerae fabB expression
Our interest in V. cholerae, an enteric bacterium related to E. coli, arose from the lack of a recognizable FabR binding site within its fabB promoter region. (Note that the original report of Heidelberg et al., 2000) that the V. cholerae contains a fabB frame-shift mutation is incorrect as demonstrated by re-sequencing, by function of the encoded protein in E. coli (Heidelberg et al., 2000; Wang and Cronan, 2003) plus the additional four V. cholerae genome sequences that were subsequently submitted to Genbank). To test if the V. cholerae FabR homologue was a functional repressor we tested its ability to functionally replace E. coli fabR in a ΔfabR strain and found that expression of the V. cholerae protein restored FabR regulation (Fig. 12A & B). In addition, the ratios of both UFA to SFA and C18:1 to C16:1 in the complemented strain were similar to those of a wild type strain (Fig. 6D). The data fulfill the criteria for functional definition of as a functional fatty acid synthesis repressor (Zhang et al., 2002). We also tested the ability of V. cholerae FabR to bind both its cognate fabA promoter region and the promoter regions of E. coli fabA and fabB (Fig. 12C & D). Gel shift assays provided direct evidence that in vitro synthesized V. cholerae FabR bound the promoter regions of E. coli fabA and fabB as avidly as did E. coli FabR. As seen for the E. coli FabR DNA binding was obtained in the absence of any regulatory ligand. Moreover, V. cholerae FabR showed the same pattern of binding as the E. coli protein to probes B1, B2, D1 and B+D derived from its fabA promoter sequence indicating that the pattern of binding to these probes did not depend on either the source of the binding protein or the operator sequence (Fig 9D).
Fig. 12. V. cholerae fabR is a functional repressor.

A. Transcription of the E. coli fabA and fabB genes under control of V. cholerae fabR as assayed by qPCR analyses. The data are from two independent experiments and the expression of each gene was quantified in triplicate.
B. Effects of V. cholerae fabR on production of unsaturated fatty acids (UFA) in E. coli. Fatty acid composition was determined by electrospray mass spectrometry. A representative result is shown.
C. & D. V. cholerae FabR protein binds to degenerate palindromic sequences within the promoter regions of E. coli fabA and fabB, respectively. The minus sign denotes no addition of FabR protein. FabR_ec and FabR_vc denote the FabRs (~ 0.6 pmol each) of E. coli and V. cholerae, respectively. A 7% native PAGE gel was run.
To test if FabR_vc bound the fabB promoter region by a sequence we had failed to recognize, a set of gel shift assays were performed in which all four promoter regions (i.e., the fabA and fabB regions of both bacteria) were tested with both FabR proteins (Fig. 13). Neither FabR_ec nor FabR_vc bound the V. cholerae fabB promoter region whereas both proteins bound the promoter regions of the E. coli fabB and fabA genes. Therefore, the possibility of a cryptic FabR binding site within the V. cholerae fabB promoter region could be eliminated.
Fig. 13. The V. cholerae fabB promoter does not bind either FabR species. The left panels are fabA probes whereas the right panels are fabB probes.

A. and B. E. coli FabR binds the promoter regions of its cognate fabA and fabB genes.
C. and D. V. cholerae FabR binds the V. cholerae fabA promoter probe but not the V. cholerae fabB probe. The decreased band intensity in gel C can be attributed to precipitation is the wells (visible at the top of the gel).
E. and F. E. coli FabR binds the V. cholerae fabA probe but not the V. cholerae fabB probe.
G. and H. V. cholerae FabR binds both E. coli fabA promoters.
Designations ec and vc denote E. coli and V. cholerae, respectively. The minus sign denotes no addition of protein; the DIG-labeled probe shifted by FabR protein is indicated with an arrow. The FabR concentrations in the right hand three lanes of each panel were (left to right) 0.1, 0.3 and 0.6 pmol. The protein samples were incubated with 0.1 pmol of DIG-labeled probe in a total volume of 20 μl.
FabR autoregulation and the fabR promoters of E. coli and V. cholerae
Although earlier genetic studies had established that E. coli fabR regulated unsaturated fatty acid synthesis (Zhang et al., 2002), the question of whether or not the fabR expression is regulated had not been addressed. Regulation of repressor levels often involves autoregulation which would require a FabR binding site to be present within the fabR promoter region (Roy et al., 2002). Although we could not find a plausible match for the consensus FabR binding site upstream of the coding sequences of the E. coli and V. cholerae fabR genes, binding sites involved in autoregulation often deviate from the consensus binding site (Collado-Vides et al., 1991; Gunsalus and Yanofsky, 1980) because weak regulation is required to allow sufficient levels of the regulatory protein to accumulate. Given this property and the degenerate nature of the FabR-binding palindrome (Fig. 1) straightforward recognition of a FabR binding site seemed unlikely. To narrow our searches for possible FabR binding sites we mapped the fabR promoters of E. coli and V. cholerae. The fabR transcriptional start sites were determined using an improved 5′-RACE method (RLM-RACE) that requires the RNA to have a 5′-triphosphate. A mixture of fabR transcripts was seen in E. coli, the longest of which was taken to be the full-length version (the two shorter products proved to be truncated fabR transcripts) (Fig. 14A) whereas only a single product was obtained for V. cholerae (Fig. 14C). Direct DNA sequencing of several 5′-RACE product clones clearly showed that in E. coli, the first nucleotide adjacent to the RLM-RACE adaptor was located 189 nucleotides upstream of the translation initiation codon (Fig. 13B) whereas in V. cholerae the first base was only 32 nucleotides upstream of its coding region (Fig. 13D). The location of the 5′-ends of the fabR transcripts of E. coli and V. cholerae allowed the fabR promoters to be defined (Fig. 13E & F). The Neutral Network Program of Promoter Prediction (http://www.fruitfly.org/seqtools/promoter.html) prediction completely matched that obtained by 5′-RACE for V. cholerae fabR (Fig. 14F), although the prediction for E. coli fabR was 12 bases downstream of the determined initiation site (Fig. 14E). Note that a C to T transition relative to the published genome sequence (27 bases upstream of the coding sequence) was found in the products of V. cholerae fabR (Fig. 14D & F). Multiple alignments disclosed that an almost identical fabR promoter region with an exception of a small gap was present in Shigella flexneri, a species very closely related to E. coli (Fig. 14E). However, even given the smaller search region enabled by the promoter mapping no plausible FabR binding site could be recognized in either promoter region suggesting that fabR expression was not autoregulated. This was confirmed by construction of strain FYJ143, a fabR::lacZ transcriptional fusion strain in which fabR was deleted (Ellermeier et al., 2002). Plasmids that overproduced the FabR proteins of E. coli, S. enterica and P. multocida were introduced and the β-galactosidase levels of these strains were compared to the strain carrying the empty vector. There was no significant effect of FabR overexpression on β-galactosidase levels (the levels varied <15%, data not shown) indicating that like fadR expression (Henry and Cronan, 1992), fabR expression showed no autoregulation of its expression.
Fig. 14. Mapping the transcriptional start sites of the fabR genes of E. coli and V. cholerae.

A. and C. Electrophoretic analyses (2.0 % agarose gel) of the fabR 5′-RACE products of E. coli and V. cholerae, respectively.
B. and D. Direct DNA sequencing of the RLM-RACE products of the fabR genes of E. coli and V. cholerae, respectively.
E. and F. Sequences of the promoter regions of fabR genes from E. coli and S. flexneri and V. cholerae, respectively. The letters “S” and “M” denote the transcriptional start site and translational initiation site, respectively. RBS denotes the predicted ribosome binding sites.
Discussion
Although a scenario in which FabR binding strictly requires unsaturated thioesters to bind its cognate promoters (Zhu et al., 2009) is attractive, our in vitro data and those of McCue et al., (McCue et al., 2001) argue that this is not the case. In the model of Zhu and coworkers (Zhu et al., 2009) accumulation of an excess of unsaturated acyl-ACPs over that required for phospholipid synthesis would inhibit transcription of fabA and fabB and thereby allow the UFA:SFA ratio of the acyl-ACP pool to return to normal. Inhibition would be relieved by dissociation of the acyl-ACP ligand from FabR and its consumption by phospholipid synthesis. Therefore, based on the model of Zhu and coworkers (Zhu et al., 2009) blocking UFA synthesis should derepress transcription of both fabA and fabB due to the lack of a pool of unsaturated acyl-ACPs plus the antagonism of unsaturated acyl-ACP binding by saturated acyl-ACPs. However, this was not the case. Upon specific inactivation of FabA with DNAC there was no increase in transcription of fabA or fabB despite the almost total blockage of UFA synthesis and the continued synthesis of saturated acyl-ACPs (Fig. 2). Thus, our in vivo data indicate that because FabR does not sense unsaturated acyl-ACP thioesters in vivo, the primary function of FabR is to report the presence of exogenous UFA (as their CoA esters) to the fabB promoter. We have not attempted to derive FabR-operator binding constants because obtaining valid dissociation constants by electrophoretic mobility shifts requires an overwhelming excess of protein over probe such that the concentration of unbound protein in the initial sample is only slightly lower than its total concentration (Cann, 1989). Given our recourse to in vitro synthesis to obtain FabR we are unable to obtain the amounts of protein needed for these analyses. Moreover, we expect that the high protein concentrations would result in FabR aggregation.
In vitro experiments showed that the E. coli and V. cholerae FabRs readily bound the intact E. coli fabA and fabB promoter regions as well as the V. cholerae fabA promoter in the absence of ligand. However, addition of unsaturated thioesters did allow binding to minimal fabA and fabB operator sequences and also gave increased binding to longer target DNA molecules. This raised the question of whether or not DNA binding by FabR necessarily inhibits transcription. Indeed, although FabR binds the yqfA promoter, the absence of FadR has no effect on yqfA expression in vivo (Fig. 12). Thus, in the fabB case it seems plausible that DNA-bound to FabR as a complex with an unsaturated acyl-CoA could block transcription more efficiently than DNA-bound FabR protein that lacked ligand, but this has yet to be tested. There are well documented examples of transcriptional regulatory systems (e.g., the E. coli arabinose and galactose operons) in which the regulatory protein is always promoter bound, but changes its properties upon ligand binding (Roy et al., 1998). The ability of FabR to bind minimal operator sequences only in the presence of an unsaturated acyl-thioester suggests that liganded FabR molecules may be more difficult for RNA polymerase to displace from the DNA than unliganded FabR molecules. Although we agree with Zhu and coworkers that FabR exists in two conformers, we believe that both conformers are bound to their cognate operators and that ligand binding probably determines the efficiency of FabR displacement by RNA polymerase. The report of Zhu and coworkers (Zhu et al., 2009) that FabR binding requires unsaturated thioesters for DNA binding in vitro could be due to the quality of their refolded protein preparations. It seems plausible that the unsaturated acyl-thioesters aided proper folding of poorly folded or aggregated FabR species as has been reported for other proteins that interact with lipids (Bogdanov and Dowhan, 1999). The data of Fig. 8 is with this scenario. In those experiments the same batch of FabR was used for parallel gel shift experiments done in the presence or absence of acyl-CoAs. In the gel lacking acyl-CoAs prominent super-shifted bands were present whereas in the gels containing acyl-CoAs, no super-shifted bands were seen. A straightforward interpretation of these observations is that acyl-CoAs act to decrease FabR aggregation.
Growth in the presence of oleate showed that regulation of fabB expression differs from that of fabA expression. This is most clearly seen when a ΔfadR strain is compared with the isogenic ΔfabR ΔfadR strain (comparison with the wild type strain is problematical due to the opposing actions of FabR and FadR on fabA and fabB expression). Addition of oleate to the medium of the ΔfadR strain resulted in increased repression of fabB expression, but had no apparent effect on fabA expression. The increased repression of fabB expression engendered by oleate addition was not seen in the ΔfabR ΔfadR strain indicating that the FadR-independent oleate sensing required FabR. This result strongly suggests that the FabR:oleoyl-CoA complex bound to the fabB promoter inhibits RNA polymerase more efficiently than the same complex bound to the fabA promoter. Note that inhibition may be due to interaction of FabR with RNA polymerase rather than by straightforward steric exclusion. Several examples are known in which regulatory proteins are bound to their cognate operators under all growth conditions and binding the small molecule ligand alters either the mode of binding or allows interaction of the regulatory protein with RNA polymerase (Roy et al., 1998). Our in vivo data are similar to those of Zhu and coworkers (Zhu et al., 2009); addition of palmitoleate gave the same levels of fabA expression in ΔfadR and ΔfabR ΔfadR stains whereas fabB expression in the ΔfadR strain was 3-fold lower than that seen in the ΔfabR ΔfadR strain. The finding that FabR binds these two promoters differently is surprising given the highly similar promoter element arrangements (Fig. 1). However, the differential binding scenario is consistent with the finding that FabR does not specifically require the fabB B2 probe FadR binding site sequences for efficient binding in vitro, a random DNA sequence suffices. In contrast, substitution of a random sequence for the FadR binding sequence of the fabA promoter resulted in significantly weaker binding (Fig. 9C).
Why does E. coli have both FabR and FadR? Our data argue that the function of FabR is to compensate for the weak FadR regulation of fabB expression in the presence of exogenous UFA (the converse argument, that FadR functions to compensate for the weak FabR regulation of fabA expression by UFA, also applies). The picture that emerges is that E. coli has yet to streamline coregulation of fabA and fabB transcription and that we are likely to observing an act in progress. Indeed, coordinated transcription of fabA and fabB is hardwired in other bacteria such as Pseudomonas aeruginosa where the genes are in a bicistronic operon (Hoang and Schweizer, 1997).
It seems possible that both FabR and FadR might bind to a given promoter. Indeed, Zhu at al., (Zhu et al., 2009) report formation of a weak FabR-FadR-fabB promoter complex. However, this cannot be assumed to occur in vivo if the reported low intracellular levels of FabR and FadR (Taniguchi et al., 2010) are accurate. Similar levels of FabR and FadR were reported 23, and 29 molecules per cell, respectively, and at such levels gene expression is stochastic (Xie et al., 2008). Thus, at any given time within a cell population fabA (or fabB) expression would be regulated by FabR in one cell subpopulation and FadR in another. This, of course, would be a dynamic situation such that at various times the regulator bound by a given promoter could be FabR or FadR or neither (thus allowing RNA polymerase to transcribe both unhindered and unaided). Unfortunately, the large number of binding sites for FadR in the E. coli genome plus the relatively modest regulatory effects of FabR and FadR would seem to preclude approach of this problem by current single molecule approaches (Xie et al., 2008). Indeed, the reported intracellular FadR levels (Taniguchi et al., 2010) seem too low to account for the observed efficient regulation the FadR controlled genes.
Prior to this study only E. coli FabR was demonstrated to be a repressor of the anaerobic unsaturated fatty acid biosynthetic pathway (Zhang et al., 2002). We have now shown that the V. cholerae FabR which is 73% identical to the E. coli protein has in vivo and in vitro properties very similar to those of E. coli FabR (Fig. 1). Moreover, expression of both the Shigella flexneri and Pasteurella multocida FabR homologues restored repressor function to an E. coli ΔfabR strain (data not shown). However, FabR regulation of UFA synthesis is asymmetric among bacteria, i.e., the fabB gene of V. cholerae is not regulated by FabR (Sequence analyses of a collection of Vibrio species with known genomes suggested that they all lack putative FabR/FadR-binding sites upstream of the fabB gene). Vibrio species have appreciably greater unsaturated fatty acid contents than does E. coli and its close relatives (Vasiurenko et al., 1996). We believe this reflects the Vibrio environmental niche, a marine environment of relatively low temperature where high levels of unsaturated fatty acids would maintain the fluidity of membrane phospholipid bilayers. Indeed, no FadR binding site is found upstream of fabB in any of the currently available Vibrio genome sequences, although a FabR binding site is invariably found within the fabA promoter regions. Since FabB is the rate-limiting step in unsaturated fatty acid synthesis (because the FabA-catalyzed reactions are reversible) (Feng & Cronan, 2009a) and because FabB overproduction In E. coli is known to result in increased UFA contents of the membrane phospholipids, we expected that the UFA content of V. cholerae would resemble those of E. coli strains that overproduce FabB (de Mendoza et al., 1983; Wang and Cronan, 2004). Indeed, this was the case. Over a wide range of growth temperatures (25–42°C) the UFA content of V. cholerae was significantly greater than that of a wild type E. coli strain and closely resembled the compositions of E. coli FabB overproduction strains (data not shown). In the case of P. multocida, a bacterium that exists in nature only in extremely close association with warm-blooded animals and thus in a thermally stable environment, a different explanation for the lack of FabR regulation of fabB is required. This bacterium and its close relatives (e.g., Haemophilus influenzae) lacks fabF, the gene that encodes the generic elongation enzyme found in most bacteria (including E. coli and the other bacteria of Fig. 4) (Wang and Cronan, 2003). Therefore, FabB is the sole P. multocida enzyme available to catalyze the elongations required to produce long chain fatty acids and thus FabB must catalyze the elongations needed to synthesize saturated as well as unsaturated chains. This state of affairs therefore precludes regulation of fabB in response to UFA.
Experimental procedures
Bacteria and growth conditions
With an exception of V. cholerae ATCC 14547 (Massengo-Tiasse and Cronan, 2008), all bacterial strains were derivatives of E. coli K-12 (Table 1). Either LB medium (10 g of tryptone, 5 g of yeast extract, and 5 g of NaCl per liter) or RB (LB with one-fifth of the yeast extract) was used to maintain E. coli strains (e.g., FYJ32). The V. cholerae strain was grown on LB medium. The chemically defined medium was medium E supplemented with 0.001% thiamine, 0.1% Vitamin-Free Casamino Acids (Difco), and 0.4% glucose as carbon source. When necessary, antibiotics were added as follows (in μg/ml): sodium ampicillin, 100; kanamycin sulfate, 50; tetracycline hydrochloride, 15 and chloramphenicol, 20. Fatty acids (Sigma) were neutralized with KOH, solubilized with Tergitol NP-40 and used at a 5 mM final concentration for induction assays. Acyl-ACP species were prepared using AasS activity as Jiang et al. (Jiang et al., 2006) previously described. Bacterial cultures were kept statically in an incubator oven at 37 °C for anaerobic growth or cultivated with vigorous shaking at 37 °C for aerobic growth unless otherwise stated. Bacterial growth was monitored by reading optical density at 600 nm.
Table 1.
Strains and plasmids used in this study
| Strain or plasmids Strains | Relevant characteristics | Origin |
|---|---|---|
| ATCC14547 | A Vibrio cholerae wild type strain | (Massengo-Tiasse and Cronan, 2008) |
| MG1655 | E. coli K-12 wild type | Lab stock |
| Topo10 | mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 rpsL endA1 | Invitrogen |
| DH5α | Δ(argF-lac)169, φ80dlacZ58(M15), ΔphoA8, glnV44, λ−, deoR481, gyrA96, recA1, endA1, hsdR17 | Lab stock |
| BL21(DE3 | T7 RNA polymerase expression strain | Novagen |
| BW25113 | A wild type strain of E. coli K-12, Δlac | CGSCa, (Baba et al., 2006) |
| UB1005 | metB, relA, gyrA | Lab stock |
| MFH8 | UB1005, fadR::Tn10 | (Henry and Cronan, 1992) |
| MFH9 | UB1005, fadR::Tn5 | (Henry and Cronan, 1992) |
| JW3935-4 | BW25113, ΔfabR751::Km | (Baba et al., 2006) |
| SI90 | MFH9, ΔfadR::Tn5 ΔfabR::Cm | (Henry and Cronan, 1992) |
| SI91 | UB1005, ΔfabR::Cm | (Henry and Cronan, 1992) |
| SI71 | pSH27/Topo10 | Lab stock |
| SI72 | pSH28/Topo10 | Lab stock |
| SI73 | pSH29/Topo10 | Lab stock |
| SI75 | pSH32/Topo10 | Lab stock |
| SI76 | pSH33/Topo10 | Lab stock |
| SI77 | pSH34/Topo10 | Lab stock |
| MC1061 | Δ(codB-lacI)3 | (Casadaban and Cohen, 1980) |
| JW2867-1 | BW25113, ΔyqfA750::Km | (Baba et al., 2006) |
| MH121 | LCD69, ΔfabA::Cm | Lab stock |
| JT173 | MC1061, cat cassette immediately downstream of fabA | Lab stock |
| JT174 | MC1061, cat cassette immediately downstream of fabB | Lab stock |
| JT175 | MC1061, FRT scar immediately downstream of fabA | Lab stock |
| JT176 | MC1061, FRT scar immediately downstream of fabB | Lab stock |
| JT180 | MC1061, fabA-lacZ fusion | JT175/pCE70, Lab stock |
| JT181 | MC1061, fabB-lacZ fusion | JT176/pCE70, Lab stock |
| FYJ30 | JT180, fabA-lacZ fusion, ΔfadR::Tn10 | P1(MFH8)×JT180b |
| FYJ31 | JT180, fabA-lacZ fusion, ΔfabR::Cm | P1(SI91)×JT180c |
| FYJ32 | JT181, fabB-lacZ fusion, ΔfadR::Tn10 | P1(MFH8)×JT181b |
| FYJ33 | JT181, fabB-lacZ fusion, ΔfabR::Cm | P1(SI91)×JT181c |
| FYJ42 | pACYC177/SI91 | This work |
| FYJ43 | pSH32/SI91 | This work |
| FYJ44 | pSH33/SI91 | This work |
| FYJ45 | pSH34/SI91 | This work |
| FYJ46 | pACYC177/FYJ31 | This work |
| FYJ47 | pSH32/FYJ31 | This work |
| FYJ48 | pSH33/FYJ31 | This work |
| FYJ49 | pSH34/FYJ31 | This work |
| FYJ50 | pACYC177/FYJ33 | This work |
| FYJ51 | pSH32/FYJ33 | This work |
| FYJ52 | pSH33/FYJ33 | This work |
| FYJ53 | pSH34/FYJ33 | This work |
| FYJ57 | BW25113, ΔfadR | (Feng and Cronan, 2010) |
| FYJ90 | FYJ31, fabA-lacZ fusion, ΔfabR::Cm, ΔfadR::Tn10 | P1(MFH8)×FYJ31b |
| FYJ91 | FYJ33, fabB-lacZ fusion, ΔfabR::Cm, ΔfadR::Tn10 | P1(MFH8)×FYJ33b |
| FYJ109 | JW2867-1 carrying pCP20ts | This work |
| FYJ110 | FYJ109, yqfA-lacZ transcription fusion | This work |
| FYJ111 | FYJ110, yqfA-lacZ transcription fusion, ΔfabR::Cm | P1(SI91)×FYJ110c |
| FYJ143 | BW25113, fabR-lacZ transcription fusion | This work |
| FYJ144 | FYJ143 carrying pACYC177 | This work |
| FYJ145 | FYJ143 carrying pSH32 | This work |
| FYJ146 | FYJ143 carrying pSH33 | This work |
| FYJ147 | FYJ143 carrying pSH34 | This work |
| Plasmids | ||
| pET28a (+) | T7 promoter-based expression vector | Novagen |
| pGEX-6p-1 | tac promoter-based expression vector | Amersham |
| pBAD24 | arabinose-induced expression vector | (Guzman et al., 1995) |
| pACYC177 | Low copy cloning vector | (Rose, 1988) |
| pCR2.1 | High copy Topo-cloning vector | Invitrogen |
| pCP20 | cat cI857 λPR flp oriT, | (Datsenko and Wanner, 2000; Ellermeier et al., 2002) |
| pKG137 | FRT lacZY+ this oriR6K, an improved version of pCE37 in FRT orientation B | (Ellermeier et al., 2002; Feng and Cronan, 2010) |
| pSH27 | pCR2.1 carrying E. coli fabR gene | Lab stock |
| pSH28 | pCR2.1 carrying S. enterica fabR gene | Lab stock |
| pSH29 | pCR2.1 carrying P. multocida fabR gene | Lab stock |
| pSH32 | pACYC177 carrying E. coli fabR gene | Lab stock |
| pSH33 | pACYC177 carrying S. enterica fabR gene | Lab stock |
| pSH34 | pACYC177 carrying P. multocida fabR gene | Lab stock |
| 6p-fabRec | pGEX-6p-1 carrying E. coli fabR gene | This work |
| 6p-fabRvc | pGEX-6p-1 carrying V. cholerae fabR gene | This work |
| 28a-fabRec | pET28a carrying E. coli fabR gene | This work |
| 28a-fabRvc | pET28a carrying V. cholerae fabR gene | This work |
| pBAD24-fabRvc | pBAD24 carrying V. cholerae fabR gene | This work |
CGSC denotes Coli Genetic Stock Center, Yale University;
Selection for tetracycline resistance;
Selection for chloramphenicol resistance.
Plasmids and DNA manipulations
To amplify the fabR genes of E. coli and V. cholerae, colony PCR was performed using the primers fabRec-F plus fabRec-R, and fabRvc-F plus fabRvc-R, respectively (Table 2). Two restriction sites, BamHI and XhoI were introduced in the primers (Table 2). After double digestion by BamHI and XhoI, the PCR products were ligated to vector pET28a (Novagen) cut with the same enzymes, to give plasmids 28a-fabRec and 28a-fabRvc (Table 1). The V. cholerae fabR gene amplified by primers fabRvc-F2 plus fabRvc-R, was inserted into pBAD24 (Guzman et al., 1995) cut with EcoRI and SalI to give pBAD24-fabRvc used for complementation. All recombinant plasmids were verified by direct DNA sequencing. A fabRec version that encoded an untagged E. coli FabR was constructed by overlap PCR. The first round of PCR was performed with 28a-fabRec as template, using primers fabRec-forward plus fabRec-reverse (Table 2) and this product was used in a second PCR reaction with the universal primer plus fabRec-reverse primer (Table 2).
Table 2.
Primers used in this study
| Primers | Primer sequences (5′-3′)a |
|---|---|
| fabRec-F | CG GGATCC ATG GGC GTA AGA GCG CAA C′ |
| fabRec-R | CCG CTCGAG TTA CTC GTC CTT CAC ATT TCC |
| fabRvc-F | CG GGATCC ATG AAA TCG TTG GGA ATC CG |
| fabRvc-R | CCG CTCGAG TTA TTC CAA CGC GCC TTT CAG |
| fabRvc-F2 | CAG GAATTC ATG AAA TCG TTG GGA ATC CG′ |
| fabRvc-CF | CG GGATCC CAT TGC AAA ACA GGT ACT CAA CC |
| fabRvc-CR | CCG CTCGAG GCG CCA GTA TTA ATG TTT TAC G |
| fabAvc_P-F | CTT GTG CAA CCG CAC GGA AT |
| fabAvc_P-R | GTT CTG CAT TAT TGG TTA CTC |
| fabAvc_BD-F | CTT AGC TGG CAC TGA TCG GAG TTG TTT GGC |
| GTA CAC GTG TTC ACT AAC ATG CAC | |
| fabAvc_BD-R | GTG CAT GTT AGT GAA CAC GTG TAC GCC AAA |
| CAA CTC CGA TCA GTG CCA GCT AAG | |
| fabAvc_B1-F | TGG CGT ACA CGT GTT CAC TAA CAT GCA CCC |
| fabAvc_B1-R | GGG TGC ATG TTA GTG AAC ACG TGT ACG CCA |
| fabAvc_B2-F | GGA GTT GTT TGG CGT ACA CGT GTT CAC TAA CAT GCA C |
| fabAvc_B2-R | GTG CAT GTT AGT GAA CAC GTG TAC GCC AAA CAA CTC C |
| fabAvc_D-F | CTT AGC TGG CAC TGA TCG GAG TTG TTT GGC |
| fabAvc_D-R | GCC AAA CAA CTC CGA TCA GTG CCA GCT AAG |
| fabAec_BD-F | TTT ATT CCG AAC TGA TCG GAC TTG TTC AGC |
| GTA CAC GTG TTA GCT ATC CTG CGT GC | |
| fabAec_BD-R | GCA CGC AGG ATA GCT AAC ACG TGT ACG CTG AAC AAG TCC GAT CAG TTC GGA ATA AA |
| fabAec_B1-F | CAG CGT ACA CGT GTT AGC TAT CCT GCG TGC |
| fabAec_B1-R | GCA CGC AGG ATA GCT AAC ACG TGT ACG CTG |
| fabAec_B2-F | GGA CTT GTT CAG CGT ACA CGT GTT AGC TAT CCT GCG TGC |
| fabAec_B2-R | GCA CGC AGG ATA GCT AAC ACG TGT ACG CTG AAC AAG TCC |
| fabAec-B2A-F | TACAGAGAAC AGC GTA CAC GTG TTA GCT ATC CTG CGT GC |
| fabAec-B2A-R | GCA CGC AGG ATA GCT AAC ACG TGT ACG CT GTTCTCTGTA |
| fabAec_D-F | TTT ATT CCG AAC TGA TCG GAC TTG TTC AGC |
| fabAec_D-R | GCT GAA CAA GTC CGA TCA GTT CGG AAT AAA |
| fabBec_BD-F | TCT ATT AAA TGG CTG ATC GGA CTT GTT CGG CGT ACA AGT GTA CGC TAT TGT GCA TTC |
| fabBec_BD-R | GAA TGC ACA ATA GCG TAC ACT TGT ACG CCG AAC AAG TCC GAT CAG CCA TTT AAT AGA |
| fabBec_B1-F | CGG CGT ACA AGT GTA CGC TAT TGT GCA TTC |
| fabBec_B1-R | GAA TGC ACA ATA GCG TAC ACT TGT ACG CCG |
| fabBec_B2-F | GGA CTT GTT CGG CGT ACA AGT GTA CGC TAT TGT GCA TTC |
| fabBec_B2-R | GAA TGC ACA ATA GCG TAC ACT TGT ACG CCG AAC AAG TCC |
| fabBec-B2A-F | TACAGAGAAC GG CGT ACA AGT GTA CGC TAT TGT GCA TTC |
| fabBec-B2A-R | GAA TGC ACA ATA GCG TAC ACT TGT ACG CC GTTCTCTGTA |
| fabBec_D-F | TCT ATT AAA TGG CTG ATC GGA CTT GTT CGG |
| fabBec_D-R | CCG AAC AAG TCC GAT CAG CCA TTT AAT AGA |
| fabBec_P-F | |
| fabBec_P-R | |
| fabBvc_P-F | CAT CGT CAA ATT GAT CGG AGA C |
| fabBvc_P-R | CGA CGT TGT TAC CGA TAC TCG |
| fabRec-P | GGT AGG GCT TAC CTG TTC TTA TAC |
| lacZ-R | GAC CAT GAT TAC GGA TTC ACT G |
| fabRec forward | TAA CTT TAA GAA GGA GAT ATA CCA ATG GGC GTA AGA GCG CAA C |
| fabRec reverse | TAT TCA TTA CTC GTC CTT CAC ATT TCC |
| fabRvc forward | TAA CTT TAA GAA GGA GAT ATA CCA ATG AAA TCG TTG GGA ATC CG |
| fabRvc reverse | TAT TCA TTA TTC CAA CGC GCC TTT CAG |
| Universal primer | GAA ATT AAT ACG ACT CAC TAT AGG GAG ACC ACA ACG GTT TCC CTC TAG AAA TAA TTT TGT TTA ACT TTA AGA AGG AGA TAT ACC A |
| yqfA-PF | GAA CTC ACG CCT GTT AGT TGA |
| yqfA_B1-F | CAG AAT TTA TTT TAG CTA ACA GGT GTT CAC TG |
| yqfA_B1-R | CAG TGA ACA CCT GTT AGC TAA AAT AAA TTC TG |
| yqfA_B2-F | CAG AAT TTA TTT TAG CTA ACA GGT GTT CAC TGG AAC TAT TCT C |
| yqfA_B2-R | GAG AAT AGT TCC AGT GAA CAC CTG TTA GCT AAA ATA AAT TCT G |
| 16S-F | GCA TAA CGT CGC AAG ACC AAA G |
| 16S-R | TTC TTC ATA CAC GCG GCA TGG |
| fabAec-F | GTT ACA CAG CAT ACT GCA TTT GC |
| fabAec-R | CAA GAG GAT TAA GAA CGT CCA AC |
| fabBec-F | CAC TGT ATC GGT AAC GCA GTA G |
| fabBec-R | GAT AAC GAA ACC GTC ACG GTG AG |
| fabRvc-GSP | CAA CAA ACG AAA TAC GTT AGG GCT G |
| fabRvc-Nested | CCG GTT CAT CCA TGT CTT TGA G |
| fabRec-GSP | CGT AGC ATT AAA CCG CTC TCA TC |
| fabRec-Nested | GAT AAA AAG AGG TGG GAG CAA TGC |
| RLM-RACE- Adaptor | GCU GAU GGC GAU GAA UGA ACA CUG CGU UUG CUG GCU UUG AUG AAA |
| RLM-RACE Outer | GCT GAT GGC GAT GAA TGA ACA CTG |
| RLM-RACE Inner | CGC GGA TCC GAA CAC TGC GTT TGC TGG CTT TGA TGA AA |
The underlined sequences are the introduced restriction sites.
Two strains JT180 (fabA-lacZ) and JT181 (fabB-lacZ) carry lacZ transcription fusions in which the promoterless lacZ gene was inserted into a FRT site located immediately downstream of the fab gene coding sequence such that the fab gene retained function (Table 1). To construct strain FYJ110 that carries the yqfA-lacZ transcription fusion temperature-sensitive plasmid pCP20 was introduced into strain JW2867-1 (yqfA750::Km) to give strain FYJ109, and this strain was subsequently transformed with pKG137 to giving strain FYJ110 (Table 1). Likewise strain FYJ143 which carries fabR-lacZ transcriptional fusion (Table 1), ΔfabR751::Km was constructed by successive electroporations with plasmids pCP20 and pKG137 (Table 2) (Ellermeier et al., 2002). The yqfA-lacZ and fabR-lacZ junctions were confirmed by PCR using two sets of primers yqfA-P plus LacZ-R and fabR-P plus LacZ-R (Table 2). Finally, strain FYJ143 was transformed with plasmids pACYC177, pSH32, pSH33 or pSH34, to give strains FYJ144, FYJ145, FYJ146 and FYJ147, respectively.
RNA isolation and RT-PCR
Bacterial cultures in mid-log phase (strains UB1005, SI91 and FYJ90) were used for total RNA isolation conducted as recently described (Feng and Cronan, 2009a). To test for RNA samples degradation the ribosomal RNAs were visualized after separation on 0.8% agarose gels. A general PCR-based assay in which total RNA served as template to test for residual DNA contamination was also done (Feng and Cronan, 2009a). The qualified RNA preparations were then used for RT-PCR analyses. Reverse transcription reaction was carried out using random primers (Feng and Cronan, 2009a, 2009b) and then the resulting cDNAs were used as templates for PCR amplification of the fabA and fabB transcripts using defined primers.
Real-time quantitative PCR
The SYBR Green method (Feng et al., 2008) of real-time quantitative PCR (qPCR) was utilized to determine the effect of V. cholerae fabR on transcription of fabA and fabB. The reactions were carried out in triplicate on a Mastercycler ep realplex (Eppendorf) with the following program: 45 cycles of 94°C for 20 s, 60°C for 20 s, and 72°C for 20 s, executed soon after a denaturing cycle at 95 °C for 15 min (Feng and Cronan, 2009a, 2009b). The internal reference gene was 16S rRNA (Table 2). The relative expression levels of fabA and fabB genes were measured using the method of 2−(ΔΔCt) (Livak and Schmittgen, 2001).
5′-RACE analysis of transcripts
The transcriptional start sites of the fabR genes of E. coli and V. cholerae were determined using the improved version of 5′-RACE kit, RLM-RACE (Ambion) as recently described (Feng and Cronan, 2009a, 2009b). Following three steps of preprocessing with calf intestine alkaline phosphatase digestion to remove 5′-phosphates from degraded mRNA, rRNA and tRNA, reverse transcription was conducted followed by nested PCR suing two rounds of PCR reactions in which 2 sets of primers, Outer Primer plus fabRvc-GSP (or fabRec-GSP) or Inner Primer plus fabRvc-Nested (or fabRec-Nested) were employed, respectively. The PCR program was a denaturing cycle at 95 °C for 5 min, followed by 35 cycles comprising of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. Following agarose gel separation and purification of the 5′-RACE products, they were inserted into the pCR2.1 TOPO vector (Invitrogen) for direct DNA sequencing. The first DNA base adjacent to the 5′-RACE adaptor was regarded as the transcription start site (Feng and Cronan, 2009a, 2009b; Zhang et al., 2007).
In vitro translation of FabR proteins
To prepare soluble and functional FabR proteins the PURExpress in vitro protein synthesis kit (New England Biolabs, a cell-free transcription/translation system in which all components are well defined) was used. The reaction system (25 μl total), was as done previously (Feng and Cronan, 2009b) with minor modifications. For preparation of nonradioactive proteins 1 μg of purified plasmid (either pET28-fabRec or pET28-fabRvc) was added together with 12.5 μl of solution A and 5 μl of solution B. To prepare radioactive proteins 1 μl of L-[35S]-methionine (10 mCi/ml, American Radio-labeled Chemicals) was added to above translation system. The synthesized proteins were stored at −80 °C.
Liquid chromatography quadrupole time-of-flight mass spectrometry
A Waters Q-Tof API-US Quad-ToF mass spectrometer connected to a Waters nano Acquity UPLC) was used to identify the FabRs prepared in vitro. The protein bands at the expected positions were cut from SDS-PAGE gel. The gel slices were destained and the proteins digested with 25 μl of Sequencing Grade Trypsin (G-Biosciences St. Louis, MO, 12.5 ng/μl in 25 mM ammonium bicarbonate) using a CEM Discover Microwave Digestor (Mathews, NC) for 15 min at 55 °C and 50W. Then, the resulting peptides were extracted using 50% acetonitrile containing 5% formic acid, dried using a Savant SpeedVac and suspended in 13 μl of 5% acetonitrile containing 0.1% formic acid. Samples of 10 μl were loaded on a Waters Atlantis C-18 column (0.03 mm particle, 0.075 mm × 150 mm) and eluted at a flow rate of 250 nL per min using a linear gradient of water/acetonitrile containing 0.1% formic acid 0–60% B in 60 min. The mass spectrometer was set for data dependent acquisition and ms/ms analysis was performed on the most abundant four peaks at any given time. Data analysis was done using the Waters Protein Lynx Global Server 2.2.5, Mascot (Matrix Sciences) and BLAST against NCBI nr database.
Size exclusion chromatography
To examine the solution structures of FabR_ec and FabR_vc, the purified [35S]-methionine labeled in vitro translation products were subjected to gel filtration analyses using a Superdex 75 column (Pharmacia) run on an Äkta fast protein liquid chromatography system (GE Healthcare) (Feng et al., 2008; Feng and Cronan, 2010). The column effluent was monitored at a flow rate of 0.3 ml/min in running buffer (20 mM Tris-HCl, 100 mM NaCl, pH 8.0), and 0.6 ml samples were collected and counted. The radioactivity of each sample was obtained by liquid scintillation counting (Jerga and Rock, 2009). When required, samples of interest were separated by 12% SDS-PAGE followed by gel drying and exposure to x-ray films (Kodak).
Electrophoretic mobility shift assays
To visualize binding of FabR protein to the two target genes, fabA and fabB, gel shift experiments were employed (Feng and Cronan, 2009b, 2010) with minor improvements. The E. coli FabR binding palindromes were synthesized using two complementary oligonucleotides (fabAec_P-F plus fabAec_P-R for fabA gene and fabBec_P-F plus fabBec_P-R for fabB gene) (Table 2). The V. cholerae fabA probe covering a predicted FabR binding site was PCR amplified with two primers, fabAec_P-F plus fabAec_P-R, and a DNA fragment containing fabB promoter region (257 bp) was obtained with a pair of primers, fabBec_P-F plus fabBec_P-R (Table 2). The probes were digoxigenin-labeled by terminal transferase incorporation of digoxigenin-ddUTP (Roche). For gel shift assays the digoxigenin-labeled DNA probes (~ 0.1 pmol) were mixed with serial dilutions of the in vitro translated FabR protein in binding buffer (20 μl total volume) (Roche) at room temperature for 15 min. The DNA/protein mixtures were loaded onto 7% native PAGE and after running the gel contents were transferred to an equilibrated, positively charged nylon membrane (Roche) by contact blotting. The luminescence signal was developed in CSPD working solution at room temperature following the incubation of the nylon membrane with an anti-digoxigenin antibody. Two hours later an exposure of the nylon membrane to an enhanced chemiluminescence (ECL, Amersham) detection system gave signal capture.
P1vir phage transduction
P1vir transductions were conducted as the protocol described by Miller (Miller, 1972) with minor modifications. Two strains (FYJ30 and FYJ31) were generated by P1vir transduction of strain JT180 with lysates grown on strains MFH8 (fadR::Tn10) or SI91 (fabR::Cm), respectively (Table 1). Similarly, the same lysates were used for P1vir transduction of strain JT181 to construct two strains, FYJ32 and FYJ33. Strains FYJ90 and FYJ91 were obtained by P1vir transduction of strains FYJ31 and FYJ33, respectively with a lysate grown on strain MFH8 and selection for tetracycline resistance (Table 1). Strain FYJ111 was generated by P1vir transduction of strain FYJ110, using a lysate grown on strain SI91 (Table 1).
β-Galactosidase assays
Mid-log phase cultures grown in either RB or minimal medium (with or without supplementation with various carbon sources) were pelleted and twice washed with Z buffer (Miller, 1972). After the optical density at 600 nm (OD600) of the resuspended bacteria was measured the cells were permeabilized by chloroform-SDS treatment and assayed for β-galactosidase activity (Feng and Cronan, 2009b; Miller, 1972). The data were recorded in triplicate for at least three independent experiments.
Synthesis of DNAC
DNAC was synthesized by N,N′-dicyclohexylcarbodiimide coupling of 3-decynoic acid and N-acetylcysteamine. N,N′-Dicyclohexylcarbodiimide (534 mg, 2.6 mmol) was dissolved in 8 ml of dry tetrahydrofuran and cooled in ice under dry nitrogen. 3-Decynoic acid (440 mg, 2.6 mmol) was added followed by 0.26 ml (290 mg, 2.4 mmol) of N-acetylcysteamine (Aldrich). The solution was stirred overnight and allowed to slowly reach room temperature. The solution was then twice filtered through columns of Celite to remove dicyclohexylurea and the DNAC product was crystallized twice from a mixture of pentane and ethyl acetate. 3-Decynoic acid was obtained by chromate-catalyzed periodate oxidation (Schmieder-van de Vondervoort et al., 2002) of 3-decynol (Aldrich). and was crystallized twice from pentane (mp 36.1–36.9 °C). This synthetic route was much more environmentally responsible than that previously used (Nunn and Cronan, 1974) and gave a product of the same quality.
Fatty acid compositional analysis
Cultures (10 ml) of E. coli MG1655 and V. cholerae ATCC 14647 grown to mid-log phase in LB medium were collected for subsequent analyses of fatty acid composition. The bacterial pellets were suspended in 3 ml of solution of chloroform/methanol (1:2, volume/volume), and then were mixed with 1 ml each of chloroform and water and centrifuged. The lower phase was extract twice with equal volume of water followed by extraction with the same volume of 1 M potassium chloride. The membrane phospholipid of the lower phase was transferred into clean tubes, dried in air and dissolved in 100 μl of methanol. Finally, the samples (20 μl each) were injected into an electrospray ionization mass spectrometer (Waters Quattro II) to determine the fatty acid composition (Sweetman et al., 1996). Methyl esters of [I-14C]-acetate labeled fatty acids were prepared and separated into saturated and unsaturated species by argentation chromatography as previously described (de Mendoza et al., 1983; Feng and Cronan, 2009a).
Bioinformatic analyses
Putative FabR binding sequences of nine γ-proteobacteria family members were searched by BLAST analyses using the known E. coli FabR binding palindromes as probes and aligned by programs ESPript 2.2 and BLAST.
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grant AI15650 from National Institute of Allergy and Infectious Diseases (NIAID). We are grateful to Dr. Peter Yau and Dr. Haijun Yao for their technical assistance in Q-TOF and EI-MS, respectively. We also thank Prof. Diego de Mendoza for helpful discussion of this work.
Footnotes
In a personal communication with the corresponding author of that paper we learned that the probe was a 46 bp fabB probe of sequence 5′-GGCTGATCGGACTTGTTCGGCGTACAAGTGTACGCTATTGTGCATT-3′. Thus in common with our 56 bp B+D fabB probe the Zhu et al probe contained both the FadR and FabR binding sites, although it lacked the extra sequences we placed upstream of the FadR binding site.
References
- Annand R, Kozlowski J, Davisson V, Schwab J. Mechanism-based inactivation of Escherichia coli β-hydroxydecanoyl thiol ester dehydrase: assignment of the imidazole nitrogen-15 NMR resonances and determination of the structure of the alkylated histidine. J Am Chem Soc. 1993;115:1088–1094. [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100050. 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch K. β-hydroxydecanoyl thioester dehydrase. In: Boyer PD, editor. The Enzymes. New York: Academic Press; 1971. pp. 441–464. [Google Scholar]
- Bloch K. The Beginnings of Enzyme Suicide. J Protein Chem. 1986;5:69–73. [Google Scholar]
- Bogdanov M, Dowhan W. Lipid-assisted protein folding. J Biol Chem. 1999;274:36827–36830. doi: 10.1074/jbc.274.52.36827. [DOI] [PubMed] [Google Scholar]
- Cann JR. Phenomenological theory of gel electrophoresis of protein-nucleic acid complexes. J Biol Chem. 1989;264:17032–17040. [PubMed] [Google Scholar]
- Casadaban MJ, Cohen SN. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol. 1980;138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- Clark DP, DeMendoza D, Polacco ML, Cronan JE., Jr Beta-hydroxydecanoyl thio ester dehydrase does not catalyze a rate-limiting step in Escherichia coli unsaturated fatty acid synthesis. Biochemistry. 1983;22:5897–5902. doi: 10.1021/bi00294a032. [DOI] [PubMed] [Google Scholar]
- Collado-Vides J, Magasanik B, Gralla JD. Control site location and transcriptional regulation in Escherichia coli. Microbiol Rev. 1991;55:371–394. doi: 10.1128/mr.55.3.371-394.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan JE, Jr, Silbert DF, Wulff DL. Mapping of the fabA locus for unsaturated fatty acid biosynthesis in Escherichia coli. J Bacteriol. 1972;112:206–211. doi: 10.1128/jb.112.1.206-211.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan JE., Jr In vivo evidence that acyl coenzyme A regulates DNA binding by the Escherichia coli FadR global transcription factor. J Bacteriol. 1997;179:1819–1823. doi: 10.1128/jb.179.5.1819-1823.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan JE, Jr, Subrahmanyam S. FadR, transcriptional co-ordination of metabolic expediency. Mol Microbiol. 1998;29:937–943. doi: 10.1046/j.1365-2958.1998.00917.x. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mendoza D, Klages Ulrich A, Cronan JE., Jr Thermal regulation of membrane fluidity in Escherichia coli. Effects of overproduction of beta-ketoacyl-acyl carrier protein synthase I. J Biol Chem. 1983;258:2098–2101. [PubMed] [Google Scholar]
- DiRusso CC, Metzger AK, Heimert TL. Regulation of transcription of genes required for fatty acid transport and unsaturated fatty acid biosynthesis in Escherichia coli by FadR. Mol Microbiol. 1993;7:311–322. doi: 10.1111/j.1365-2958.1993.tb01122.x. [DOI] [PubMed] [Google Scholar]
- Ellermeier CD, Janakiraman A, Slauch JM. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene. 2002;290:153–161. doi: 10.1016/s0378-1119(02)00551-6. [DOI] [PubMed] [Google Scholar]
- Feng Y, Li M, Zhang H, Zheng B, Han H, Wang C, Yan J, Tang J, Gao GF. Functional definition and global regulation of Zur, a zinc uptake regulator in a Streptococcus suis serotype 2 strain causing streptococcal toxic shock syndrome. J Bacteriol. 2008;190:7567–7578. doi: 10.1128/JB.01532-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Cronan JE. Escherichia coli unsaturated fatty acid synthesis: complex transcription of the fabA gene and in vivo identification of the essential reaction catalyzed by FabB. J Biol Chem. 2009a;284:29526–29535. doi: 10.1074/jbc.M109.023440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Cronan JE. A new member of the Escherichia coli fad regulon: transcriptional regulation of fadM (ybaW) J Bacteriol. 2009b;191:6320–6328. doi: 10.1128/JB.00835-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Cronan JE. Overlapping repressor binding sites result in additive regulation of Escherichia coli fadH by FadR and ArcA. J Bacteriol. 2010;192:4289–4299. doi: 10.1128/JB.00516-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Matsuoka H, Hirooka K. Regulation of fatty acid metabolism in bacteria. Mol Microbiol. 2007;66:829–839. doi: 10.1111/j.1365-2958.2007.05947.x. [DOI] [PubMed] [Google Scholar]
- Gunsalus RP, Yanofsky C. Nucleotide sequence and expression of Escherichia coli trpR, the structural gene for the trp aporepressor. Proc Natl Acad Sci U S A. 1980;77:7117–7121. doi: 10.1073/pnas.77.12.7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry MF, Cronan JE., Jr Escherichia coli transcription factor that both activates fatty acid synthesis and represses fatty acid degradation. J Mol Biol. 1991;222:843–849. doi: 10.1016/0022-2836(91)90574-p. [DOI] [PubMed] [Google Scholar]
- Henry MF, Cronan JE., Jr A new mechanism of transcriptional regulation: release of an activator triggered by small molecule binding. Cell. 1992;70:671–679. doi: 10.1016/0092-8674(92)90435-f. [DOI] [PubMed] [Google Scholar]
- Hoang TT, Schweizer HP. Fatty acid biosynthesis in Pseudomonas aeruginosa: cloning and characterization of the fabAB operon encoding beta-hydroxyacyl-acyl carrier protein dehydratase (FabA) and beta-ketoacyl-acyl carrier protein synthase I (FabB) J Bacteriol. 1997;179:5326–5332. doi: 10.1128/jb.179.17.5326-5332.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerga A, Rock CO. Acyl-Acyl carrier protein regulates transcription of fatty acid biosynthetic genes via the FabT repressor in Streptococcus pneumoniae. J Biol Chem. 2009;284:15364–15368. doi: 10.1074/jbc.C109.002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Chan CH, Cronan JE. The soluble acyl-acyl carrier protein synthetase of Vibrio harveyi B392 is a member of the medium chain acyl-CoA synthetase family. Biochemistry. 2006;45:10008–10019. doi: 10.1021/bi060842w. [DOI] [PubMed] [Google Scholar]
- Kass LR. The antibacterial activity of 3-decynoyl-n-acetylcysteamine. Inhibition in vivo of β-hydroxydecanoyl thioester dehydrase. J Biol Chem. 1968;243:3223–3228. [PubMed] [Google Scholar]
- Leesong M, Henderson BS, Gillig JR, Schwab JM, Smith JL. Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: two catalytic activities in one active site. Structure. 1996;4:253–264. doi: 10.1016/s0969-2126(96)00030-5. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Magnuson K, Jackowski S, Rock CO, Cronan JE., Jr Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol Rev. 1993;57:522–542. doi: 10.1128/mr.57.3.522-542.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massengo-Tiasse RP, Cronan JE. Vibrio cholerae FabV defines a new class of enoyl-acyl carrier protein reductase. J Biol Chem. 2008;283:1308–1316. doi: 10.1074/jbc.M708171200. [DOI] [PubMed] [Google Scholar]
- McCue L, Thompson W, Carmack C, Ryan MP, Liu JS, Derbyshire V, Lawrence CE. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res. 2001;29:774–782. doi: 10.1093/nar/29.3.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1972. [Google Scholar]
- Nunn WD, Cronan JE., Jr Unsaturated fatty acid synthesis is not required for induction of lactose transport in Escherichia coli. J Biol Chem. 1974;249:724–731. [PubMed] [Google Scholar]
- Rose RE. The nucleotide sequence of pACYC177. Nucleic Acids Res. 1988;16:356. doi: 10.1093/nar/16.1.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Garges S, Adhya S. Activation and repression of transcription by differential contact: two sides of a coin. J Biol Chem. 1998;273:14059–14062. doi: 10.1074/jbc.273.23.14059. [DOI] [PubMed] [Google Scholar]
- Roy S, Sahu A, Adhya S. Evolution of DNA binding motifs and operators. Gene. 2002;285:169–173. doi: 10.1016/s0378-1119(02)00413-4. [DOI] [PubMed] [Google Scholar]
- Schmieder-van de Vondervoort L, Bouttemy S, Padrón J, Bras JL, Muzart J, Alsters P. Chromium catalyzed oxidation of (homo-)allylic and (homo-)propargylic alcohols with sodium periodate to ketones or carboxylic acids. Synlett. 2002;2:243–246. [Google Scholar]
- Schwab J, Klassen J. Steric course of the allylic rearrangement catalyzed by β-hydroxydecanoylthioester dehydrase. Mechanistic implications. J Am Chem Soc. 1984;106:7217–7722. [Google Scholar]
- Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T. Cell-free translation reconstituted with purified components. Nat Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- Sweetman G, Trinei M, Modha J, Kusel J, Freestone P, Fishov I, Joseleau-Petit D, Redman C, Farmer P, Norris V. Electrospray ionization mass spectrometric analysis of phospholipids of Escherichia coli. Mol Microbiol. 1996;20:233–238. doi: 10.1111/j.1365-2958.1996.tb02504.x. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiurenko ZP, Padchenko AG, Ruban NM, Alekseenko VV, Dobroshtan EV, Gurleva GG, Opanasenko IS. The cellular fatty acid composition of bacteria in the family Vibrionaceae. Mikrobiol Z. 1996;58:38–44. [PubMed] [Google Scholar]
- Wang H, Cronan JE. Haemophilus influenzae Rd lacks a stringently conserved fatty acid biosynthetic enzyme and thermal control of membrane lipid composition. J Bacteriol. 2003;185:4930–4937. doi: 10.1128/JB.185.16.4930-4937.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Cronan JE. Functional replacement of the FabA and FabB proteins of Escherichia coli fatty acid synthesis by Enterococcus faecalis FabZ and FabF homologues. J Biol Chem. 2004;279:34489–34495. doi: 10.1074/jbc.M403874200. [DOI] [PubMed] [Google Scholar]
- Xie XS, Choi PJ, Li GW, Lee NK, Lia G. Single-molecule approach to molecular biology in living bacterial cells. Annu Rev Biophys. 2008;37:417–444. doi: 10.1146/annurev.biophys.37.092607.174640. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Marrakchi H, Rock CO. The FabR (YijC) transcription factor regulates unsaturated fatty acid biosynthesis in Escherichia coli. J Biol Chem. 2002;277:15558–15565. doi: 10.1074/jbc.M201399200. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Zhu K, Frank MW, Rock CO. A Pseudomonas aeruginosa transcription factor that senses fatty acid structure. Mol Microbiol. 2007;66:622–632. doi: 10.1111/j.1365-2958.2007.05934.x. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Rock CO. Transcriptional regulation in bacterial membrane lipid synthesis. J Lipid Res. 2009;50(Suppl):S115–119. doi: 10.1194/jlr.R800046-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Choi KH, Schweizer HP, Rock CO, Zhang YM. Two aerobic pathways for the formation of unsaturated fatty acids in Pseudomonas aeruginosa. Mol Microbiol. 2006;60:260–273. doi: 10.1111/j.1365-2958.2006.05088.x. [DOI] [PubMed] [Google Scholar]
- Zhu K, Zhang YM, Rock CO. Transcriptional regulation of membrane lipid homeostasis in Escherichia coli. J Biol Chem. 2009;284:34880–34888. doi: 10.1074/jbc.M109.068239. [DOI] [PMC free article] [PubMed] [Google Scholar]