

Abstract
Microglia, the immune cells of the central nervous system, constantly survey the parenchyma in the healthy brain to maintain homeostasis. When a disturbance, such as cell death, results in ATP release in vivo, microglial processes respond by utilizing P2Y12 purinergic receptors to trigger extension toward the site of damage. Processes ultimately surround the injury site, preventing the spread of harmful cellular constituents and assisting with tissue repair. In contrast to the healthy brain, many neurodegenerative diseases, including Parkinson’s disease, are characterized by the presence of neuroinflammation. Yet, the ability of microglia to respond to tissue damage under pro-inflammatory conditions has not been well studied. To assess the ability of microglia to respond to tissue injury and localized cell death in the context of Parkinson’s disease, we performed confocal imaging of acute brain slices from mice with microglia-specific green fluorescent protein expression. Microglia in coronal slices containing the substantia nigra extend processes toward a mechanical injury in a P2Y12 receptor-dependent manner. However, microglia in mice treated for 5 days with 20 mg/kg/day 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) show significantly reduced process displacement toward the injury compared to microglia in control animals. Pre-treatment of slices from MPTP-injected mice with the A2A receptor-selective antagonist preladenant restores the ability of activated microglia to respond to tissue damage. These data support the hypothesis that chronic inflammation impedes microglial motility in response to further injury, such as cell death, and suggest that some aspects of the neuroprotection observed with adenosine A2A receptor antagonists may involve direct or indirect actions at microglia.
Keywords: Parkinson’s disease, microglia, adenosine A2A receptor, preladenant, acute brain slice, imaging, neuroinflammation
Introduction
Parkinson’s disease (PD)3, the second most common neurodegenerative disorder in the United States, is characterized by tremors, bradykinesia, rigidity, and postural instability. The cellular hallmarks of the disease are loss of dopaminergic neurons originating in the substantia nigra (SN) and subsequent loss of dopamine in the striatum (Kish et al., 1988; Rinne, 1991). Another important feature of PD is the presence of neuroinflammation. For example, some pro-inflammatory cytokines, such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and others, can be found at higher levels in cerebrospinal fluid samples from PD patients compared to age-matched controls (Mogi et al., 1994a; Mogi et al., 1996; Mogi et al., 1994b). Further supporting the involvement of inflammation, meta-analyses of several studies show that use of non-steroidal anti-inflammatory drugs (NSAIDs), and specifically ibuprofen, are associated with lower risk for developing PD (Gagne and Power, 2010; Gao et al., 2011). Moreover, activated microglia, the brain’s resident immune cells, can be detected in brains of living PD patients [with positron emission tomography (PET) imaging] and in post-mortem samples from people who suffered from the disease (Gerhard et al., 2006; McGeer et al., 1988). Finally, activated microglia have been observed in post-mortem samples from animal models of PD, such as monkeys intoxicated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and in humans accidentally exposed to MPTP even years after the toxin has been removed (Barcia et al., 2004; Kanaan et al., 2008; Langston et al., 1999; McGeer et al., 2003).
The ability of microglia to perform immune functions such as cytokine secretion and reactive oxygen species generation when activated renders microglia as potential contributors to PD pathology by compromising neuronal survival (Block et al., 2007). However, microglia also perform many other functions in the brain that are not directly linked to the immune response (Kettenmann et al., 2013). For example, the “resting” microglia in the healthy brain have highly motile processes that can detect disturbances of the brain parenchyma, such as rupture of brain capillaries or cell death that occur throughout life (Davalos et al., 2005; Nimmerjahn et al., 2005). The response to injury in vivo is mediated by ATP release by damaged cells, activation of P2Y12 receptors on microglia, and directional process extension to surround the damaged area and promote tissue repair (Davalos et al., 2005; Haynes et al., 2006). Interestingly, microglia that are in an activated state downregulate P2Y12 receptors and upregulate adenosine A2A receptors (Haynes et al., 2006; Orr et al., 2009), the latter of which are indirectly activated by ATP after its rapid breakdown to adenosine (Zimmermann, 2000). However, unlike the ability of ATP to induce process extension in resting microglia, ATP (and adenosine) induces process retraction in activated microglia (Orr et al., 2009). This raises a question as to how activated microglia, such as those found in PD, detect and respond to the neuronal death that is characteristic of the disease.
To study the ability of activated microglia to respond to tissue damage in a PD-related context, we developed an assay that allowed us to examine microglial motility in acute slices containing the SN from MPTP-treated mice in response to mechanically induced tissue injury. Using this preparation, we demonstrate that microglia in slices from MPTP-treated mice show a diminished capacity to extend their processes to the site of damage. Blockade of A2A receptors restored process extension to the damaged area in slices from MPTP-treated mice. Our findings suggest that microglia in PD might display a delayed response to the ongoing cell death, which could promote disease progression. Moreover, these data show that A2A receptor antagonists have the capacity to restore normal function of microglial processes, which could be relevant to their clinical utility.
Materials and Methods
Animals
All procedures involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committee at Emory University. CX3CR1GFP/GFP mice, which exhibit enhanced green fluorescent protein (GFP) expression from the microglia-specific CX3C- type chemokine receptor 1 (CX3CR1) promoter (Jung et al., 2000), were purchased from Jackson Laboratories and bred in-house with C57Bl/6 mice to generate mice heterozygous for the GFP knock-in gene (CX3CR1GFP/+ mice) to be used for slice preparation and imaging. To achieve microglial activation in vivo, mice were injected with either lipopolysaccharide (LPS, E. coli strain K-235, Sigma L2143) or MPTP (Sigma, cat. #M0896). The LPS injection paradigm consisted of a single treatment of 2 mg/kg intraperitoneally (i.p.), and preparation of slices or isolation of tissues two days later. MPTP-treated mice received daily subcutaneous (s.c.) injections of 20 mg/kg (free base) of the neurotoxin for 5 consecutive days for a total dose of 100 mg/kg. We chose a lower dose than what has been commonly used [30 mg/kg/day; Jackson-Lewis and Przedborski (2007)] to reduce the high mortality we observed with 30 mg/kg in CX3CR1GFP/+ mice. The total dose of 100 mg/kg MPTP administered here is known to induce statistically significant dopaminergic neuronal loss [Seniuk et al. (1990); see Figure 5]. Tissues for immunohistochemistry and imaging were collected 4-6 days after the conclusion of the MPTP treatment. We chose this time point to capture the tissue early in the processes of cell death, rather than at a terminal stage after dopaminergic cells have already been lost.
Brain slicing and imaging
Acute coronal brain slices that contained the substantia nigra were prepared from CX3CR1GFP/+ mice at a thickness of 200 μm as described before (Gyoneva and Traynelis, 2013). After at least 1 hr of rest to allow microglia to recover from the slicing procedure, the slices were moved to the pre-warmed stage (32 °C) of an inverted Olympus IX51 confocal microscope equipped with a disc scanning unit and imaged through a 20x dry objective (NA 0.50) over time with constant perfusion with oxygenated artificial cerebrospinal fluid (aCSF). The slices were centered on the stage to include the SN in the imaging field and held in place by a ring with nylon threads positioned on top of the slice. Imaging consisted of obtaining 31 optical sections through the slices (1 μm step) every 30-60 s for 20 min through a Hamatsu Orca-ER camera with the IPlab software. Following recording of baseline motility for 20 min, we induced localized tissue injury in the substantia nigra by lowering a blunted 30-gauge needle (referred to as “rod”). The rod was carefully positioned over the substantia nigra pars compacta (SNc) and lowered at a rate of 100 μm/s with a closed loop motorized micromanipulator (SD Instruments, model MC1000e) for 180 μm into the tissue and left in place until the conclusion of the experiment. A second 20-min recording was set up as soon as the injury was created in order to capture the microglial response to injury in the same slice. For some experiments, the P2Y12 receptor antagonist clopidogrel (2 μM final concentration from DMSO stock, Tocris 2490) or the adenosine A2A receptor antagonist preladenant [5 μM final concentration from DMSO stock, synthesized at Dept. of Chemistry, Emory University, using procedures described by Neustadt et al. (2007)] were included in the perfusion solution for the duration of the experiment (baseline recording, induction of injury, response to injury).
To assess the extent of damage to dopaminergic neurons induced by the mechanical injury, slices were prepared from tyrosine hydroxylase (TH)-GFP animals (kindly provided by Dr. David Weinshenker, Emory University) which express GFP from the TH promoter, and injured as described above. The slices were then fixed overnight in 4% paraformaldehyde (PFA) and mounted on glass slides. The GFP signal in dopaminergic neurons in the SN was imaged with a Leica SP8 multiphoton microscope through a 10x dry objective (NA 0.30) to obtain optical sections through the slices every 4.3 μm. These sections were used to generate 3D reconstructions of the slices with ImageJ software (National Institutes of Health) and identify the location of the injury. After calculating the volume of the injury, the number of TH-positive neurons that could be located within the injured tissue was estimated from non-damaged areas in injured slices (n = 8) and sham-injured slices (n = 3).
Analysis of microglial motility
The optical sections along the z-axis at each time point of the time-lapse recordings (baseline and response to injury) from CX3CR1GFP/+ mice were used to generate 2D maximum intensity projections, which were later used to quantify microglial movement with the Imaris software v7.6 (Bitplane AG, Switzerland). The software detected objects with diameter larger than 2 μm, which mostly represent microglial processes. (Cell bodies, identified as objects larger than 5 μm-diameter in eleven randomly selected recordings, represented 5.25% ± 0.55% of all objects). All objects were tracked over time with an Autoregressive Motion GapClose algorithm (max distance of 5 μm) to measure the displacement and velocity of each object at each time point in both the x- and y-directions. The net displacement for each track was calculated with the Pythagorean theorem,
and net velocity taken as net distance travelled per time step. The average (mean) displacement and velocity were then calculated for all tracks in a recording.
Paired baseline and injury recordings were obtained for each slice. The sign of the average displacement and velocity vectors in injury recordings was manually adjusted to reflect movement towards (positive) or away (negative) from the injury based on the signs of the average x- and y-components. Direction of movement was also adjusted for baseline recordings, which do not have an injury site, by considering the positive direction to be the same as in the subsequent injury recording from the same slice. Finally, the fraction of tracks with displacement longer than 5 μm gives an indication of the uniformity of movement – a large fraction of tracks with long displacement would suggest the presence of many processes moving over long distances rather than random motion with frequent changes in direction. There are no significant differences in the fluorescence intensities between paired (control-treatment) recordings, indicating that changes in response reflect changes in motility rather than inability of the software to detect moving objects (data not shown).
Reverse transcriptase polymerase chain reaction (RT-PCR)
Total cellular RNA was isolated from acute brain slices from CX3CR1GFP/+ mice using the PureLink RNA Mini Kit (Invitrogen) as described before (Gyoneva and Traynelis, 2013). Briefly, the tissue was homogenized and passed through silica columns to bind the RNA. The eluted RNA (50 ng starting material) was then used for RT-PCR in order to determine IL-1β expression with the SuperScript III One-step RT-PCR system with Platinum Taq DNA Polymerase (Invitrogen). Amplification conditions and primer sequences are described elsewhere (Gyoneva and Traynelis, 2013).
Immunohistochemistry
Changes in TH, Iba1, and adenosine A2A receptor expression were detected with immunohistochemistry of free-floating brain sections. The brains of MPTP- or saline-treated mice were collected 5 days following the final MPTP injection, drop-fixed in 4% PFA overnight, and cryoprotected through passages in 15% and 30% sucrose overnight, and sliced on a cryostat to prepare 40 μm-thick coronal slices. For staining, all solutions were prepared in PBS, and all incubations were performed with stirring. All washes consisted of three 10-min incubations in PBS unless otherwise indicated.
For TH and Iba1 staining, slices containing the SN or striatum were treated with 3% hydrogen peroxide for 10 min, blocked with 10% normal goat serum + 0.15% Triton X-100 for 1 hr, and incubated in rabbit anti-TH antibody (Millipore AB152, 1:1000 dilution) or rabbit anti-Iba1 antibody (Wako 019-19741, 1:1000 dilution) overnight at 4°C. After washing out the primary antibody, biotinylated goat anti-rabbit secondary antibody (Vector Labs BA-1000, 1:200 dilution) was applied for 1 hr in blocking buffer. Following a wash step, the signal was visualized using the avidin-biotin complex system (Vectastain ABC Kit) and DAB substrate (Vector Labs) according to manufacturer’s instructions. The slices were mounted on coverslips, dehydrated in ethanol, cleared with xylene, and coverslipped with Cytoseal (Richard-Allan Scientific).
To detect A2A receptors, slices were blocked in 10% normal donkey serum + 1 % bovine serum albumin (BSA) + 0.3% triton X for 1 hr and incubated in mouse anti-A2A antibody (Millipore 05-717, 1:1000 dilution) overnight at room temperature. Slices were then washed and incubated with donkey anti-mouse Texas Red-conjugated secondary antibody (Jackson ImmunoResearch, 1:200 dilution) for 2 hr, washed again, mounted on coverslips, allowed to airdry and coverslipped with Vectashield (Vector Labs). For visualization of A2A receptor-stained sections, slices were imaged with an Olympus IX51 confocal microscope. Slices from saline-treated mice containing the striatum were used to determine imaging conditions because of the constitutive A2A receptor expression there. The Texas Red fluorophore was detected by excitation at 590 nm. Microglia were visualized by detecting the GFP signal with excitation at 480 nm. Because GFP is genetically encoded in the CX3CR1GFP/+ mice, no staining was necessary.
Some 200 μm-thick slices were prepared as for imaging, but used for staining in order to quantify the extent of the mechanical tissue damage. After induction of injury, the slices were fixed in 4% PFA overnight. Slices were treated with 1% sodium borohydride for 20 min before washing five times in PBS and blocking for 1 hr in 1% normal horse serum + 0.3% Triton X-100 + 1% BSA. The primary anti-NeuN antibody (Millipore MAB377, 1:2000 dilution) was applied overnight. After washing off unbound antibody, the slices were incubated with secondary horse anti-mouse antibody at 1:200 dilution in 1% normal horse serum + 0.3% triton X + 1% BSA for 90 min. Then, the signal was visualized by incubation in ABC as described above, but for 90 min. After two washes in PBS and a wash in Tris, the slices were added to DAB (Sigma) for 10 min before mounting and coverslipping.
Quantification of immunohistochemistry
To detect changes in TH-positive terminals in the striatum, the average absorbance from the DAB signal was calculated for regions of interest containing the striatum with ImageJ software. Changes in microglial morphology were analyzed in three ways with ImageJ. First, to calculate the average cell body area of microglia, Iba1-stained images were thresholded to remove the fine microglial processes. The remaining cell bodies were detected as particles larger than 50 μm2 (to eliminate debris and unspecific objects), and their area was measured by the software. Second, the whole area occupied by Iba1 immunoreactivity (cell bodies and processes) was calculated as a fraction of the imaged area. Finally, microglia were assigned a score of 1 to 4 based on their morphology: 1 represented cells with resting morphology, 2 – hyperramified, bushy cells, 3 – cells with only a few processes remaining, and 4 – completely amoeboid cells (Walker et al., 2014). The percentage of each morphological phenotype was calculated from the total number of cells in an image. For all types of quantification, 2-4 sections per animal were analyzed to obtain an average response for each animal and then averaged across groups (3 saline- and 4 MPTP-treated mice).
Statistical analysis
All statistical tests were performed in SigmaPlot v11.0. The details of the tests used, sample sizes, and specific probability values are indicated in the text and figure legends as necessary. In general, slice recordings were analyzed with two-way repeated measures analysis of variance (ANOVA) to compare the baseline dynamics and response to injury in the same slice. Slices from different treatments were compared with Tukey’s post hoc test. Slices from at least three animals were used for each treatment, with each slice regarded as an independent sample. Data about Iba1 and TH immunoreactivity and A2A receptor expression were analyzed with two-tailed unpaired Student’s t tests. Values from slices from the same animal were averaged to compare individual saline- or MPTP-treated mice. Results were considered to be significantly different if p < 0.05.
Results
Microglia in acute brain slices respond to localized mechanical damage
Microglia respond in several important ways to tissue damage, all of which work together to resolve the injury. These include extension of processes to the site of injury, phagocytosis of cellular debris, and secretion of factors that limit damage and facilitate recovery. ATP release (for example, from the damaged cells) can modulate all of these actions through P2Y receptors, rendering it a central stimulus that initiates many of the helpful responses to microglia (Koizumi et al., 2013). For the current study, we focused on the initial response of microglia to ATP through process extension as an assessment of the ability of microglia to respond to injury.
The ability of microglia to detect tissue damage and extend processes to the site of damage is commonly studied in vivo with two-photon microscopy. However, the brain regions primarily affected in Parkinson’s disease, the substantia nigra and striatum, are beyond the limits of light penetration in the brain, and are thus not amenable to in vivo imaging with conventional light microscopy methods. To study microglial motion in a PD-relevant context, we prepared acute brain slices containing the substantia nigra from CX3CR1GFP/+ mice that exhibit microglia-specific GFP expression in the brain [Figure 1A; Jung et al. (2000)]. The location of the substantia nigra was straightforward to identify in the slices from the high proportion of microglia in this brain region (Lawson et al., 1990) when the slices were examined under GFP excitation (Figure 1B). Despite any microglial activation that might be caused by the slicing procedure itself, microglia in slices extend their processes following localized or bath application of ATP (Avignone et al., 2008; Gyoneva and Traynelis, 2013), and surround an ablated area following laser-induced tissue damage in a manner similar to resting microglia in vivo (Krabbe et al., 2012).
Figure 1. Induction of tissue damage in SNc-containing acute brain slices.

Coronal slices (200 μm-thick) that contain the substantia nigra were prepared from CX3CR1GFP/+ mice that have microglia-specific GFP expression. A, Approximate outlines of the SNc and SNr in a brain slice generated based on microglial density. B, The SNr is easily identified by the high proportion of GFP-positive microglia. C, To induce tissue damage, a stainless steel rod is lowered 180 μm into the SNc with a micromanipulator at constant velocity. The rod is drawn approximately to scale. Scale bar: 200 μm. D-E, Quantification of tissue damage. Slices prepared from CX3CR1GFP/+ mice were immunostained for NeuN (D) or prepared from TH-GFP mice (E) to determine the approximate size of the damaged area in the SN. The location of the injury is indicated by an arrow (D) or a circle (E). Scale bar: D, 1 mm for inset and 0.25 mm for magnified image. E, 200 μm.
In this study, we modeled the response of microglia to localized damage, representative of the death of only a few dopaminergic neurons, by lowering a thin rod into the SNc with a micromanipulator (Figure 1C). Staining the slices for the neuronal marker NeuN allowed us to visualize the extent of damage the injury was creating, indicating that the damage had an approximate diameter of 100-150 μm (Figure 1D). Preparing slices from TH-GFP animals confirmed that the injury was located within the SNc and had an approximate radius of 63.1 μm and height of 180 μm, resulting in ~2.25×106 μm3 volume (Figure 1E; also see Movie 1 for 3D reconstruction of SN). Based on cell density of the surrounding non-injured SNc (n = 8 slices) and non-injured sham slices (n = 3), we estimate that the rod damaged ~10 TH-positive cells.
We then performed time-lapse confocal imaging to capture the ramified morphology of microglia as well as assess process motion in three dimensions. The slices were imaged for 20 min to record the baseline motility of microglia. Similar to microglia in the cortex in vivo (Davalos et al., 2005; Nimmerjahn et al., 2005), microglia in the midbrain constantly extend and retract their processes (Movie 2). We next induced tissue damage as described above and performed a second imaging session in the same slice to record microglial response to the damage (Movie 3). Unlike the seemingly random motion of microglia before damage induction, microglia in corresponding injury recordings quickly extended their processes in the direction of damage (Movie 3; Figure 2). Moreover, the cell bodies remained mostly stationary over the recording period, which is in agreement to the response of cortical microglia to mechanical or laser damage in vivo (Davalos et al., 2005).
Figure 2. Response of microglia to tissue injury in acute brain slices.

Acute brain slices from CX3CR1GFP/+ were imaged with confocal microscopy over time before and after induction of tissue injury in the same slice. For image analysis, the optical stacks at each time point were converted to 2D maximum intensity projections. A, A portion from a representative slice showing microglial response to the damage immediately after (t = 0 min) and 20 min after the injury. Location of the injury is indicated with a solid white arc. The approximate border to the SNc and SNr is represented with a dashed line. Arrows point to microglial cells with processes that moved in the direction of the injury. The same cells are enlarged in B.
To quantify the response, we used an automated object tracking software that detects objects larger than 2 μm, ~95% of which are microglial processes (see Materials and Methods, Analysis of microglial motility). Plotting all tracks from the same starting point confirmed that the overwhelming majority of objects moved in the direction of the injury (84% with positive displacement along the x axis; Figure 3A). To assess the involvement of ATP signaling through P2Y12 receptors in this response, we included the selective P2Y12 receptor antagonist clopidogrel (Savi et al., 2001) in the perfusion solution for the duration of the experiment (baseline recording, injury, response to injury). Addition of clopidogrel appeared to abolish the directional process extension of microglia to the injury (Figure 3B).
Figure 3. Quantification of microglial response to injury in acute brain slices.

Slices from CX3CR1GFP/+ mice were imaged with confocal microscopy for 20 min before induction of injury and 20 min after injury, and 2D projections of the optical stacks at each time point were used for analysis by tracking moving objects larger than 2 μm in diameter. To confirm the involvement of P2Y12 receptors in the response to injury, the slices were treated with 2 μM of the selective P2Y12 receptor antagonist clopidogrel or DMSO vehicle. A, The image shows all tracks detected with Imaris for a baseline (left) and an injury (right) recording from a representative DMSO-treated slice; both panels are on the same scale. B, All tracks for a baseline (left) and an injury (right) recording from a representative clopidogrel-treated slice. The tracks are color-coded according to their duration; tracks with longer duration move in the direction of the injury. Scale bar: 5 μm. C-E, The average displacement of all moving objects (C), the average velocity of movement (D), and the fraction of tracks with longer than 5 μm displacement (E) were calculated for baseline (BL) recordings before injury and after the induction of the injury in the same slice. Numbers of slices for each condition are shown in parentheses. Statistics: two-way repeated measures ANOVA and Tukey’s post hoc test. *, p < 0.05.
We quantified the extent of the response in three different ways: the overall object displacement in the direction of the injury (Figure 3C), the instantaneous velocity for the duration of the imaging (Figure 3D), and the fraction of objects that had displacements larger than 5 μm (in any direction; Figure 3E). All three measures were significantly increased in the recordings following injury (two-way repeated measures ANOVA and Tukey’s test comparing baseline to injury: p < 0.05 for displacement, velocity, fraction; 7 DMSO-treated slices). Importantly, clopidogrel treatment prevented microglial process extension to the site of damage (two-way repeated measures ANOVA and Tukey’s test comparing injury with and without clopidogrel: p < 0.05 for displacement, velocity, fraction; 7 DMSO- and 6 clopidogrel-treated slices; Figure 3). These data show that microglia in acute brain slices use the ATP/P2Y12 receptor pathway to respond to tissue damage, which is the same pathway that microglia in vivo employ (Davalos et al., 2005; Haynes et al., 2006). These data also show that acute brain slices faithfully reproduce the ability of microglia to respond to tissue damage.
Activated microglia have a reduced capacity to respond to tissue damage
We have previously shown that activated microglia respond to ATP with process retraction in vitro, which is in contrast to the process extension displayed by resting microglia (Orr et al., 2009). To determine if this occurs in tissues and affects the ability of microglia to respond to damage, we induced general microglial activation by injecting CX3CR1GFP/+ mice with the bacterial endotoxin LPS at a dose (2 mg/kg i.p.) that is known to induce neuroinflammation (Orr et al., 2009). We subsequently prepared slices two days later. To confirm microglial activation in the brain, we isolated mRNA from brain slices at different time points after the slicing procedure. There was no visible change in expression in the pro-inflammatory marker IL-1β in slices prepared from PBS-treated (control) mice, but clear induction in IL-1β transcription following LPS treatment (Figure 4A). Microglia in slices from mice treated with PBS responded to mechanical damage with significantly increased displacement and velocity in the direction of injury, and a significantly increased fraction of tracks with displacement over 5 μm (two-way repeated measures ANOVA and Tukey’s test comparing baseline to injury: p < 0.05 for displacement, velocity, fraction; 9 PBS-treated slices; Figure 4B-D). LPS-induced microglial activation did not affect the baseline motility of microglia in terms of the three parameters analyzed (two-way repeated measures ANOVA and Tukey’s test comparing slices from PBS- and LPS-injected mice: displacement: p = 0.635; velocity: p = 0.823; fraction: p = 0.678; 9 PBS- and 10 LPS-treated slices; Figure 4B-D). However, in slices prepared from LPS-treated mice, microglia had a significantly reduced response to the injury (two-way repeated measures ANOVA and Tukey’s test comparing slices from PBS- and LPS-injected mice: displacement: p < 0.05 for displacement, velocity, fraction; 9 PBS- and 10 LPS-treated slices; Figure 4B-D). Thus, activated microglia in tissue appear to have an impaired ability to sense ATP released at the site of damage and/or extend their processes to the damaged area.
Figure 4. Microglial response to tissue damage in slices from LPS-treated animals.

CX3CR1GFP/+ mice were injected with 2 mg/kg i.p. LPS or PBS, and slices were prepared 2 days later. A, Microglial activation was assessed by measuring IL-1β mRNA expression with RT-PCR with RNA extracted from slices at different time points after cutting. The cutting procedure does not induce IL-1β expression, but LPS injection 48 hr prior to slice preparation increases IL-1β expression. B-D, Quantification of microglial motility in slices from LPS-treated mice by tracking objects in time lapse recordings. The average displacement of all moving objects (B), the mean instantaneous velocity (C), and the fraction of tracks with longer than 5 μm displacement (D) were calculated for baseline (BL) recordings before injury and after the induction of the injury in the same slice. Numbers of slices for each condition are shown in parentheses. Statistics: two-way repeated measures ANOVA and Tukey’s post hoc test. *, p < 0.05.
MPTP treatment impairs microglial response to tissue damage
The inability of LPS-activated microglia to respond in the normal fashion to tissue damage raised the question of how the activated microglia found in the brains of PD patients will react to the ongoing cell death that is part of the disease. In an attempt to address this question, we examined microglial behavior in the substantia nigra of slices from MPTP-treated mice. We injected CX3CR1GFP/+ mice with 20 mg/kg MPTP s.c. once a day for five days, a treatment regimen that is similar to the one that induces a relatively slow and progressive loss of dopaminergic neurons over at least 25 days (Seniuk et al., 1990; Tatton and Kish, 1997). We wanted to examine microglial motility at earlier stages of MPTP-induced toxicity, before all dopaminergic SN neurons are lost. Even at 4-6 days after MPTP treatment (Jackson-Lewis and Przedborski, 2007), we could detect loss of TH-positive dopaminergic neurons in the SN and their terminals in the striatum (Figure 5A). This was confirmed by measuring TH immunoreactivity in the striatum, which is significantly decreased following MPTP treatment (Student’s t test, p = 0.001; 3 saline- and 4 MPTP-treated mice; Figure 5B). However, detectable TH immunoreactivity remained in the SN, indicating the presence of viable neurons in the SN. Following MPTP injection, microglia displayed an activated morphology in both the SN and striatum (Figure 5C). Quantification of Iba1 staining in the SN revealed cells with larger cell bodies (Student’s t test, p = 0.0149; 3 saline- and 4 MPTP-treated mice; Figure 5D) and thicker processes, which led to significantly increased area occupied by Iba1 immunoreactivity (Student’s t test, p = 0.0112; 3 saline- and 4 MPTP-treated mice; Figure 5E). Scoring of microglial morphology on a scale of 1 (resting) to 4 (amoeboid) confirmed that MPTP treatment had a significant effect on the distribution of cells within the different morphological subtypes [two-way ANOVA, F(3,20) = 95.64, p < 0.0001; 3 saline- and 4 MPTP-treated mice; Figure 5F].
Figure 5. Characterization of MPTP treatment in CX3CR1GFP/+ mice.

CX3CR1GFP/+ mice were treated with 20 mg/kg s.c. MPTP or saline once daily for 5 days, and brains were isolated for immunohistochemistry 5 days after the final MPTP injection. A, Staining of dopaminergic neurons with anti-TH antibody shows decreased immunoreactivity in the SN and striatum after MPTP treatment. Scale bar: 200 μm. B, Quantification of TH immunoreactivity in the striatum. The average absorbance of the TH signal, visualized with DAB staining, is significantly decreased following MPTP treatment. Statistics: two-tailed Student’s t test comparing average signals for each animal. *, p < 0.05. C, Microglia, identified with anti-Iba1 antibody, displayed activated phenotype in the SN and striatum after MPTP treatment. Approximate borders between different brain regions are shown as dashed lines. Scale bar: 50 μm. D-F, Quantification of changes in microglial morphology shows a shift of microglia to a more activated phenotype. The size of microglial cell bodies was determined from thresholded images that displayed only the cell body (D). The area occupied by Iba1 immunoreactivity in the substantia nigra was calculated as the fraction of the image that was occuplied by Iba1 immunoreactivity above a pre-determined threshold (E). To quantify overall morphology, individual cells were assigned phenotypes ranging from resting (score 1) to amoeboid (score 4). The percentage of cells for each morphological phenotype was calculated from all cells examined (F). Figure shows representative slices from 3 saline- and 4 MPTP-treated mice. Statistics: two-tailed Student’s t test comparing average signals for each animal. *, p < 0.05.
After confirming microglial activation in the SN of MPTP-treated mice, we tested their ability to respond to mechanically induced tissue injury designed to damage only a few neurons. We performed time-lapse recordings with slices from saline- or MPTP-treated CX3CR1GFP/+ mice before and after injury (Movies 4 and 5, respectively), and quantified microglial responses in terms of displacement toward the site of damage (Figure 6A), instantaneous velocity (Figure 6B), and fraction of objects with displacements longer than 5 μm (Figure 6C). All three measures were significantly increased after damage in saline-treated mice (two-way repeated measures ANOVA and Tukey’s test comparing baseline to injury: p < 0.05 for displacement, velocity, fraction; 11 saline-treated slices; Figure 6). Microglial activation by MPTP did not detectably affect the baseline dynamics (short extensions and retractions) of microglia (two-way repeated measures ANOVA and Tukey’s test comparing slices from saline- and MPTP-injected mice: displacement: p = 0.853; velocity: p = 0.897; fraction: p = 0.309; 11 saline- and 9 MPTP-treated slices; Figure 6; Movie 4). However, the displacement and velocity of microglial processes in the direction of injury were significantly reduced in slices from MPTP-treated mice (two-way repeated measures ANOVA and Tukey’s test comparing slices from saline- and MPTP-injected mice: p < 0.05 for displacement and velocity; 11 saline- and 9 MPTP-treated slices; Figure 6A, B; Movie 5). The fraction of tracks with displacements longer than 5 μm was not affected by MPTP treatment (two-way repeated measures ANOVA and Tukey’s test comparing slices from saline- and MPTP-injected mice, p = 0.197; 11 saline- and 9 MPTP-treated slices, Figure 6C), implying that some processes moved over longer distances, but this motion was likely random and not in the direction of the injury. These findings indicate that nigral microglia in MPTP-treated mice did not extend processes toward the region of damage, consistent with activated microglia in LPS-treated animals (Figure 4).
Figure 6. Microglial response to tissue damage in slices from MPTP-treated animals.

CX3CR1GFP/+ mice were injected with 20 mg/kg s.c. MPTP or saline once daily for 5 days, and slices were prepared for imaging 4-7 days later. 2D projections of the optical stacks at each time point were used for analysis by tracking moving objects larger than 2 μm in diameter. The average displacement of all moving objects (A), the mean instantaneous velocity (B), and the fraction of tracks with longer than 5 μm displacement (C) were calculated for baseline (BL) recordings before injury and after the induction of the injury in the same slice. Numbers of slices for each condition are shown in parentheses. Statistics: two-way repeated measures ANOVA and Tukey’s post hoc test. *, p < 0.05.
Antagonism of adenosine A2A receptors restores microglial responses to tissue injury
Our previous findings indicate that adenosine A2A receptors are upregulated in LPS-activated microglia and mediate microglial responses to ATP. Specifically, adenosine, generated from ATP breakdown, induces process retraction and migration away from the ATP source in primary cultured cortical microglia (Orr et al., 2009). Here, we examined whether adenosine A2A receptors mediate the inability of nigral microglia in brain slices to respond to tissue damage. We first determined if A2A receptors are expressed in MPTP-treated mice by performing immunohistochemistry using both DAB and fluorescent (with a Texas Red-conjugated secondary antibody) detection methods. In saline-treated mice, much of the A2A receptor immunoreactivity was found both as diffuse neuropil staining as well as in long, thin structures that resembled blood vessels (Figure 7B). Following MPTP treatment, we detected apparent increase in A2A receptor expression in the form of diffuse staining that was more pronounced in the substantia nigra pars reticulata (SNr) compared to surrounding brain regions (Figure 7A, B). The average Texas Red fluorescence (representing A2A receptor expression) in the SNr was significantly higher in MPTP-treated mice (Student’s t test, p = 0.0208; 3 saline- and 4 MPTP-treated mice; Figure 7C), indicating that MPTP treatment induced A2A receptor upregulation. Yet, even after upregulation in the SNr, the signal remained lower than the signal in the striatum, which shows constitutive A2A receptor expression (Figure 7D, E). While our analysis shows a significant change in immunoreactivity, the low level of expression in the SNr compared to the striatum should be considered a caveat for this finding.
Figure 7. Changes in adenosine A2A receptor expression in MPTP-treated mice.

CX3CR1GFP/+ mice were treated with 20 mg/kg s.c. MPTP or saline once daily for 5 days, and brains were isolated for analysis 5 days later. A, Adenosine A2A receptor expression in the substantia nigra was detected with immunofluorescence. Microglia were identified without staining from the expression of GFP from the CX3CR1 promoter. Approximate borders between SNc and SNr are given with dashed lines based on microglial cell density. Scale bar: 30 μm. B, Adenosine A2A receptor expression in the substantia nigra as detected with DAB precipitation. Scale bar: 200 μm. C, Upregulation of A2A receptor expression in the SNr following MPTP treatment was quantified by measuring the average Texas Red fluorescence signal. D, For comparison, the A2A receptor signal in the striatum is shown. Even after upregulation in the SN, A2A receptor is not expressed at the levels of the striatum. Scale bar: 30 μm. E, Quantification of A2A receptor expression in the striatum following MPTP treatment by measuring the average Texas Red fluorescence signal. Statistics: two-tailed Student’s t test. *, p < 0.05.
Because of the apparent presence of A2A receptor immunoreactivity in PD-relevant brain regions (SN and straitum), we examined the ability of A2A receptor antagonists to modulate microglial response to tissue damage in slices from MPTP-treated mice. In order to do this, we included the selective A2A receptor antagonist preladenant (5 μM) in the perfusion solution for the duration of the experiment, before and after induction of mechanical damage. The inclusion of the antagonist during the baseline recording did not significantly affect the baseline extensions and retractions of microglial processes in the absence of injury (two-way repeated measures ANOVA and Tukey’s test comparing slices from MPTP-injected mice before and after preladenant: displacement: p = 0.451; velocity: p = 0.495; fraction: p = 0.009; 5 DMSO- and 8 preladenant-treated slices; Figure 8). This is consistent with the lack of difference in baseline motility of microglia induced by MPTP activation (Figure 6). Yet, microglia in slices from MPTP-treated mice that were perfused with preladenant showed significant increases in their displacement toward injury, velocity, and fraction of tracks with displacement longer than 5 μm (two-way repeated measures ANOVA and Tukey’s test comparing DMSO and preladenant: p < 0.05 for displacement, velocity, fraction; 5 DMSO- and 8 preladenant-treated slices; Figure 8). That is, preladenant restored process motility in MPTP-treated animals near to the level seen in control animals.
Figure 8. Effect of A2A receptor antagonism on microglial response to tissue damage in slices from MPTP-treated animals.
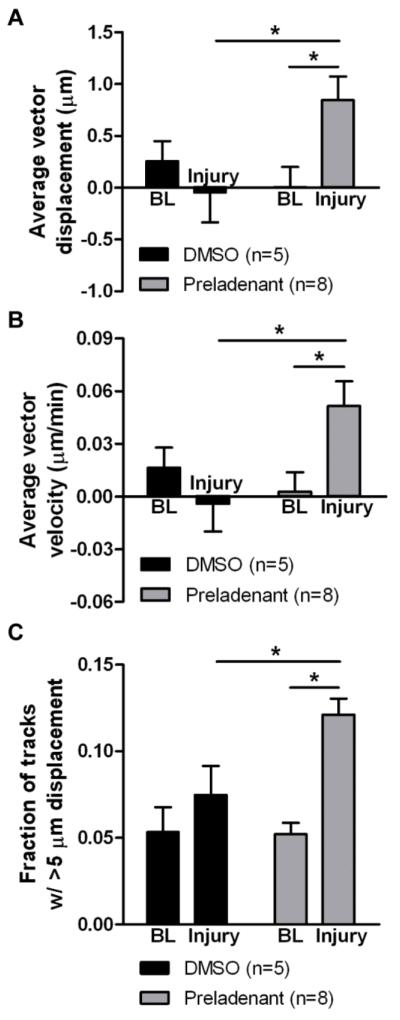
CX3CR1GFP/+ mice were injected with 20 mg/kg s.c. MPTP once daily for 5 days, and slices were prepared for imaging 4-7 days later. The perfusion solution contained either the selective A2A receptor antagonist preladenant (5 μM) or DMSO vehicle. 2D projections of the optical stacks at each time point were used for analysis by tracking moving objects larger than 2 μm in diameter. The average displacement of all moving objects (A), the mean instantaneous velocity (B), and the fraction of tracks with longer than 5 μm displacement (C) were calculated for baseline (BL) recordings before injury and after the induction of the injury in the same slice. Numbers of slices for each condition are shown in parentheses. Statistics: two-way repeated measures ANOVA and Tukey’s post hoc test. *, p < 0.05.
Discussion
The goal of this study was to examine the ability of activated microglia to respond to tissue disturbances under pro-inflammatory conditions that model aspects of PD pathogenesis. Using either LPS or MPTP to induce neuroinflammation, we show that activated microglia in the SN display reduced process extension to the site of localized damage. Importantly, antagonism of adenosine A2A receptors restored the ability of activated microglia to respond to tissue damage in the MPTP model of PD. The implications of these findings are discussed below.
Activated microglia in Parkinson’s disease
Even though the presence of activated microglia in PD brains is supported by several lines of evidence (see Introduction), the mechanism by which microglia might become activated is not clear. In general, there are three main pathways that can result in microglial activation. First, the sudden appearance of pathogen-associated molecular patterns or intracellular constituents (ATP, nucleic acids, etc.) commonly induce microglial conversion to a phagocytic phenotype in an attempt to clear the parenchyma of abnormal particles (Hanisch, 2002; Kreutzberg, 1996). Second, certain cell-cell contacts between neurons and microglia (such as the CD200 receptor-ligand pair) serve as “calming signals” that keep microglial reactivity low; the removal of these signals, for example as a result of neuronal death, disinhibits microglia, facilitating the adoption of a more reactive state (Hanisch, 2002). Both of these mechanisms could be involved in PD through the death of dopaminergic neurons and release of cellular constituents that are not efficiently cleared by microglia. Third, the inflammation following systemic infections could in some cases lead to neuroinflammation and microglial activation [for reviews, see Cunningham (2013); Perry et al. (2007); Perry et al. (2003)]. Once microglia achieve an activated state, they secrete pro-inflammatory factors (such as cytokines and reactive oxygen species) that can damage neurons through multiple pathways (Block and Hong, 2005; Block et al., 2007; Tansey and Goldberg, 2010). The subsequent neuronal death can further activate microglia, leading to a self-perpetuating cycle of microglial activation and cell death. In this way, microglia could contribute to the progression of neurodegenerative diseases, including PD (Block et al., 2007; Gao and Hong, 2008; Long-Smith et al., 2009; Tansey and Goldberg, 2010). Once initiated, this pro-inflammatory environment likely affects various microglial functions, including motility.
Here we show that activated microglia have a reduced capacity to respond to tissue damage in the substantia nigra of acute brain slices using a model of both a direct (LPS) and indirect (MPTP) activation of microglia (Figure 4, 6). While the injury paradigm we used is non-physiological, it allowed us to injure a small, defined area of the brain at a specific time and record the subsequent microglial response to the death of only a few neurons in real time (Figure 1). The containment of the damaged area by microglial processes is thought to prevent the spread of damage and promote tissue healing. Delayed containment is associated with expansion of the injury site (Hines et al., 2009). While these findings were observed in healthy mice, a similar process might be occurring in mice undergoing active cell death in the SNc. The delayed response of activated microglia to tissue damage or cell death could prevent the efficient clearance of tissue debris, resulting in leakage of debris into the surrounding brain parenchyma and induction of detrimental signaling pathways in nearby cells that could impair their function. Therefore, our findings suggest that altered motility of activated microglia in the MPTP model of PD might represent yet another mechanism by which microglia contribute to neurodegeneration in mice and humans.
Neuroprotective properties of A2A receptor antagonists
There is a large body of evidence suggesting that inhibiting adenosine A2A receptors is beneficial in Parkinson’s disease. Epidemiological studies show an inverse association between the consumption of caffeine, a non-selective adenosine receptor antagonist, and the risk for developing PD (Ascherio et al., 2001; Ross et al., 2000). Mouse models of PD confirm the human findings, and indicate that adenosine A2A receptors mediate the neuroprotective effects of caffeine (Xiao et al., 2006; Yu et al., 2008). Finally, selective A2A receptor antagonists mimic the effects of caffeine (Chen et al., 2001; Morelli et al., 2010). As a result, several selective A2A receptor antagonists have entered clinical trials for PD, including preladenant, which has been used here (Barkhoudarian and Schwarzschild, 2011; Hauser et al., 2011; Schwarzschild et al., 2006).
Adenosine A2A receptors are expressed on striatal neurons where they appear to oppose the effects of dopamine D2 receptor activation (Ferre et al., 1993). Thus, the majority of the neuroprotective properties of A2A receptor antagonists have been attributed to actions in neuronal receptors (Carta et al., 2009; Xiao et al., 2006). While it is likely that the ability of A2A receptors to affect motor symptoms of PD occurs through actions on neuronal receptors, it remains an open question as to whether block of glial A2A receptors might mediate some of the neuroprotective effects [e.g., neuronal health; Yu et al. (2008)]. A2A receptors are highly expressed on glial cells in the substantia nigra of healthy monkeys, and to a lower extent in the SN of healthy rats (Bogenpohl et al., 2012). Moreover, A2A receptors were upregulated in the substantia nigra following treatment with 20 mg/kg/day MPTP for 5 days in CX3CR1GFP/+ mice (Figure 7). A2A receptor upregulation has been reported before in the striatum and the SN of MPTP-treated mice (Boison et al., 2010; Singh et al., 2009), and some of this upregulation is in glial cells (Boison et al., 2010).
We have previously shown that activation with LPS increases A2A receptor expression in primary mouse microglia (Orr et al., 2009). However, based on immunohistochemistry analysis, we were unable to conclusively localize A2A receptors to microglia cell bodies in the substantia nigra of MPTP-treated mice (Figure 7). Instead, in addition to possible expression in fine microglial processes, A2A receptors may also be expressed in other cell types, such as astrocytes and/or neurons. The different pattern of A2A receptor activation seen in isolated cells and in tissues could be explained by the different type of stimulus used (LPS vs. MPTP) as well as the region from which the microglia are derived. Yet, the selective A2A receptor antagonist preladenant was able to restore microglial response to tissue damage in the MPTP model of PD (Figure 8). Although diffuse microglial expression of A2A may mediate the effects of preladenant, it is plausible that A2A receptor expression on other cells within the substantia nigra, for example astrocytes, causes the release of substances that can then modulate microglial motility. The exact cellular localization of A2A receptors following MPTP treatment and the mechanism of action of preladenant in modulating microglial motility in slices from MPTP-treated mice need further investigation.
Possibility for differential populations of microglia in PD
The neuronal populations most affected in PD are midbrain neurons, specifically dopaminergic neurons projecting from the substantia nigra to the striatum [Figure 5; Bernheimer et al. (1973); Kish et al. (1988)]. This is closely mirrored by the state of microglial activation. Microglia in the midbrain/substantia nigra display higher degree of activation in both patients with PD [in post-mortem samples and assessed with PET imaging; Gerhard et al. (2006)], and in animal models of the disease (Tansey and Goldberg, 2010). In addition to the SN, microglia in the striatum also display signs of activation in animal models of PD [Figure 5; Tansey and Goldberg (2010)]. Moreover, striatal microglia have the capacity to influence dopaminergic neuronal survival through their actions on damaged or degenerating terminals. In contrast to microglia in the SN and the striatum, microglia in the cortex did not display signs of overt activation following MPTP treatment (Figure 5). Thus, it is possible that microglial populations in distinct brain regions could have unique functional properties in PD, including motility patterns. Elucidating the specific differences between these microglia could lead to important insights into PD onset and progression.
Conclusions
We employed acute brain slices from MPTP-treated mice to study microglial response to tissue damage under pro-inflammatory conditions. Microglia in MPTP-treated slices had reduced process extension to a site of tissue damage, suggesting that they might not be as efficient in responding to and containing the damage as microglia in healthy mice. The A2A receptor antagonist preladenant restored the ability of microglia in slices from MPTP-treated mice to extend processes in the direction of the injury, reversing the effects of MPTP. Thus, our findings suggest that at least part of the neuroprotective properties of A2A receptor antagonists might be explained by modulation of microglial motility in response to cell death, but further work is needed to characterize these direct or indirect glial effects of preladenant.
Supplementary Material
Highlights.
Confocal imaging of brain slices allows the study of microglial motion in tissues
Microglia in the MPTP model of Parkinson’s disease show signs of activation
Activated microglia display a delayed response to tissue damage in brain slices
Adenosine A2A receptor antagonism reverses the activation-induced delay in response
Acknowledgements
Funding was provided by NINDS NRSA (F31NS076215, S.G.), NIEHS Toxicology institutional training grant (T32ES12870, S.G.), NIH Pharmacological Sciences institutional training grant (T32GM008602, S.G.), a pilot grant from the Emory University Udall Center for Parkinson’s Disease Research (NIH/NINDS P50-NS071669, S.F.T), the Alzheimer’s Disease Research Center (NIHP50 AG025688, S.F.T.), the Yerkes Primate Center NIH base grant (RR00165, Y.S.), and the Emory University Integrated Cellular Imaging Microscopy Core. Thanks are also due to Susan Jenkins for technical assistance. Finally, we thank Dr. Malu Tansey for helpful discussions and suggestions while preparing the manuscript and Dr. David Weinshenker for providing TH-GFP mice to us.
Footnotes
Abbreviations: aCSF, artificial cerebrospinal fluid; ANOVA, analysis of variance; BSA, bovine serum albumin; CX3CR1, CX3C-type chemokine receptor 1; GFP, enhanced green fluorescent protein; IL, interleukin; i.p., intraperitoneal; s.c., subcutaneous; LPS, lipopolysaccharide; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NSAIDs, non-steroidal anti-inflammatory drugs; PD, Parkinson’s disease; PET, positron emission tomography; PFA, paraformaldehyde; RT-PCR, reverse transcriptase polymerase chain reaction; SN(c/r), substantia nigra (pars compacta/pars reticulata); TH, tyrosine hydroxylase; TNF, tumor necrosis factor; DAB, 3,3′-diaminobenzidine.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no competing financial interests.
References
- Ascherio A, et al. Prospective Study of Caffeine Consumption and Risk of Parkinson’s Disease in Men and Women. Annals of Neurology. 2001;50:56–63. doi: 10.1002/ana.1052. [DOI] [PubMed] [Google Scholar]
- Avignone E, et al. Status Epilepticus Induces a Particular Microglial Activation State Characterized by Enhanced Purinergic Signaling. Journal of Neuroscience. 2008;28:9133–9144. doi: 10.1523/JNEUROSCI.1820-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcia C, et al. Evidence of Active Microglia in Substantia Nigra Pars Compacta of Parkinsonian Monkeys 1 Year After MPTP Exposure. Glia. 2004;46:402–409. doi: 10.1002/glia.20015. [DOI] [PubMed] [Google Scholar]
- Barkhoudarian MT, Schwarzschild MA. Preclinical jockeying in the translational track of adenosine A2A receptors. Experimental Neurology. 2011;228:160–164. doi: 10.1016/j.expneurol.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheimer H, et al. Brain Dopamine and the Syndromes of Parkinson and Huntington: Clinical, Morphological and Neurochemical Correlations. Journal of the Neurological Sciences. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong J-S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Progress in Neurobiology. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, et al. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature Reviews. Neuroscience. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bogenpohl JW, et al. Adenosine A2A Receptor in the Monkey Basal Ganglia: Ultrastructural Localization and Colocalization With the Metabotropic Glutamate Receptor 5 in the Striatum. Journal of Comparative Neurology. 2012;520:570–589. doi: 10.1002/cne.22751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, et al. Adenosine signaling and function in glial cells. Cell Death and Differentiation. 2010;17:1071–1082. doi: 10.1038/cdd.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta AR, et al. Inactivation of neuronal forebrain A2A receptors protects dopaminergic neurons in a mouse model of Parkinson’s disease. Journal of Neurochemistry. 2009;111:1478–1489. doi: 10.1111/j.1471-4159.2009.06425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J-F, et al. Neuroprotection by Caffeine and A2A Adenosine Receptor Inactivation in a Model of Parkinson’s Disease. The Journal of Neuroscience. 2001;21:RC143. doi: 10.1523/JNEUROSCI.21-10-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C. Microglia and Neurodegeneration: The Role of Systemic Inflammation. Glia. 2013;61:71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- Davalos D, et al. ATP mediates rapid mecroglial response to local brain injury in vivo. Nature Neuroscience. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Ferre S, et al. The Striatopallidal Neuron: A Main Locus for Adenosine-Dopamine Interactions in the Brain. Journal of Neuroscience. 1993;13:5402–5406. doi: 10.1523/JNEUROSCI.13-12-05402.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagne JJ, Power MC. Anti-inflammatory drugs and risk of Parkinson disease: A meta-analysis. Neurology. 2010;74:995–1002. doi: 10.1212/WNL.0b013e3181d5a4a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H-M, Hong J-S. Why neurodegenerative diseases are progressice: uncontrolled inflammation drives disease progression. Trends in Immunology. 2008;29:357–365. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, et al. Use of ibuprofen and risk of Parkinson disease. Neurology. 2011;76:863–869. doi: 10.1212/WNL.0b013e31820f2d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiology of Disease. 2006;21:404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Gyoneva S, Traynelis SF. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. Journal of Biological Chemistry. 2013 doi: 10.1074/jbc.M113.458901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch U-K. Microglia as a Source and Target of Cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Hauser RA, et al. Preladenant in patients with Parkinson’s disease and motor fluctuations: a phase 2, double blind, randomised trial. Lancet Neurology. 2011;10:221–229. doi: 10.1016/S1474-4422(11)70012-6. [DOI] [PubMed] [Google Scholar]
- Haynes SE, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nature Neuroscience. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Hines DJ, et al. Microglia Processes Block the Spread of Damage in the Brain and Require Functional Chloride Channels. Glia. 2009;57:1610–1618. doi: 10.1002/glia.20874. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Przedborski S. Protocol for the MPTP mouse model of Parkinson’s disease. Nature Protocols. 2007;2:141–151. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- Jung S, et al. Analysis of Fractalkine Receptor CX3CR1 Function by Targeted Deletion and Green Fluorescent Protein Reporter Gene Insertion. Molecular and Cellular Biology. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, et al. Age and Region-Specific Responses of Microglia, but not Astrocytes, Suggest a Role in Selective Vulnerability of Dopamine Neurons After 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine Exposure in Monkeys. Glia. 2008;56:1199–1214. doi: 10.1002/glia.20690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, et al. Microglia: New Roles for the Synaptic Stripper. Neuron. 2013;77:10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- Kish SJ, et al. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. New England Journal of Medicine. 1988;318:876–880. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- Koizumi S, et al. Purinergic Receptors in Microglia: Functional Modal Schifts of Microglia Mediated by P2 and P1 Receptors. Glia. 2013;61:47–54. doi: 10.1002/glia.22358. [DOI] [PubMed] [Google Scholar]
- Krabbe G, et al. Activation of serotonin receptors promotes microglial injury-induced motility but attenuates phagocytic activity. Brain Behav. Immun. 2012;26:419–428. doi: 10.1016/j.bbi.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends in Neurosciences. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Langston J, et al. Evidence of Active Nerve Cell Degeneration in the Substantia Nigra of Humans Years after 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Exposure. Annals of Neurology. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Lawson L, et al. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Long-Smith CM, et al. The influence of microglia on the pathogenesis of Parkinson’s disease. Progress in Neurobiology. 2009;89:277–287. doi: 10.1016/j.pneurobio.2009.08.001. [DOI] [PubMed] [Google Scholar]
- McGeer P, et al. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- McGeer PL, et al. Presence of Reactive Microglia in Monkey Substantia Nigra Years after 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Administration. Annals of Neurology. 2003;54:599–604. doi: 10.1002/ana.10728. [DOI] [PubMed] [Google Scholar]
- Mogi M, et al. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neuroscience Letters. 1994a;180:147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- Mogi M, et al. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neuroscience Letters. 1996;211:13–16. doi: 10.1016/0304-3940(96)12706-3. [DOI] [PubMed] [Google Scholar]
- Mogi M, et al. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neuroscience Letters. 1994b;165:208–210. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- Morelli M, et al. Pathophysiological roles for purines: adenosine, caffeine and urate. Progress in Brain Research. 2010;183:183–208. doi: 10.1016/S0079-6123(10)83010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neustadt BR, et al. Potent, selective, and orally active adenosine A2A receptor antagonists: Arylpiperazine derivatives of pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines. Bioorganic & Medicinal Chemistry Letters. 2007;17:1376–1380. doi: 10.1016/j.bmcl.2006.11.083. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, et al. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Orr AG, et al. Adenosine A2A receptor mediates microglial process retraction. Nature Neuroscience. 2009;12:872–878. doi: 10.1038/nn.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, et al. Systemic infections and inflamamtion affect chronic neurodegeneration. Nature Reviews. Immunology. 2007;7:161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- Perry VH, et al. The impact of systemic infection on the progression of neurodegenerative disease. Nature Reviews. Neuroscience. 2003;4:103–112. doi: 10.1038/nrn1032. [DOI] [PubMed] [Google Scholar]
- Rinne J. Nigral degeneration in Parkinson’s disease in relation to clinical features. Acta Neurol Scand. 1991;84:87–90. doi: 10.1111/j.1600-0404.1991.tb05026.x. [DOI] [PubMed] [Google Scholar]
- Ross GW, et al. Association of Coffee and Caffeine Intake with the Risk of Parkinson Disease. JAMA. 2000;283:2674–2679. doi: 10.1001/jama.283.20.2674. [DOI] [PubMed] [Google Scholar]
- Savi P, et al. P2Y12, a New Platelet ADP Receptor, Target of Clopidogrel. Biochemical and Biophysical Research Communications. 2001;283:379–383. doi: 10.1006/bbrc.2001.4816. [DOI] [PubMed] [Google Scholar]
- Schwarzschild MA, et al. Targeting adenosine A2A receptors in Parkinson’s disease. Trends in Neurosciences. 2006;29:647–654. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Seniuk N, et al. Dose-dependent destruction of the coeruleus-cortical and nigral-striatal projections by MPTP. Brain Research. 1990;527:7–20. doi: 10.1016/0006-8993(90)91055-l. [DOI] [PubMed] [Google Scholar]
- Singh S, et al. Effect of caffeine on the expression of cytochrome P450 1A2, adenosine A2A receptor and dopamine transporter in control and 1-methyl 4-phenyl 1, 2, 3, 6-tetrahydropyridine treated mouse striatum. Brain Research. 2009;1283:115–126. doi: 10.1016/j.brainres.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiology of Disease. 2010;37:510–518. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton N, Kish S. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- Walker FR, et al. Dynamic structural remodelling of microglia in health and disease: A review of the models, signals and the mechanisms. Brain, Behavior, and Immunity. 2014 doi: 10.1016/j.bbi.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Xiao D, et al. Forebrain Adenosine A2A Receptors Contribute to L-3,4- Dihyrdoxyphenylalanine-Induced Dyskinesia in Hemiparkinsonian Mice. Journal of Neuroscience. 2006;26:13548–13555. doi: 10.1523/JNEUROSCI.3554-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, et al. Adenosine A2A Receptor Antagonists Exert Motor and Neuroprotective Effects by Distinct Cellular Mechanisms. Annals of Neurology. 2008;63:338–346. doi: 10.1002/ana.21313. [DOI] [PubMed] [Google Scholar]
- Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn- Schmiedeberg’s Archives of Pharmacology. 2000;362:299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.