

p66Shc, but not p52Shc or p46Shc, is regulated by the Nrf2-ARE system, indicative of p66Shc as an antioxidant gene. p66Shc serves as an antioxidant protein in the cytoplasm, protecting cells from ROS toxicity and maintaining expression of other ARE-regulated genes. Finally, p66Shc is essential for erythroid differentiation.
Abstract
The mammalian Shc family, composed of p46, p52, and p66 isoforms, serves as an adaptor protein in cell growth and stress response. p66Shc was shown to be a negative lifespan regulator by acting as a prooxidant protein in mitochondria; however, the regulatory mechanisms of p66Shc expression and function are incompletely understood. This study provides evidence for new features of p66Shc serving as an antioxidant and critical protein in cell differentiation. Unique among the Shc family, transcription of p66Shc is activated through the antioxidant response element (ARE)–nuclear factor erythroid 2–related factor 2 (Nrf2) pathway in K562 human erythroleukemia and other cell types after treatment with hemin, an iron-containing porphyrin. Phosphorylated p66Shc at Ser-36, previously reported to be prone to mitochondrial localization, is increased by hemin treatment, but p66Shc remains exclusively in the cytoplasm. p66Shc knockdown inhibits hemin-induced erythroid differentiation, in which reactive oxygen species production and apoptosis are significantly enhanced in conjunction with suppression of other ARE-dependent antioxidant genes. Conversely, p66Shc overexpression is sufficient for inducing erythroid differentiation. Collectively these results demonstrate the isoform-specific regulation of the Shc gene by the Nrf2-ARE pathway and a new antioxidant role of p66Shc in the cytoplasm. Thus p66Shc is a bifunctional protein involved in cellular oxidative stress response and differentiation.
INTRODUCTION
Reactive oxygen species (ROS) are constantly generated in a wide variety of cellular metabolic pathways and immunological responses (Lander, 1997; Lambeth, 2004; Balaban et al., 2005). Oxidative stress is caused by metabolic imbalance between the formation and detoxification of oxidants and ROS, and those produced in excess of cellular antioxidant capacity cause oxidative stress that is critically associated with various human disorders, including cancer and neurodegeneration. To cope with oxidative stress, cells evolved antioxidant defense systems regulated by several key signaling pathways for detoxification and removal of these oxidants and ROS (Ray et al., 2012). Cells activate transcription of an array of antioxidant detoxification genes under oxidative stress conditions, such as heme oxygenase-1, NAD(P)H quinone oxidoreductase-1 (NQO1), and glutathione S-transferases (GSTs), through nuclear factor erythroid 2–related factor 2 (Nrf2)–dependent antioxidant response element (ARE) activation (Ray et al., 2012; Takaya et al., 2012). Along with these metabolic enzymes, ferritin, the major iron storage multimeric protein, which is composed of H and L subunits (Arosio et al., 2009), is transcriptionally up-regulated by the Nrf2-ARE system (Tsuji et al., 2000; Tsuji, 2005; Hintze and Theil, 2005; MacKenzie et al., 2008b), thereby buffering and storing intracellular free iron in ferritin shells to minimize iron-catalyzed production of hydroxyl radical (MacKenzie et al., 2008a). The regulatory mechanism of Nrf2 in response to oxidative stress has been extensively studied, in which Keap1, a cytoplasmic Nrf2-binding protein and a sensor of electrophiles and oxidative stress, is subject to either oxidation or formation of electrophilic adducts (Zhang and Hannink, 2003; Kobayashi et al., 2004), which allows Nrf2 to dissociate from Keap1, cross the nuclear membrane, and bind ARE enhancer sequences (Itoh et al., 1999; Motohashi and Yamamoto, 2004; Hayes and McMahon, 2009), in balance with the availability of the repressor Bach1 (Sun et al., 2002, 2004).
The mammalian ShcA family consists of three adapter proteins, p66Shc, p52Shc, and p46Shc (Wills and Jones, 2012). Expression of p66Shc and p52Shc/p46Shc isoforms is regulated by an alternative promoter, and p52Shc and p46Shc isoforms are synthesized from alternative translational initiation sites (Ventura et al., 2002; Figure 1A). Therefore p66Shc and p52Shc isoforms have a common C-terminal region equivalent to the full-length p46Shc that contains a phosphotyrosine binding domain, a proline-rich collagen homology (CH) domain-1, and a Src homology 2 domain (Migliaccio et al., 2006; Gertz and Steegborn, 2010). In addition to these common domains, both p52Shc and p66Shc isoforms have extended and overlapping N-terminal regions containing a cytochrome c binding domain and a second CH domain (CH2) that is unique to p66Shc (Migliaccio et al., 2006; Gertz and Steegborn, 2010), allowing p66Shc to gain distinct functions from p52Shc and p46Shc. p66Shc was reported to be a negative lifespan regulatory protein when localized in mitochondria (Migliaccio et al., 1999), and p52Shc and p46Shc are adaptor proteins in receptor tyrosine kinase signaling pathways (Pelicci et al., 1992). p66Shc-knockout mice showed resistance to apoptosis induced by such stressors as ultraviolet (UV) light and H2O2 and exhibited ∼30% extension in lifespan (Migliaccio et al., 1999). This was explained by less production of ROS in p66Shc-deficient mice, suggesting that p66Shc is a prooxidant protein and may play a detrimental role in aging-related diseases. p66Shc phosphorylation at Ser-36 in the CH2 domain by protein kinase Cβ (Pinton et al., 2007) or JNK (Le et al., 2001) has been reported to play a prominent role in translocation and/or prooxidant function of p66Shc in mitochondria, where it oxidizes cytochrome c to form H2O2 (Giorgio et al., 2005). Conversely, it was demonstrated that p66Shc also exhibits antiapoptotic effects through protection from hypoxia-induced cytotoxicity in human breast cancer and stem cell models (Sansone et al., 2007). In addition, it was recently reported that reevaluation of the p66Shc function using a larger number of p66Shc-knockout mice showed no significant difference in longevity between wild-type and p66Shc-knockout mice, leading to the conclusion that p66Shc is not a longevity protein (Ramsey et al., 2013). Furthermore, p66Shc-knockout mice are short lived under such natural conditions as a large outdoor enclosure, food competition, and natural temperatures (Giorgio et al., 2012), suggesting that p66Shc may have antioxidant activity in some stress conditions.
FIGURE 1:

Hemin induces p66Shc gene expression in K562 cells. (A) Schematic representation of the human ShcA gene. A close-up view of the −300–base pair p66Shc promoter region is shown in Figure 2B. (B) K562 cells were treated with 0, 10, 20, and 40 μM hemin for 24 h, and whole-cell lysates were used for Western blot with a Shc family antibody. Long and short exposures of the blot are shown along with the GAPDH Western as a loading control. (C) Time course of 20 μM hemin treatment for p66Shc mRNA levels in K562 cells. Real-time PCR was performed. Results from three independent experiments are presented as relative mRNA expression and normalized by GAPDH. (D) K562 cells were treated with 0, 10, 20, and 40 μM hemin for 24 h before total RNA isolation. Real-time PCR was performed to measure p66Shc mRNA levels. Results from three independent experiments are presented as relative mRNA expression (hemin 0 as 1.0) and normalized by GAPDH. p values are calculated using Student's t test. Asterisk indicates significantly different from nontreatment sample. *p = 0.003, **p < 0.001. (E) K562 cells were treated with 40 μM hemin (He), 20 μM sulforaphane (SUL), 10 μM arsenite (As), or 80 μM t-BHQ for 24 h. p66Shc mRNA expression was measured by real-time PCR, and results were normalized by GAPDH. *p < 0.01, **p < 0.05 (n > 3). (F) K562 cells were transfected with a pGL3 luciferase reporter plasmid containing the −450 to +60 5′-regulatory region of the human p66Shc gene (pGL3 p66Shc-450/+60 plasmid), and hemin was added to a final concentration of 0, 10, 20, and 40 μM for 24 h. The firefly luciferase activity was normalized by Renilla luciferase activity and presented relative to nontreatment sample. p values were calculated using Student's t test. Asterisk indicates significantly different from nontreatment sample. *p = 0.028, **p < 0.001. All error bars represent mean ± SD (n = 6).
K562 erythroleukemic cells carrying the chimeric bcr-abl gene, which expresses a constitutively active abl tyrosine kinase (Lozzio and Lozzio, 1975), have been used as an in vitro chronic myelogenous leukemia model (McGahon et al., 1994). K562 cells can be differentiated into the erythroid lineage with various chemotherapeutic anticancer agents, such as actinomycin D, cytosine arabinoside, and cisplatin (Rowley et al., 1981). Hemin, an iron-containing porphyrin, is also a potent inducer of K562 erythroid differentiation (Benz et al., 1980). We and others reported that hemin is a potent transcriptional activator of ferritin H and L genes through the ARE-Nrf2 system (Hintze and Theil, 2005; Iwasaki et al., 2006). In this study, we find that p66Shc, but not p52 or p46, is transcriptionally activated during hemin-induced erythroid differentiation of K562 and cytokine-dependent TF-1 cells. We identify an ARE in the human p66Shc gene to which Nrf2 binds after treatment with hemin. p66Shc phosphorylation at Ser-36, previously reported to trigger p66Shc translocation to mitochondria and become an active prooxidant protein (Pinton et al., 2007), is induced by hemin treatment, but p66Shc exclusively stays in the cytoplasm in these cell types. Forced expression of p66Shc induces erythroid differentiation without hemin treatment, whereas p66Shc knockdown inhibits hemin-induced K562 erythroid differentiation, in which ROS production is significantly enhanced in conjunction with decreased expression of other ARE-dependent antioxidant genes such as ferritin and NQO1. These results provide evidence for the first time that p66Shc in the cytoplasm serves as an antioxidant protein that is important for cellular defense against oxidative stress and erythroid differentiation.
RESULTS
Hemin induces p66Shc expression
As a first step toward understanding the regulation of the Shc family in cell differentiation, we treated K562 human erythroleukemic cells with hemin and studied the expression of p66, p52, and p46Shc. As shown in Figure 1B, p66Shc protein expression was induced significantly by treatment with hemin at 20 and 40 μM for 24 h, whereas expression of p52Shc or p46Shc proteins was unchanged or slightly decreased (short exposure in Figure 1B). To explore the mechanism of p66Shc up-regulation by hemin, we first measured p66Shc mRNA levels. Quantitative real-time (qRT)-PCR showed that hemin induced p66Shc mRNA in a dose- and time-dependent manner (Figure 1, C and D). In addition to hemin, arsenite and electrophiles such as sulforaphane and tert-butylhydroquinone (t-BHQ) also induced p66Shc mRNA expression (Figure 1E). To further characterize the mechanism through which p66Shc mRNA was induced by hemin, we cloned the −450 to +60 base pair 5′-regulatory region of the human Shc gene and tested the possibility of transcriptional regulation in luciferase reporter assays. pGL3p66Shc-450/+60 plasmid was transiently transfected into K562 cells and treated with 0–40 μM hemin, 10 μM arsenite, and 80 μM t-BHQ for 24 h, which significantly increased luciferase expression (Figure 1F and Supplemental Figure 1C). Similar results were obtained in Jurkat human T lymphocytes treated with hemin, arsenite, and t-BHQ (Supplemental Figure 1, A and B) and cytokine-dependent human TF-1 cells (Supplemental Figure 2A). These results suggest that p66Shc expression is induced at the transcriptional level by hemin treatment through uncharacterized enhancers in the −450 to +60 base pair 5′-regulatory region of the p66Shc gene.
p66Shc is transcriptionally regulated by the Nrf2-ARE pathway
Two alternative promoters regulate transcription of the Shc gene, giving rise to three isoforms of Shc protein, p66Shc and p52/p46Shc (Figure 1A; Ventura et al., 2002). All three isoforms share a common C-terminal region, but p66Shc and p52Shc have an extended and overlapping N-terminal region (Ray et al., 2012). Given the fact that hemin activates the Nrf2-ARE signaling pathway (Iwasaki et al., 2006) and induces accumulation of Nrf2 in the K562 nucleus (Figure 2A), we searched for potential ARE sequences in the −450 to +60 base pair p66Shc 5′-regulatory region. According to a consensus ARE sequence and its similarity to the AP1-binding site (Wasserman and Fahl, 1997), we found a 12–base pair match out of 16 base pairs of the ARE sequence in the proximal CAAT box and another AP1-like ARE sequence located ∼280 base pairs upstream from the transcription start site (Figure 2B). To investigate the functional involvement of these ARE enhancers in the transcriptional activation of p66Shc, we assessed binding of the Nrf2 transcription factor, a major ARE activating protein, to these ARE sites by chromatin immunoprecipitation (ChIP) assay in K562 cells treated with 20, 40, and 75 μM hemin for 12 h. Indeed, Nrf2 was recruited to the proximal ARE and AP1-like ARE sites, whereas it was marginal to a 2.3-kb upstream non-ARE site in the p66Shc 5′-regulatory region or β-globin promoter (Figure 2C). Similar results were obtained in TF-1 cells (Supplemental Figure 2, B and C). Furthermore, protein pull-down assays using biotinylated double-strand oligonucleotides demonstrated that both the p66Shc ARE and the AP1-like sequences bind Nrf2, but the ARE sequences showed much higher Nrf2 binding in vitro (Figure 2D). To further validate our observation, we introduced mutations in the ARE and AP1-like sequences in pGL3 p66Shc-450/+60 luciferase plasmid (Figure 2E) and measured luciferase expression after hemin treatment. As shown in Figure 2E, luciferase expression induced by hemin treatment in wild-type p66Shc reporter was significantly diminished in these two mutant reporter plasmids. It was shown that the methylation status of a CpG site (GCGGAAGCC) located 64 base pairs downstream from the transcription start site (TSS) in the p66Shc gene determines Nrf2 binding to the CpG site and p66Shc expression (Du et al., 2013). Note that our p66Shc luciferase reporters (−450 to +60 base pairs) do not contain this CpG site, and therefore the ARE and AP1-like sites we identify here are different from the +64 CpG site.
FIGURE 2:

Nrf2 binds the human p66Shc promoter containing the ARE and AP1-like sequences. (A) K562 cells were treated with 20 μM hemin for 36 h, and nuclear (Nuc.) and cytoplasmic (Cyto.) fractions were used for Western blot with Nrf2, lamin B1, or LDH antibody. Lamin B1 and LDH are nuclear and cytoplasmic fraction markers, respectively. (B) Schematic of the 5′-promoter region of the human p66Shc gene, which contains an ARE and an AP1-like sequence. n = any base; M = A or C; R = A or G; Y = C or T. Asterisks indicate matched bases. (C) K562 cells treated with 0, 20, 40, and 75 μM hemin for 12 h were subjected to ChIP assays with control IgG or Nrf2 antibody, followed by real-time PCR using p66Shc primers spanning the ARE, AP1-like, a 2.3-kb upstream 5′-promoter region, and a β-globin promoter region. Data were normalized by input DNA and are shown as mean ± SD (n = 3). (D) Nuclear extracts of K562 cells treated with 20 μM hemin for 36 h were used for pull-down assay. Nrf2 binding to a biotinylated double-strand p66Shc ARE (−107 to −82 base pairs) or a p66Shc AP1-like (−291 to −266 base pairs) probe was detected by Western blot using an Nrf2 antibody. A human ferritin H ARE (–4433 to –4412 base pairs) probe was used as a positive control and no DNA probe in the binding reaction as a negative control. Coomassie brilliant blue (CBB) staining is shown for verification of equal loading. (E) K562 cells were transfected with wild-type or mutant pGL3 p66Shc-450/+60 plasmid and incubated with 0, 20, and 40 μM hemin for 24 h. Firefly luciferase activity was normalized by Renilla luciferase activity and presented as relative to nontreated wild-type p66Shc-450/+60 plasmid. Top, asterisks indicate mutated nucleic acid. Bottom chart, asterisks indicate significantly different from nontreated cells transfected with wild-type p66Shc-450/+60 plasmid. *p < 0.005, **p < 0.001 (n > 3).
Consistently, Nrf2 knockdown specifically blocked hemin-induced p66Shc mRNA (Figure 3A) and protein expression without affecting p52Shc or p46Shc expression (Figure 3B). Conversely, p66Shc protein expression was enhanced by Nrf2 overexpression with or without hemin treatment (Figure 3C). Furthermore, RNA polymerase II was recruited to the p66Shc transcription start site (TSS) after hemin treatment for 12 h and was diminished by Nrf2 knockdown (Figure 3D), supporting our conclusion that the functional AREs in the p66Shc 5′-regulatory region are responsible for isoform-specific transcriptional activation of the ShcA locus by Nrf2 after hemin treatment.
FIGURE 3:

Nrf2 knockdown blocks hemin-induced transcriptional activation of the p66Shc gene. (A) K562 cells were treated with 0 and 40 μM hemin for 24 h after Nrf2 knockdown using Nrf2-targeted siRNA-1 or -2 (si Nrf2-1 or si Nrf2-2). Real-time PCR was performed for p66Shc and Nrf2 mRNA expression. Results are presented as relative mRNA expression (nontargeted control siRNA [si Control], no hemin as 1.0) and normalized by GAPDH. p values were calculated using Student's t test. Asterisk indicates significant difference; *p = 0.01, **p = 0.045. Data are shown as mean ± SD (n > 4). (B) K562 cells transfected with a si Control (si Ctrl) or si Nrf2-1 were treated with 0, 10, 20, or 40 μM hemin for 24 h. Top, Western blots using anti-Shc and anti-GAPDH antibodies. Bottom, Nrf2 mRNA levels measured by real-time PCR after normalization with GAPDH mRNA. Data are shown as mean ± SD (n > 4). (C) K562 cells were transfected with pCMV or pCMV-Nrf2 plasmid and treated with 0, 20, and 40 μM hemin for 24 h. Whole-cell lysate (Total) and nuclear fraction (Nuc.) were used for Shc and GAPDH Western blot and Nrf2 and lamin B1 Western blot, respectively. (D) K562 cells were treated with 0 and 50 μM hemin for 12 h after Nrf2 knockdown, and ChIP assays were performed with control IgG or an anti–RNA polymerase II antibody (RNAPII), followed by real-time PCR using primers spanning the p66Shc transcriptional start site. Data were normalized by input DNA and are shown as mean ± SD (n = 3). p values were calculated using Student's t test. Asterisk indicates significant difference; *p = 0.002, **p = 0.005.
Mitochondrial accumulation of p66Shc after phosphorylation at Ser-36 is cell-type specific
Some stress conditions induce phosphorylation of p66Shc at Ser-36, leading to mitochondrial localization and activation of p66Shc as a prooxidant protein (Migliaccio et al., 1999; Le et al., 2001; Pinton et al., 2007). To test whether hemin treatment induces phosphorylation of p66Shc at Ser-36, we immunoprecipitated (IP) cell lysates from hemin-treated or nontreated K562 cells with anti-Shc antibody followed by Western blot with anti–phospho-Ser-36 p66Shc antibody (Shc IP/Western was necessary to detect Ser-36–phosphorylated p66Shc due to many nonspecific bands in direct Western blots of cell lysates with our phospho–Ser-36 p66Shc antibody). This approach revealed that hemin treatment for 24 h induced p66Shc expression along with phosphorylation at Ser-36; however, the phosphorylation did not trigger p66Shc translocation to mitochondria (Figure 4A). To rule out the possibility of missing appropriate time points in our experiments to detect p66Shc mitochondrial localization, we performed time course treatments (0, 3, 6, 12, 24, and 48 h at 20 μM hemin) and carried out IP/Western to detect Ser-36–phosphorylated p66Shc in mitochondria and the cytoplasm. As shown in Figure 4B, 20 μM hemin increased p66Shc protein expression and phosphorylation at Ser-36 at 24 and 48 h, but p66Shc was detected only in the cytoplasmic fraction. Similar results were obtained in hemin-treated TF-1 cells (Supplemental Figure 3, A and B). However, as reported previously in some cell types, we observed that a small fraction of p66Shc was translocated to mitochondria in hemin-treated SH-SY5Y human neuroblastoma cells (Supplemental Figure 3, C and D). These results suggest that hemin induces p66Shc expression and Ser-36 phosphorylation but that mitochondrial translocation of p66Shc is cell-type dependent and indicate that both phosphorylated and nonphosphorylated forms of p66Shc localize exclusively in the cytoplasm in K562 and TF-1 erythroid cells.
FIGURE 4:

Hemin induces phosphorylation of p66Shc at Ser-36 but no mitochondrial translocation in K562 cells. (A) Endogenous p66Shc phosphorylation at Ser-36 was detected in whole-cell lysate (Total) and mitochondrial (Mito.), cytoplasmic (Cyto.), and nuclear (Nuc.) fractions of K562 cells treated with 0 and 20 μM hemin for 24 h. IPs with anti-Shc antibody were subjected to Western blots with anti–phospho-Ser-36 p66Shc antibody (IP/WB). Equivalent protein loading of IP samples was assessed by probing the blot with anti-rabbit IgG. PBR, LDH, and lamin B1 were detected by Western blots for mitochondrial, cytoplasmic, and nuclear fraction markers. (B) Phospho–Ser-36 p66Shc protein levels were measured in K562 cells treated with 20 μM hemin for 0, 3, 6, 12, 24, and 48 h. Mitochondrial and cytoplasmic fractions were used for p66Shc IP/WB. Equal protein loading of IP samples was assessed by probing the blot with anti-rabbit IgG. TOM20 and LDH are markers of mitochondria and cytoplasm, respectively. The ratio indicates pS36-p66Shc/p66Shc in cytoplasmic fractions (0 h as 1.0).
p66Shc knockdown enhances accumulation of ROS and apoptosis after hemin treatment
Mitochondrial p66Shc plays a prooxidant and proapoptotic role in mediating production of ROS (Giorgio et al., 2005). Although p66Shc protein in this study was found to be exclusively cytoplasmic, we next examined whether p66Shc is involved in hemin-induced oxidative stress and apoptosis. First, we performed p66Shc knockdown with small interfering RNA (siRNA) and detected apoptosis markers by measuring caspase 3 activity and annexin V binding after hemin treatment. Under the specific p66Shc-knockdown condition, without affecting the expression of p52Shc and p46Shc (Figure 5A), p66Shc knockdown in hemin-treated K562 cells induced caspase 3 activation (Figure 5B) and increased annexin V binding (Figure 5D) that was completely blocked by pretreatment with N-acetyl cysteine (NAC; Figure 5, C and D). To characterize the status of oxidative stress in these cells, we measured intracellular and mitochondrial ROS production using chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) and MitoSox Red probes after treatment of K562 cells with 20 μM hemin for 0–72 h. Under p66Shc knockdown, 20 μM hemin treatment significantly enhanced production of ROS detected by two different chemical ROS probes (Figure 5E), suggesting that p66Shc is an antioxidant protein. Consistently, p66Shc knockdown increased the amount of oxidized cellular proteins in hemin-treated cells (carbonylated proteins were detected by Western blotting with a dinitrophenol antibody) and was completely blocked by NAC pretreatment (Figure 5F). To further verify these results, we cotransfected siRNA-resistant p66Shc expression plasmids (wild-type and serine 36–to–alanine mutant [S36A]) with control or p66Shc siRNA and treated them with hemin. We first confirmed the expression of transfected p66Shc and resistance to siRNA. The Western blot in Figure 5G demonstrates that the endogenous p66Shc was knocked down but transfected p66Shc was resistant to p66Shc siRNA. Next we measured hemin-induced caspase 3 activity in each sample, in which knocking down p66Shc activated hemin-induced caspase 3 activity, which was suppressed by siRNA-resistant p66Shc overexpression (Figure 5H). In this experiment we did not observe statistically significant difference between wild-type p66Shc and S36A p66Shc (Figure 5H), suggesting that phosphorylation of p66Shc at Ser-36 is not essential for p66Shc-mediated inhibition of apoptosis. Collectively these results indicate that p66Shc plays an antioxidant role, which is consistent with the fact that a battery of Nrf2-regulated genes is involved in cellular antioxidant defense mechanisms (Hayes and McMahon, 2009).
FIGURE 5:

p66Shc knockdown enhances ROS production and induces apoptosis through oxidative cell damage. (A) Whole-cell lysates isolated from K562 cells transfected with p66Shc-specific siRNA-1 or -2 (si p66Shc-1 or si p66Shc-2) and treated with 20 μM hemin for 24 h were subjected to Western blot with anti-Shc antibody, with GAPDH as a protein loading control. Short exposure (top) and long exposure (middle) of the same Shc Western blot. (B) Caspase 3 activity was measured using a luminogenic substrate containing the DEVD sequence as described in Materials and Methods. K562 cells were treated with 0 and 20 μM hemin for 2 d after p66Shc knockdown using si p66Shc-1 or si p66Shc-2. p values were calculated using Student's t-test. Asterisk indicates significant difference; *p < 0.001. Data are shown as mean ± SD (n = 3). (C, D) si Control or p66Shc siRNA-1 (si p66Shc-1) was transfected into K562 cells; then cells were pretreated with 0 or 2 mM NAC before 0 or 20 μM hemin treatment for 2 d. (C) Caspase 3 assays were performed. p values were calculated using Student's t test. Asterisk indicates significant difference; *p < 0.001 (si p66Shc +He –NAC vs. all other conditions). Data are shown as mean ± SD (n = 3). (D) Annexin V assays were performed. Representative flow cytometry plot. The number indicates the percentage of early apoptotic cell populations (annexin V positive and Fixable Viability Dye eFluor 780 negative). (E) K562 cells transfected with si p66Shc-1 were stimulated with 20 μM hemin for 0, 12, 24, 48, and 72 h, and ROS production was measured by flow cytometry using CM-H2DCFDA and MitoSox Red. Top, representative histogram; bottom, statistical charts (fold increase). Asterisk indicates significantly different from nontreated si Control sample. *p < 0.05. All error bars represent mean ± SD (n > 3). (F) K562 cells transfected with si Control (si Ctrl) or si p66Shc-1 were pretreated with 0 or 2 mM NAC before 0 or 20 μM hemin treatment for 2 d. Total proteins were incubated with DNPH and analyzed by Western blot using anti-DNP antibody to detect carbonylated proteins. Coomassie brilliant blue (CBB) staining was used for verification of equal protein loading. (G, H) K562 cells were transfected with si Control or si p66Shc-1 and pcDNA3.1His empty vector (pcDNA), si p66Shc-1-resistant pcDNA3.1His-p66Shc wild type (WT), or -S36A mutant (S36A). G) Whole-cell lysates were used for Western blot with a Shc or GAPDH antibody. (H) K562 cells were treated with 0 and 20 μM hemin for 2 d after transfection, and caspase 3 assays were performed. Asterisk indicates significantly different from si p66Shc/pcDNA/+hemin sample. *p < 0.001, **p = 0.003. All error bars represent mean ± SD (n > 3). NS, no significant difference.
p66Shc regulates expression of other ARE-dependent antioxidant genes
We showed that knocking down p66Shc is sufficient to increase cellular oxidative stress (Figure 5). This led us to test whether p66Shc is important for expression of other antioxidant genes regulated by Nrf2. To this end, we measured expression of several Nrf2-regulated ARE-dependent antioxidant genes—the heavy and light subunits of ferritin (ferritin H and L, respectively), NQO1, GSTπ, and thioredoxin-1 (Trx)—in hemin-treated K562 cells after knocking down p66Shc. p66Shc knockdown reproducibly decreased induction of these ARE-dependent genes in K562 cells treated with 20 μM hemin for 1–3 d (Figure 6A). Consistently, mRNA levels of these genes were decreased in p66Shc-deficient cells (Figure 6B). We then asked whether the decrease in expression of these ARE-regulated antioxidant genes is due to diminished Nrf2 nuclear translocation under p66Shc knockdown. Of interest, nuclear accumulation of Nrf2 was slightly increased under the p66Shc-deficient condition with or without hemin treatment; however, its binding ability to the ferritin ARE in vitro was decreased compared with control siRNA transfection under hemin treatment (Figure 6C). Indeed, ChIP assays for Nrf2 binding to ferritin and NQO1 AREs in K562 cells confirmed the decreased Nrf2 binding to these ARE sites (Figure 6D). These results suggest that p66Shc deficiency impairs Nrf2 binding to ARE and its transcription function and that p66Shc regulates expression of some other ARE-dependent antioxidant gene expression at the transcriptional level, which may ultimately contribute to the overall antioxidant function of p66Shc.
FIGURE 6:
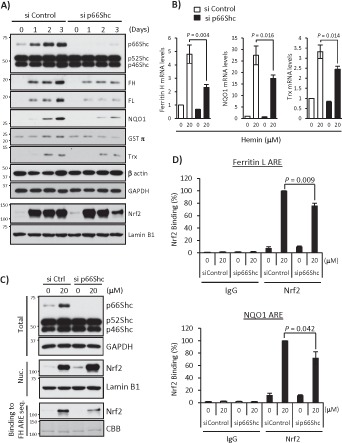
p66Shc deficiency impairs expression of other ARE-dependent antioxidant genes. (A) si Control– or p66Shc siRNA-1–transfected K562 cells were treated with 20 μM hemin for 1, 2, and 3 d. Whole-cell lysates were used in Western blots for Shc, ferritin H (FH), ferritin L (FL), NQO1, GSTπ, thioredoxin-1 (Trx), β-actin, and GAPDH, and nuclear extracts were used for Nrf2 and lamin B1. (B) siRNA-transfected K562 cells were treated with 20 μM hemin for 36 h, and real-time PCR was performed for ferritin H, NQO1, and Trx mRNA. Data are presented as relative mRNA expression (si Control/day 0 is defined as 1.0) after normalization with RPL13A (ribosomal protein L13A). Data are shown as mean ± SD (n = 3). (C) Shc and GAPDH Western blots and Nrf2 and lamin B1 Western blots were performed using whole-cell lysates (Total) and nuclear extracts (Nuc.) of siRNA-transfected K562 cells treated with 20 μM hemin for 36 h, respectively. Nuclear extracts were also used for pull-down assay. Nrf2 binding to biotinylated double-strand ferritin H ARE (−4067 to −4046 base pairs) probe was detected by Western blot using an Nrf2 antibody. Coomassie brilliant blue (CBB) staining is shown for verification of equal loading. (D) siRNA-transfected K562 cells treated with 20 μM hemin for 36 h were subjected to ChIP assays with control IgG or Nrf2 antibody, followed by real-time PCR using ferritin L and NQO1 primers spanning the ARE region. Data were normalized by input DNA and are shown as mean ± SD (n = 3).
p66Shc is involved in hemin-induced erythroid differentiation of K562 cells
Hemin is a potent inducer of erythroid differentiation in K562 cells, so we asked whether hemin-induced p66Shc expression via the Nrf2-ARE system is involved in erythroid differentiation. To address this issue and understand the role of p66Shc in cell differentiation, we knocked down p66Shc in K562 cells, treated them with 20 μM hemin, and measured fetal hemoglobin synthesis to monitor erythroid differentiation. Direct visualization of hemoglobin content in the cell pellets (unpublished results) and hemoglobin benzidine staining demonstrated that p66Shc knockdown strongly inhibited hemoglobin synthesis compared with control siRNA–transfected cells (Figure 7, A and B). Furthermore, in Western blots with antibodies for fetal hemoglobins and specific to γ-globin, p66Shc knockdown inhibited expression of these globins after hemin treatment (Figure 7C). Consistently, p66Shc overexpression (both wild type and S36A mutant) enhanced hemin-induced fetal hemoglobin synthesis (Figure 7D). To further characterize the role of p66Shc in the absence of hemin treatment, we overexpressed p66Shc wild type and S36A mutant in K562 cells. γ-Globin expression was induced by p66Shc wild type as well as S36A mutant p66Shc (Figure 7E), suggesting that overexpression of p66Shc is sufficient for inducing erythroid differentiation. These results also suggest that Ser-36 phosphorylation of p66Shc is dispensable for induction of erythroid differentiation.
FIGURE 7:

p66Shc is involved in hemin-induced K562 erythroid differentiation. (A, B) K562 cells transfected with si Control or p66Shc siRNA were treated with 20 μM hemin for 2 d. (A) Benzidine cytochemical staining. (B) Quantified data. Four different areas were randomized, and >1000 cells were counted in each condition. Benzidine-positive cells: 8.4% (si Control/day 0), 7.0% (si p66Shc-1/day 0), 92.6% (si Control/day 2), 69.4% (si p66Shc-1/day 2). (C) K562 cells transfected with si Control or si p66Shc-1 were treated with 20 μM hemin for 0–5 d, and Western blots were performed with fetal hemoglobins (Hb Fetal), γ-globin, and GAPDH antibodies. (D) p66Shc expression plasmids were transfected into K562 cells, and cells were treated with 20 μM hemin for 24 h. Western blots were performed with Shc, Hb Fetal, and GAPDH antibodies. (E) p66Shc expression plasmids were transfected into K562 cells, and Western blots were performed with Shc, γ-globin, and GAPDH antibodies.
DISCUSSION
In this study we provide evidence that p66Shc plays an antioxidant role in cellular defense against ROS toxicity and hemin-induced erythroid differentiation. We also demonstrate that expression of p66Shc, but not p52Shc or p46Shc, is transcriptionally activated through the ARE-Nrf2 pathway in hemin-treated K562 erythroleukemic cells. This is not specific to K562 cells or hemin treatment but is also observed in cytokine-dependent TF-1 cells, SH-SY5Y cells, and human Jurkat T-cells treated with sulforaphane, sodium arsenite, or t-BHQ, potent activators of the ARE-Nrf2 pathway (Motohashi and Yamamoto, 2004; Takaya et al., 2012). Given the fact that an array of genes regulated by the ARE-Nrf2 axis is involved in cellular antioxidant and detoxification functions (Hayes and McMahon, 2009), our finding of p66Shc regulated by the ARE-Nrf2 seems to be consistent with the antioxidant function of the p66Shc protein. However, the majority of studies using the knockout mouse model and cellular functional approaches of p66Shc demonstrate that p66Shc plays prooxidative and proapoptotic roles. For instance, p66Shc-knockout mice exhibited 30% lifespan extension and better handling of ROS and oxidative stress induced by paraquat (Migliaccio et al., 1999). Furthermore, p66Shc-deficient mouse fibroblasts showed less production of ROS in nutrient- or serum-starvation conditions (Nemoto and Finkel, 2002) and lower susceptibility to apoptosis induced by H2O2 treatment or UV light exposure (Migliaccio et al., 1999). What are the reasons for the apparently opposite p66Shc functions observed between this study and many other studies? One reason may be the difference in the types of oxidative stressor used. Paraquat, H2O2, and UV light cause ROS production and oxidative cell damage but generally are not potent activators of Nrf2 or transcription of Nrf2-regulated antioxidant genes (Prestera et al., 1993; Leiser and Miller, 2010). Therefore these agents used in previous studies caused oxidative cell damage probably without physiologically relevant induction of p66Shc expression via the ARE-Nrf2 pathway. In contrast, hemin, as well as arsenite and t-BHQ, are strong activators of Nrf2 (Pi et al., 2003; Iwasaki et al., 2006). Thus cells responded to hemin- or arsenite-induced expression of p66Shc and other Nrf2-regulated antioxidant detoxification genes as shown in this study. Blocking hemin-induced p66Shc expression by siRNA apparently caused an imbalance in the cellular antioxidant defense mechanism, resulting in accumulation of ROS and increased susceptibility to apoptosis by hemin (Figure 5). In conjunction with the issue of oxidative stressor types, differences in the expression levels of p66Shc in various tissues and cells may also contribute to the complication of the p66Shc function. It was previously shown that expression levels of p66Shc mRNA vary significantly in different types of cells, in which the degree of DNA methylation in the p66Shc promoter inversely correlates with expression levels of p66Shc (Ventura et al., 2002). We also observed highly varied p66Shc protein expression levels in different types of culture cells, such as >10-fold higher p66Shc expression in NIH3T3 mouse fibroblasts and SH-SY5Y cells than K562, TF-1, or Jurkat cells used in this study (unpublished observations). Some cell types used in previous studies, such as mouse fibroblasts expressing abundant p66Shc protein, may have a system to allow a fraction of p66Shc to be constitutively localized in mitochondria, where p66Shc is involved in ROS production. In contrast, cells expressing relatively low levels of p66Shc, such as K562 or TF-1 cells, may not have a system for mitochondrial shuttling of p66Shc. In these cell types, changes in p66Shc expression levels may have a significant effect on their antioxidant cell defense mechanisms.
Previous elegant studies also demonstrated that p66Shc phosphorylation at Ser-36, the unique site to p66Shc among the Shc family, was induced by some stress conditions and that p66Shc phosphorylation seems to be prerequisite for mitochondrial localization and subsequent prooxidant activity (Migliaccio et al., 1999; Pinton et al., 2007). However, in our study, neither unphosphorylated nor Ser-36–phosphorylated p66Shc was detectable in mitochondria in K562 and TF-1 cells after hemin treatment (Figure 4 and Supplemental Figure 3B). In contrast, as reported previously in some cell types, a small fraction of p66Shc was translocated to mitochondria in hemin-treated SH-SY5Y cells (Supplemental Figure 3, C and D). This cell type–specific response suggests that there may be additional regulatory mechanisms of p66Shc shuttling between cytoplasm and mitochondria besides Ser-36 phosphorylation of p66Shc. Prolyl isomerase 1 (Pin1) induces conformational changes of proteins containing phospho-Ser-Pro (or phospho-Thr-Pro) bonds through cis-trans isomerization (Lu and Zhou, 2007). It was demonstrated that Pin1 preferentially interacts with Ser-36–phosphorylated p66Shc and facilitates p66Shc translocation to mitochondria (Pinton et al., 2007). We wondered whether K562 cells are Pin1 deficient; however, our preliminary Western blots showed that Pin1 is expressed in K562 cells, as well as in SH-SY5Y and Jurkat cells, and that Pin-1 expression levels are relatively similar in various cell types (unpublished observations). Although further investigation will be necessary to understand the role of phospho-p66Shc at Ser-36, p66Shc may primarily play an antioxidant role when localized in the cytoplasm but serve as a prooxidant protein when translocated to mitochondria and subsequent interaction with cytochrome c (Giorgio et al., 2005).
Our findings appear to be supported by recent work showing that p66Shc-knockout mice are short lived under such natural condition as a large outdoor enclosure, food competition, and natural temperatures (Giorgio et al., 2012). By contrast, p66Shc may have an opposite role in mice under specific pathogen-free conditions, as Migliaccio et al. (1999) reported lifespan extension in p66Shc-knockout mice, although a recent reevaluation of p66Shc's role in mouse longevity does not support the previous conclusion (Ramsey et al., 2013). An additional p66Shc study showing its cytoprotective role in hypoxia conditions (Sansone et al., 2007) may be consistent with the results of this study. However, our work here and that by other groups are not sufficient to address how and why p66Shc serves as an antioxidant as well as a prooxidant. Further investigation will be necessary to understand the bifunctional roles of p66Shc in oxidative stress.
We previously reported that hemin is a potent activator of the Nrf2-ARE pathway, leading to transcriptional activation of ferritin and other ARE-regulated antioxidant genes, including NQO1 (Iwasaki et al., 2006). In this study, p66Shc knockdown inhibited hemin-induced expression of these ARE-regulated genes (Figure 6). To understand the mechanism by which p66Shc deficiency suppresses other ARE-regulated antioxidant genes, we investigated the possibility that p66Shc is a positive regulator of Nrf2, by comparing Nrf2 nuclear accumulation after hemin treatment between control and p66Shc-knockdown K562 cells. Under almost complete knockdown of p66Shc, Nrf2 accumulation was not impaired but slightly increased in the nucleus after hemin treatment compared with control siRNA–transfected cells (Figure 6C). However, expression of ferritin, NQO1, and Trx genes was reproducibly suppressed due to Nrf2 dysfunction of ARE binding under p66Shc knockdown (Figure 6). These results suggest that p66Shc may not be a direct Nrf2 regulator but that p66Shc deficiency causes hemin-mediated severe oxidative stress (demonstrated by increased ROS production and protein oxidation shown in Figure 5). The overwhelming oxidative stress likely suppressed the transcription of these ARE-regulated antioxidant genes, probably through Nrf2 oxidation or some unknown negative modifications of Nrf2, and ultimately blocked hemin-induced erythroid differentiation (Figure 7). It was reported that Nrf2 is involved in K562 erythroid differentiation through binding and activation of an ARE in the γ-globin gene (Macari and Lowrey, 2011). Our results, summarized in Figure 8, are consistent with the positive regulatory role of Nrf2 in erythroid differentiation, in which we propose p66Shc as an essential Nrf2-regulated antioxidant gene.
FIGURE 8:

Graphic summary. Transcription of the human p66Shc, but not p52Shc or p46Shc, is regulated by Nrf2. Phosphorylation of p66Shc at Ser-36, known as an active mitochondrial form, was induced in K562 cells treated with hemin but still localized in the cytoplasm. p66Shc serves as an antioxidant protein by maintaining up-regulation of other ARE-dependent genes (NQO1, Trx, ferritin H and L, γ-globin) and is also involved in erythroid differentiation. The present work demonstrates the isoform-specific regulation of the Shc gene by the Nrf2-ARE system and a new antioxidant function of cytoplasmic p66Shc involved in cellular oxidative stress response and differentiation.
In summary, our results demonstrate the isoform-specific regulation of the Shc gene by the Nrf2-ARE system and a new antioxidant role of p66Shc in the cytoplasm. p66Shc is a bifunctional protein involved in cellular oxidative stress response and differentiation.
MATERIALS AND METHODS
Cell culture and reagents
K562 and TF-1 human erythroleukemia and Jurkat human T-cell leukemia cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). They were cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS; Mediatech, Herndon, VA), 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 2 mM l-glutamine, and 4.5 g/l glucose. Jurkat and TF-1 culture media contained 1 mM sodium pyruvate, and TF-1 cells were cultured in the presence of 2 ng/ml granulocyte-macrophage colony-stimulating factor. SH-SY5Y human neuroblastoma cells were also purchased from ATCC. They were cultured in 1:1 mixture of MEM with nonessential amino acids and Ham's F12 medium containing 10% FBS. They were maintained at 37°C in a humidified 95% air, 5% carbon dioxide incubator. For K562 erythroid differentiation, exponentially growing cells were exposed to hemin (Fluka-Sigma-Aldrich, St. Louis, MO). Hemin and sodium arsenite (Thermo Fisher Scientific, Waltham, MA) were dissolved in 100 μM NaOH and distilled water, respectively. t-BHQ (Sigma-Aldrich, St. Louis, MO) was dissolved in dimethyl sulfoxide for 1 M solution and further diluted to 10 mM with distilled water before addition to the culture media. Sulforaphane (Sigma-Aldrich) was also dissolved in dimethyl sulfoxide for 100 mM solution.
Antibodies and plasmids
Antibodies used in this work were purchased as follows: anti-Shc (610081) and anti-GSTπ (610718) from BD Biosciences (Franklin Lakes, NJ); anti–phospho-Ser-36-Shc (6E10) (sc-81520), anti-ferritin H (sc-25617), anti-NQO1 (sc-32793), anti-Nrf2 (sc-13032x), anti–γ-globin (sc-21756), anti-rabbit immunoglobulin G (IgG; sc-2027), anti–RNA polymerase II (sc-899x), anti-Tom20 (sc-11415), anti-Tom40 (sc-11414), and anti-Trx (sc-20146) from Santa Cruz Biotechnology (Santa Cruz, CA); anti–lamin B1 from Cell Signaling Technology (Danvers, MA); anti–lactate dehydrogenase (LDH; AB1222), anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; MAB374) from Chemicon (Temecula, CA); anti–hemoglobin F (374824) from Calbiochem (San Diego, CA); anti-dinitrophenol (DNP) and anti–ferritin L (F5012) from Sigma-Aldrich; and anti–β-actin (AM1829b) from Abgent (San Diego, CA). Anti-peripheral benzodiazepine receptor (PBR) antibody was a kind gift from Janet Morgan, Roswell Park Cancer Institute, Buffalo, NY (Morgan et al., 2004). pCMV-Nrf2 plasmid was constructed by cloning the full-length Nrf2 fragment into the BamHI-XhoI site of pCMV plasmid. The Nrf2 fragment was obtained from pOTB7Nrf2 plasmid (4548874; Invitrogen, Carlsbad, CA). pcDNA3.1His-p66Shc wild type and S36A mutant were provided by Toren Finkel, National Institutes of Health, Bethesda, MD (Nemoto and Finkel, 2002), through Addgene (Cambridge, MA). The p66Shc promoter region (from −450 to +60 base pairs) was amplified by PCR using K562 genomic DNA as a template with forward primer 5′-ATAGAGAGGGTGGGGGAATTCGAGT-3′ and reverse primer 5′-CAGCTCAGACCCAGACAGTTTCAGG-3′. The PCR product was cloned into pGL3-Basic plasmid vector (Promega, Madison, WI). p66Shc ARE and AP1-like mutations were introduced into pGL3 p66Shc-450/+60 luciferase reporter plasmid using QuikChange II Site-Directed Mutagenesis (Agilent Technologies, Santa Clara, CA). siRNA-resistant p66Shc expression plasmids (wild type and S36A mutant) were constructed from pcDNA3.1His-p66Shc wild-type and S36A mutant plasmids by QuikChange II Site-Directed Mutagenesis. The primers used for mutagenesis were as follows. p66Shc mutant ARE: forward, 5′-CAGCCCCTCTGAGCCTGTGAGTTGATACTGTCCTCACGATTG-3′, and reverse, 5′-CAATCGTGAGGACAGTATCAACTCACAGGCTCAGAGGGGCTG-3′; p66Shc mutant AP1-like: forward, 5′-CCTGCGTGTGTCCGGTTTCACGTGGCCCAGACCTCCTATC-3′, and reverse, 5′-GATAGGAGGTCTGGGCCACGTGAAACCGGACACACGCAGG-3′; and p66Shc siRNA–resistant p66Shc: forward, 5′-CTGCCTGGGGACGATAGCCCTACGACACTGTGCTCCTTC-3′, and reverse, 5′-GAAGGAGCACAGTGTCGTAGGGCTATCGTCCCCAGGCAG-3′.
Plasmid DNA transfection and luciferase reporter assay
K562 cells were transiently transfected by electroporation (Gene Pulser X-Cell; Bio-Rad, Hercules, CA) with pGL3 p66Shc-450/+60 luciferase reporter plasmid and pLightSwitch EMPTY_3UTR Renilla luciferase plasmid (S890005; SwitchGear Genomics, Menlo Park, CA).Three micrograms of pGL3 p66Shc-450/+60 plasmid and 100 ng of pLightSwitch EMPTY_3UTR plasmid were cotransfected to 1 × 107 K562 cells and incubated for 24 h. Cells were divided into 12-well plate and then treated with hemin, sodium arsenite, or t-BHQ for 24 h and subjected to luciferase assays using a Dual-Luciferase Assay System (E1960; Promega).
Western blotting
Equal amounts of whole-cell lysates or mitochondria, nuclear, and cytosolic fractions were loaded on 7.5 or 12.5% SDS–PAGE (Mini-Protean Tetra Cell; Bio-Rad), and separated proteins were transferred to polyvinylidene difluoride membranes (Thermo Scientific, Rockford, IL) by Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad). After blocking with Tris-buffered saline (TBS)–0.1% Tween 20 containing 5% skim milk or 1% BSA, membranes were incubated with a primary antibody at 4°C overnight. After washing with TBS–0.1% Tween 20, the membranes were incubated with a secondary antibody conjugated with horseradish peroxidase (HRP) at room temperature for 2 h and washed with TBS–0.1% Tween 20. HRP signals were visualized using HyGLO detection reagent (Denville Scientific, Metuchen, NJ), ECL Prime (GE Healthcare Life Sciences, Piscataway, NJ), or SuperSignal (Thermo Scientific). The isolation of subcellular fractions was carried out with kits for isolation of mitochondria and nuclear/cytoplasmic fractions (Active Motif, Carlsbad, CA). For detection of Ser-36–phosphorylated p66Shc, 200–300 μg of whole-cell lysates in midRIPA buffer (25 mM Tris, pH 7.4, 1% NP-40, 0.5% sodium deoxycholate, 15 mM NaCl) was immunoprecipitated with anti-Shc antibody plus protein A+G agarose (Calbiochem), followed by Western blotting with anti–phospho-Ser-36 p66Shc antibody (Santa Cruz Biotechnology) and detection with CanGet Signal solutions (Toyobo, Osaka, Japan).
ROS measurement
K562 cells were washed with magnesium- and calcium-free Hank's balanced salt solution (HBSS(-)) and incubated for 30 min in a 37°C/5% carbon dioxide incubator with 10 μM CM-H2DCFDA or 5 μM MitoSox Red (Invitrogen). After labeling, cells were washed once with HBSS(-), resuspended in HBSS(-), and analyzed on a FACS LSRII (BD Biosciences).
Annexin V and LIVE/DEAD staining
K562 cells were washed with phosphate-buffered saline (PBS) and stained with Fixable Viability Dye eFluor780 (eBioscience, San Diego, CA) for 30 min on ice. After that, the dye was removed, and cells were reincubated with Pacific Blue-Annexin V (BioLegend, San Diego, CA) in annexin V binding buffer (BioLegend) for 15 min at room temperature and analyzed on a FACS LSRII.
siRNA transfection
K562 cells (1 × 107) were electroporated with 200 pmol of siRNA by Gene Pulser X-Cell in 100 μl of siRNA transfection medium (sc-36868; Santa Cruz Biotechnology). After electroporation, cells were incubated at 37°C for 10 min, transferred into fresh culture medium, and incubated at 37°C for 24–48 h. p66Shc-1 siRNA: sense, 5′-CGAUAGUCCCACUACCCUGUU-3′, and antisense, 5′-CAGGGUAGUGGGACUAUCGUU-3′ (custom order; Thermo Scientific); p66Shc-2 siRNA: sense: 5′-GGAUGAGCAACCUGAGGCUTT-3′, and antisense, 5′-AGCCUCAGGUUGCUCAUCCGG-3′ (custom order; Qiagen, Valencia, CA); Nrf2 siRNA-1: sense, 5′-UGGAGUAAGUCGAGAAGUAUU-3′, and antisense, 5′-PUACUUCUCGACUUACUCCAUU-3′ (J-003755-11; Dharmacon, Lafayette, CO); Nrf2 siRNA-2: sense, 5′-CAUUGAUGUUUCUGAUCUATT-3′, and antisense, 5′-UAGAUCAGAAACAUCAAUGGG-3′ (Sl03246950; Qiagen); and nontargeting siRNA (si Control; 1027281; Qiagen).
qRT-PCR
Reverse transcription of 500 ng of total RNA was performed with the iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer's protocols. cDNA was subjected to SYBR Green qPCR with iQ SYBR Green Supermix or iTaq Universal SYBR Green Supermix (Bio-Rad) in a CFX96 Real-Time PCR System (Bio-Rad) with specific primer pairs for p66Shc (forward, 5′-CAGAGGTCCAACCAGGGTAA-3′, and reverse, 5′-CTCATTCCGGAGTGGATTGT-3′), GAPDH (forward, 5′-GAGTCAACGGATTTGGTCGT-3′, and reverse, 5′-TTGATTTTGGAGGGATCTCG-3′), ferritin H (forward, 5′-ACTGATGAAGCTGCAGAACC-3′, and reverse, 5′-GTCACCCAATTCTTTGATGG-3′), NQO1 (forward, 5′-GAAGAGCACTGATCGTACTGGC-3′, and reverse, 5′-GGATACTGAAAGTTCGCAGGG-3′), Nrf2 (forward, 5′-TCAGCGACGGAAAGAGTATGA-3′, and reverse, 5′-CCACTGGTTTCTGACTGGATGT-3′), thioredoxin-1 (forward, 5′-GTGAAGCAGATCGAGAGCAAG-3′, and reverse, 5′-CGTGGCTGAGAAGTCAACTACTA-3′), and RPL13A (forward, 5′-CCTGGAGGAGAAGAGGAAAGAGA-3′, and reverse, 5′-TTGAGGACCTCTGTGTATTTGTCAA-3′). Relative expression was quantified using the 2−ΔΔCt method by CFX Manager Software (Bio-Rad).
Chromatin immunoprecipitation assay
ChIP assays were performed according to a previously described fast ChIP method (Nelson et al., 2006) with minor modifications. Briefly, K562 or TF-1 cells were fixed with 1.42% formaldehyde for chromatin cross-linking for 15 min at room temperature, and the cross-linking reaction was stopped with 125 mM glycine. Cells were suspended with IP buffer (150 mM NaCl, 50 mM Tris, pH 7.5, 5 mM EDTA, 0.5% NP-40, 1% Triton X-100) and sonicated to shear chromatin DNA for 10 s, repeated 12 times. The chromatin DNA samples were incubated with 0.25 μg of normal IgG or specific antibodies and protein A agarose beads/salmon sperm DNA (16-157; Millipore, Billerica, MA) at 4°C for overnight. Protein A agarose beads conjugated with antibody/protein/DNA complexes were washed with IP buffer five times and de-cross-linked by proteinase K. Genomic DNA was subjected to SYBR Green qPCR with iQ SYBR Green Supermix or iTaq Universal SYBR Green Supermix in a CFX96 Real-Time PCR System by using the following primer pairs. p66Shc ARE: forward, 5′-CTACCCCCTGCTCCAGTTC-3′, and reverse, 5′-GGAGCTAAGGGGAGAGACCA-3′; p66Shc AP1-like: forward, 5′-GTACTCCTGCCTGGCTTCCT-3′, and reverse, 5′-AGGGAAGGATGGAAGAGTGG-3′; p66Shc TSS: forward, 5′-CGATTGGTCTCTCCCCTTAG-3′, and reverse, 5′-CAGCTCAGACCCAGACAGTT-3z; p66Shc 2.3-kb: forward, 5′-CTGGCTTTCCCTTCCCAGGACCAAC-3′, and reverse, 5′-GCCAGGAGGAATGCCCAGACAAGAA-3′; β-globin promoter: forward, 5′-AGGACAGGTACGGCTGTCATC-3′, and reverse, 5′-TTTATGCCCAGCCCTGGCTC-3′; ferritin L ARE: forward, 5′-TAGTTCTGAGGGTCCCACCA-3′, and reverse, 5′-GGTATCTGGGGTCCTTGTTG-3′; and NQO1 ARE: forward, 5′-CCCAGGGAAGTGTGTTGTAT-3′, and reverse, 5′-CAAGAGAGTCCAGGGATCAA-3′) (Huang et al., 2013). Real-time PCR data were normalized by the present input method.
Streptavidin-agarose pull-down assay
We incubated 20- to 60-μg nuclear extracts with 3–4 μg of biotinylated DNA probe and streptavidin-agarose beads (15942-050; Invitrogen) in PBS containing protease inhibitor (539131; Calbiochem) for 2 h at room temperature. After incubation, the beads were washed with PBS three times and Nrf2 detected by Western blot with the anti-Nrf2 antibody. Specific 5′- biotinylated double-strand p66Shc ARE (sense, 5′-TCTGAGCCTGTGAGTCAGCACTGTCC-3′, and antisense, 5′-GGACAGTGCTGACTCACAGGCTCAGA-3′), p66shc AP1-like (sense, 5′-CGGTTTTGAGTGACCCAGACCTCCTA-3′, and antisense, 5′-TAGGAGGTCTGGGTCACTCAAAACCG-3′), and ferritin H ARE (sense, 5′-GGAGTGCTGAGTCACGGTGGGA-3′, and antisense, 5′-TCCCACCGTGACTCAGCACTCC-3′) probes were used. Single-strand sense and antisense oligonucleotides were obtained from Sigma-Aldrich and annealed by incubation at 95°C for 5 min and then allowed to cool gradually to room temperature before pull-down assay.
Caspase activity assay
Caspase assays were performed using the Caspase 3/7 Glo assay kit (Promega). Cells were lysed with midRIPA buffer, and 2 μg of protein was incubated with 20 μl of Caspase Glo reagent for 30 min at room temperature. The luminescence of each samples were measured in a GloMax20/20 Luminometer (Promega) with preset program of the Promega caspase protocol.
Carbonylated protein assay
Carbonylated proteins were visualized as an indicator of oxidized protein by Western blot with anti-DNP antibody. Briefly, 200 μg of whole-cell lysates precipitated with 10% trichloroacetic acid (TCA) was treated with 2 M HCl containing 10 mM 2,4-dinitrophenylhydrazine (DNPH) at room temperature for 1 h. The mixture was then reprecipitated by 10% TCA, and washed with ethanol-ethyl acetate (1:1) mixture three times, and the final precipitates were dissolved in 8 M Urea. DNPH-labeled protein samples, 2.5 μg, were subjected to Western blot with anti-DNP antibody.
Statistical analysis
Experimental data are expressed as the mean ± SD and statistically analyzed using one-way analysis of variance and Student's t test (Fisher's least significant difference). p < 0.05 was considered statistically significant. SPSS Statistics 21 software (IBM, Armonk, NY) was used for analysis.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grants R01GM088392 and R01GM095550 from the National Institute of General Medical Sciences to Y.T. We thank Janet Dow, Flow Cytometry and Cell Sorting Facility at the North Carolina State University College of Veterinary Medicine, for excellent technical support.
Abbreviations used:
- ARE
antioxidant response element
- DNP
dinitrophenol
- Nrf2
nuclear factor erythroid 2–related factor 2
- t-BHQ
tert-butylhydroquinone
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-11-0666) on May 7, 2014.
The authors declare that they have no conflict of interest.
REFERENCES
- Arosio P, Ingrassia R, Cavadini P. Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta. 2009;1790:589–599. doi: 10.1016/j.bbagen.2008.09.004. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Benz EJ, Jr, Murnane MJ, Tonkonow BL, Berman BW, Mazur EM, Cavallesco C, Jenko T, Snyder EL, Gorget BG, Hoffman R. Embryonic-fetal erythroid characteristics of a human leukemic cell line. Proc Natl Acad Sci USA. 1980;77:3509–3513. doi: 10.1073/pnas.77.6.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Jiang Y, Zheng Z, Zhang Z, Chen N, Ma Z, Yao Z, Terada L, Liu Z. Feedback loop between p66(Shc) and Nrf2 promotes lung cancer progression. Cancer Lett. 2013;337:58–65. doi: 10.1016/j.canlet.2013.05.016. [DOI] [PubMed] [Google Scholar]
- Gertz M, Steegborn C. The Lifespan-regulator p66Shc in mitochondria: redox enzyme or redox sensor. Antioxid Redox Signal. 2010;13:1417–1428. doi: 10.1089/ars.2010.3147. [DOI] [PubMed] [Google Scholar]
- Giorgio M, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Giorgio M, et al. The p66Shc knocked out mice are short lived under natural condition. Aging Cell. 2012;11:162–168. doi: 10.1111/j.1474-9726.2011.00770.x. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Hintze KJ, Theil EC. DNA and mRNA elements with complementary responses to hemin, antioxidant inducers, and iron control ferritin-L expression. Proc Natl Acad Sci USA. 2005;102:15048–15052. doi: 10.1073/pnas.0505148102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang BW, Ray PD, Iwasaki K, Tsuji Y. Transcriptional regulation of the human ferritin gene by coordinated regulation of Nrf2 and protein arginine methyltransferases PRMT1 and PRMT4. FASEB J. 2013;27:3763–3774. doi: 10.1096/fj.12-226043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki K, Mackenzie EL, Hailemariam K, Sakamoto K, Tsuji Y. Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and role of Ref-1 during erythroid differentiation of K562 cells. Mol Cell Biol. 2006;26:2845–2856. doi: 10.1128/MCB.26.7.2845-2856.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997;11:118–124. [PubMed] [Google Scholar]
- Le S, Connors TJ, Maroney AC. c-Jun N-terminal kinase specifically phosphorylates p66ShcA at serine 36 in response to ultraviolet irradiation. J Biol Chem. 2001;276:48332–48336. doi: 10.1074/jbc.M106612200. [DOI] [PubMed] [Google Scholar]
- Leiser SF, Miller RA. Nrf2 signaling, a mechanism for cellular stress resistance in long-lived mice. Mol Cell Biol. 2010;30:871–884. doi: 10.1128/MCB.01145-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozzio CB, Lozzio BB. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood. 1975;45:321–334. [PubMed] [Google Scholar]
- Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- Macari ER, Lowrey CH. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood. 2011;117:5987–5997. doi: 10.1182/blood-2010-10-314096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008a;10:997–1030. doi: 10.1089/ars.2007.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie EL, Ray PD, Tsuji Y. Role and regulation of ferritin H in rotenone-mediated mitochondrial oxidative stress. Free Radic Biol Med. 2008b;44:1762–1771. doi: 10.1016/j.freeradbiomed.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGahon A, Bissonnette R, Schmitt M, Cotter KM, Green DR, Cotter TG. BCR-ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood. 1994;83:1179–1187. [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Pelicci PG. Apoptosis and aging: role of p66Shc redox protein. Antioxid Redox Signal. 2006;8:600–608. doi: 10.1089/ars.2006.8.600. [DOI] [PubMed] [Google Scholar]
- Morgan J, Oseroff AR, Cheney RT. Expression of the peripheral benzodiazepine receptor is decreased in skin cancers in comparison with normal skin. Br J Dermatol. 2004;151:846–856. doi: 10.1111/j.1365-2133.2004.06198.x. [DOI] [PubMed] [Google Scholar]
- Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Pawson T, Pelicci PG. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- Pi J, Qu W, Reece JM, Kumagai Y, Waalkes MP. Transcription factor Nrf2 activation by inorganic arsenic in cultured keratinocytes: involvement of hydrogen peroxide. Exp Cell Res. 2003;290:234–245. doi: 10.1016/s0014-4827(03)00341-0. [DOI] [PubMed] [Google Scholar]
- Pinton P, et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science. 2007;315:659–663. doi: 10.1126/science.1135380. [DOI] [PubMed] [Google Scholar]
- Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Chemical and molecular regulation of enzymes that detoxify carcinogenes. Proc Natl Acad Sci USA. 1993;90:2965–2969. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey JJ, et al. The influence of Shc proteins on life span in mice. J Gerontol A Biol Sci Med Sci. 2013 doi: 10.1093/gerona/glt198. doi: 10.1093/gerona/glt198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley PT, Ohlsson-Wilhelm BM, Farley BA, LaBella S. Inducers of erythroid differentiation inK562 human leukemia cells. Exp Hematol. 1981;9:32–37. [PubMed] [Google Scholar]
- Sansone P, Storci G, Giovannini C, Pandolfi S, Pianetti S, Taffurelli M, Santini D, Ceccarelli C, Chieco P, Bonafe M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells. 2007;25:807–815. doi: 10.1634/stemcells.2006-0442. [DOI] [PubMed] [Google Scholar]
- Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci USA. 2004;101:1461–1466. doi: 10.1073/pnas.0308083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21:5216–5224. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaya K, Suzuki T, Motohashi H, Onodera K, Satomi S, Kensler TW, Yamamoto M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic Biol Med. 2012;53:817–827. doi: 10.1016/j.freeradbiomed.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y. JunD activates transcription of the human ferritin H gene through an antioxidant response element during oxidative stress. Oncogene. 2005;24:7567–7578. doi: 10.1038/sj.onc.1208901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Coordinate transcriptional and translational regulation of ferritin inresponse to oxidative stress. Mol Cell Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Luzi L, Pacini S, Baldari CT, Pelicci PG. The p66Shc longevity gene is silenced through epigenetic modifications of an alternative promoter. J Biol Chem. 2002;277:22370–22376. doi: 10.1074/jbc.M200280200. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci USA. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills MK, Jones N. Teaching an old dogma new tricks: twenty years of Shc adaptor signalling. Biochem J. 2012;447:1–16. doi: 10.1042/BJ20120769. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.