

Abstract
The ICR-derived glomerulonephritis (ICGN) mouse is a chronic kidney disease (CKD) model that is characterized histologically by glomerulosclerosis, vascular sclerosis and tubulointerstitial fibrosis, and clinically by proteinuria and anemia, which are common symptoms and pathological changes associated with a variety of kidney diseases. Previously, we performed a quantitative trait locus (QTL) analysis to identify the causative genes for proteinuria in ICGN mice, and found a deletion mutation of the tensin 2 gene (Tns2nph, MGI no: 2447990). Interestingly, the congenic strain carrying the Tns2nph mutation on a C57BL/6J (B6) genetic background exhibited milder phenotypes than did ICGN mice, indicating the presence of several modifier genes controlling the disease phenotype. In this study, to identify the modifier/resistant loci for CKD progression in Tns2-deficient mice, we performed QTL analysis using backcross progenies from susceptible ICGN and resistant B6 mice. We identified a significant locus on chromosome (Chr) 2 (LOD = 5.36; 31 cM) and two suggestive loci on Chrs 10 (LOD = 2.27; 64 cM) and X (LOD = 2.65; 67 cM) with linkage to the severity of tubulointerstitial injury. One significant locus on Chr 13 (LOD = 3.49; approximately 14 cM) and one suggestive locus on Chr 2 (LOD = 2.41; 51 cM) were identified as QTLs for the severity of glomerulosclerosis. Suggestive locus in BUN was also detected in the same Chr 2 region (LOD = 2.34; 51 cM). A locus on Chr 2 (36 cM) was significantly linked with HGB (LOD = 4.47) and HCT (LOD = 3.58). Four novel epistatic loci controlling either HGB or tubulointerstitial injury were discovered. Further genetic analysis should lead to identification of CKD modifier gene(s), aiding early diagnosis and providing novel approaches to the discovery of drugs for the treatment and possible prevention of kidney disease.
Introduction
ICR-derived glomerulonephritis (ICGN) mice are an inbred strain that exhibit glomerulosclerosis characterized by marked glomerular hypertrophy, mesangial expansion, foot process effacement of podocytes and thickening of the glomerular basement membrane. The ICGN mouse is also a chronic kidney disease (CKD) model that presents common symptoms and pathological changes associated with a variety of kidney diseases, such as albuminuria, hypoproteinemia, hyperlipidemia, anemia and systemic edema and eventual end-stage renal failure [1], [2]. In ICGN mice, nephrotic syndrome occurs at the suckling stage, which is unique in spontaneously nephropathic strains of mice or rats [3]. Although glomerular alteration is observed from two weeks after birth, renal function is preserved until three to four months after birth, when ICGN mice begin to show tubulointerstitial fibrosis [4]. This pathological process in ICGN mice conforms well with the clinical presentation of glomerular disease in humans [5].
Proteinuria is intimately involved in the dysfunction of podocytes or slit diaphragm, and glomerulosclerosis (GS) sequentially starts with podocytopenia. A line of evidence suggests that podocytes act as the main component of this barrier as mutations in a number of podocyte-specific genes (NPHS1, NPHS2, CD2AP, ACTN4, TRPC6, PLCE1, MYH9) responsible for GS have been identified [6], [7]. Therefore, podocyte loss is a common determining factor for progression toward many types of kidney disease [8], [9]. Previously, we performed a QTL analysis to isolate the causative genes for ICGN phenotypes, and identified a major QTL on chromosome (Chr) 15 that is identical to a single recessive locus causing albuminuria, and found a deletion mutation of the tensin 2 gene (Tns2, also known as Tenc1). This mutation (Tns2nph, MGI no: 2447990) leads to the loss of tensin 2 in podocytes, parietal epithelial cells and tubular epithelial cells [10]. The tensin (Tns) family of multidomain scaffold proteins (including Tns1, Tns2, Tns3 and Tns4) bind to the cytoplasmic tail of β-integrins, and localize to adhesions that anchor stress fibers in cells [11]. Therefore, Tns2 is thought to be a component of the actin cytoskeletal structures linking actin filaments with focal adhesions and plays a role as an intracellular signal transduction mediator through integrin [11]. It has been reported that Tns2 may be involved in the expression of nephrin (Nphs1) and synaptopodin (Synpo) in podocytes [12]. However, the mechanism by which Tns2 deficiency leads to CKD is unclear.
In humans, the prevalence rates of CKD vary considerably by race, sex and age [13]. Multiple studies, such as genome-wide association studies, have identified many genetic factors that participate in kidney disease, although it is estimated that a vast amount of genetic factors for CKD remain to be identified [14]. CKD has a complex etiology resulting from the combination of multiple genetic and environmental factors, each with small effects. Epistatic interactions among susceptibility modifier loci make many of the loci difficult to detect in genome-wide association studies [15], [16]. Several reports using mouse models of nephropathy have demonstrated similar phenomena in that the effects of nephropathy-inducing factors were attenuated in other genetic backgrounds [17], [18], [19]. Recently, we have shown that congenic strains carrying the Tns2nph mutation on a B6 or 129×1/SvJ (129) genetic background exhibited milder phenotypes than do ICGN mice [20], [21]. In contrast, other researchers have reported DBA/2J (D2) congenic mice with Tns2nph were susceptible to CKD at a level similar to ICGN mice [22]. This suggests that B6 and 129 mice have resistant genes to CKD induced by Tns2nph. In the present study, we performed a genome-wide linkage analysis in (B6×ICGN) F1×ICGN backcross mice carrying Tns2nph to identify the CKD resistance genes and elucidate the mechanism by which Tns2 deficiency leads to CKD.
Materials and Methods
Animals
ICGN mice were purchased from Osaka National Institute of Biomedical Innovation. C57BL/6J (B6: Charles River) mice were backcrossed to ICGN mice. Tns2 (nph/nph) N2 backcross progenies were obtained by mating ICGN male mice to Tns2 (+/nph) F1 hybrid female mice heterozygous for Tns2nph. Backcross mice homozygous for Tns2nph were born in accordance with Mendelian inheritance and at an equal sex ratio. B6-Tns2nph congenic mice were generated as described previously [20]. The animal room was air-conditioned at 22±4°C, maintained at 40–60% relative humidity, and mice were maintained under a 12 hr light-dark cycle. A standard laboratory diet, CE-2 (Nihon Clea, Tokyo, Japan), and tap water were available ad libitum. In this study, 16-week-old mice were selected for all analyses as the ICGN mice show severe albuminuria and anemia, which are the end stages of a variety of pathological conditions. A humane end point was applied when the mice with severe anemia became moribund.
Ethical Statement
All research and experimental protocols were adhered to the Regulation for the Care and Use of Laboratory Animals, Hokkaido University and approved by the President of Hokkaido University following to the review of the Institutional Animal Care and Use Committee (Approval ID: No. 110226).
Phenotypic Analysis
Blood samples were collected from the postcava under isoflurane anesthesia. Hemoglobin concentration (HGB), hematocrit (HCT) and blood urea nitrogen (BUN) were measured as described previously [20].
Formalin-fixed and paraffin-embedded kidney blocks were sectioned at a thickness of 2 µm, and stained with Periodic acid-Schiff (PAS). In this study, two types of histopathological scores were adopted to quantitate the severities of renal damage. A glomerular index, in which the severities were based on glomerular injury, was calculated from the scores for twenty glomeruli randomly selected from each sample. The severity of glomerular injury was scored as follows: 0, no abnormality or nearly normal; 1, expansion of the mesangial matrix; 2, severe expansion of the mesangial matrix; 3, hyalinosis or collapse (Fig. S1). First, the highest and lowest scores were removed to exclude bias in random selection. The sum of the eighteen glomerular scores was then taken as the glomerular index. However, if there were glomeruli that scored 3 points, the index was the sum of 36 and the number of glomeruli that scored 3 points. This calculation method means that a kidney with hyalinosis or collapse has a more severe glomerular injury than a kidney with only mesangial expansion. The tubular index was the sum of two histopathological parameters, urinary cast and tubule dilation. The severity of tubule dilation was performed using scale 0 to 2∶0, no abnormality; 1, mild; 2, severe. One point was then added to the tubular index if a urinary cast was detected (Fig. S2). These analyses were performed by two independent observers. There were remarkable congruences in both blinded scores [the interclass correlation coefficients (ICCs) = 0.96].
Genotyping Analysis
We used 246 (male = 124, female = 122) Tns2 (nph/nph) N2 backcross mice for a genome-wide scan. Extraction of genomic DNA from tail tips was performed by a standard method. Tns2 genotypes were determined by PCR using genomic DNA from the tail tips. Tns2nph homozygous mice were identified using the primers AGACACCACCAGCACCTTCT and CGATCCAGCTCCTGTCTTTC, which distinguish the Tns2nph allele by the 8-base deletion in exon 18 of Tns2 [10]. For QTL analysis, a total of 92 informative microsatellite markers and coat color were used for the genotyping analysis, as listed in Table S1. The map positions of the microsatellite markers were based on information from the Mouse Genome Informatics of Jackson Laboratory (MGI; http://www.informatics.jax.org/, MGI_4.41). PCR was carried out with a cycling sequence of 95°C for 5 min (one cycle), followed by 35 cycles consisting of denaturation at 95°C for 30 sec, primer annealing at 55°C for 30 sec, and extension at 72°C for 30 sec. Amplified samples were electrophoresed with 12% polyacrylamide gels and stained with ethidium bromide. The stained gels were then visualized and photographed under an ultraviolet lamp.
QTL Analysis and Statistics
QTL analysis was performed using the Map Manager QTXb20 software program that uses a maximum likelihood algorithm with “interval mapping” and “simultaneous search”, and permits better localization of loci and exclusion mapping [23]. Recombination frequencies (%) were converted into genetic distance (cM) using the Kosambi map function. This program provides linkage data as a likelihood ratio statistic (LRS) score. For each Chr, the LRS values were calculated by 10000 random permutations of the trait values relative to the genotypes of the marker loci. Genome-wide significance thresholds were calculated in terms of LRS by 10000 random permutation tests based on the established guidelines. The logarithm of odds (LOD) score was calculated by dividing the LRS value by 4.61. LOD values of the suggestive and significant thresholds for each trait are summarized in Table 1. Significant linkage was determined according to the traditional threshold (LOD score >3.3) [24]. Statistical analyses were performed using the Stat-View program (SAS Institute Inc. Cary, USA). Spearman’s rank correlation analysis was used to find the correlation between two sets of tested traits.
Table 1. QTL identified for nephropathic traits.
| Chr | Peak (cM) | Nearest marker | LOD | Phenotype | ICGN/ICGNa | B6/ICGNa | Resistance allele |
| 2 | 31 | D2Mit369 | 5.36 | Tubule | 1.0 | 0.4 | B6b ** |
| 2 | 36 | D2Mit380 | 4.47 | HGB (g/dL) | 12.6 | 13.2 | B6c |
| 3.58 | HCT (%) | 43.1 | 45.2 | B6c | |||
| 2 | 51 | D2Mit66 | 2.41 | Glomeruli | 30.2 | 25.9 | B6b ** |
| 2.34 | BUN (mg/dL) | 38.2 | 31.3 | B6c | |||
| 10 | 64 | D10Mit271 | 2.27 | Tubule | 0.8 | 0.4 | B6b * |
| 13 | 14 | D13Mit60 | 3.49 | Glomeruli | 30.4 | 24.9 | B6b ** |
| X | 67 | DXMit186 | 2.65 | Tubule | 0.8 | 0.4 | B6b * |
Data resent the mean of the phenotype.
Mann-Whitney’s U test,
**P<0.001;
*P<0.01.
Student’s T test, P<0.001.
Results
1. Phenotypic Characterization of Tns2nph Backcross Mice
To verify the effect of genetic background on susceptibility to CKD induced by Tns2nph, we produced a total of 246 (ICGN×B6) F1×ICGN backcross mice and performed blood testing and histopathological analysis at the beginning of a genome-wide linkage analysis. Blood samples collected at 16 weeks old were analyzed. In CKD, a high BUN level and low HGB signify renal impairment and renal anemia, respectively, which is also a common symptom in ICGN mice (Fig. 1A, right). In backcross progenies, wide spectrums of BUN and HGB were observed from the various degrees of B6 type to that of ICGN types (Fig. 1A). These results imply the existence of multiple modifier loci for CKD caused by Tns2nph. In addition, the distribution of BUN in backcross males, as compared with females, was statistically more severe (P<0.001, Mann-Whitney’s U test).
Figure 1. Hemoglobin concentration and BUN and kidney injury in backcross mice.

(A, left) Distributions of HGB and BUN in (ICGN×B6) F1×ICGN backcross progenies. (A, right) Distributions of HGB and BUN in B6-Tns2nph, ICGN, (ICGN×B6) F1, backcross mice. (B) Glomerular index and tubular index distributions in (ICGN×B6) F1×ICGN backcross progenies. B6con, B6-Tns2nph; BC, backcross mice; NS, no significant; asterisk, P<0.001.
Figure 1B shows the box plot for glomerular index and the histogram for tubular index. The scores histologically evaluate glomerular injury and tubulointerstitial injury, respectively. The backcross mice showed wide range of the histopathological severity, from B6 types to ICGN types (see also Fig. S1, S2, S3). There were also significant sexual dimorphism in both scores, with males exhibiting a higher level of severity than females (glomerular index P<0.001, tubular index P<0.05, Mann-Whitney’s U test). These differences were presumably due to the protective effect of estrogen in the female glomeruli [25].
The symptom severity of all tested traits exhibited significant correlations with each other according to Spearman’s rank correlation analysis (P<0.01). However, the Spearman’s rank correlation coefficients were comparatively smaller for BUN combinations (|r| = 0.27 ∼ 0.48) than for the other sets (|r| = 0.4 ∼ 0.65).
2. Genome-wide Linkage Analysis for the Mapping of CKD-Resistant Genes in Backcross Mice
To identify the modifier genes for CKD, we carried out a genome-wide linkage analysis using Tns2nph backcross mice. HGB, HCT, BUN, glomerular index and tubular index at 16 weeks of age were used as indicators for CKD. Table 1 summarizes the microsatellite markers linked to the phenotype, LOD score, interval and phenotypic values of each genotype. The QTL analysis of the tubular index, which shows the intensity of tubulointerstitial injury, identified one significant locus on Chr 2 and two suggestive loci on Chrs 10 and X, respectively (Fig. 2A, Table 1). The significant QTL had a peak LOD score of 5.36 and explained 10% of the variance. Regarding the other nephropathic traits (HGB, HCT, BUN and glomerular index), significant or suggestive loci were also detected on Chr 2 (Fig. 2B, Table 1). Within these QTLs, each trait exhibited CKD resistance associated with the ICGN/B6 genotype.
Figure 2. Linkage of nephropathy to chromosome 2.

(A) Genome-wide linkage analysis of the tubular index. (B) LOD plots show the linkage of nephropathic traits to chromosome 2. HGB (solid line) and tubular index (dotted line) yielded significant LOD scores. The approximate 95% confidential intervals: 19.6–56.4 cM (tubule), 26–73 cM (glomeruli), 26–55 cM (HGB), 23–59 cM (HCT), 27–72 cM (BUN). (C) LOD curve and bootstrap histogram of the QTL on chromosome 2 for tubular index.
The QTL analysis of the glomerular index, which shows the severity of GS, identified one significant locus (peak LOD = 3.49) on Chr 13 (Fig. 3A). Grouping backcross mice by their genotype at the marker near the linkage peak, D13Mit60 (14.44 cM), ICGN/B6 heterozygous mice showed statistically milder glomerular injury than did ICGN/ICGN homozygous mice (P<0.001, Mann-Whitney’s U test, Fig. 3B). Although the linkage of the tubular index to Chr 13 was similar to that of glomerular index, the peak LOD score was only 1.73. No suggestive linkages were observed for hematological phenotypes on Chr 13 (Fig. 3A).
Figure 3. LOD score curves on chromosome 13.
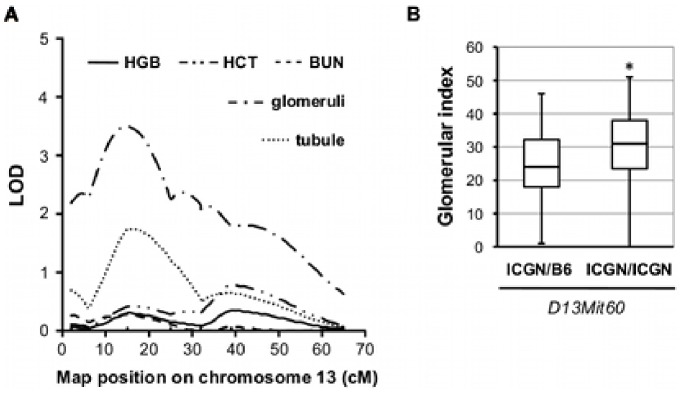
(A) A significant QTL for glomerular index was detected on chromosome 13. The approximate 95% confidential intervals: 0–31.5 cM. (B) Glomerular index at the peak LOD score (D13Mit60). The resistance phenotype associated with ICGN/B6 genotype (P<0.001, Mann-Whitney’s U test).
Epistatic interactions were searched between all of the tested markers by MapManager QTXb20 (P<0.000001). We detected three significant interactions in the tubular index associated with markers on Chrs 2 and 10, which showed significant or suggestive linkages to tubular index (P<0.01, ANOVA test, Fig. 4). Genotype variations in the tubular index at D2Mit380 (40.89 cM), D2Mit293 (17.24 cM) and D10Mit271 (72.31 cM) were only expressed in the presence of the ICGN/ICGN genotype at D19Mit33 (51.76 cM), the ICGN/ICGN genotype at D2Mit102 (57.65 cM) and the ICGN/B6 genotype at D3Mit129 (80.49 cM), respectively. The B6 allele at D19Mit33 also suppressed the genotypic effect of D2Mit380 in HGB. D2Mit293 (17.24 cM), which lies outside the 95% confidence interval for the QTL of the tubular index (19.6–56.4 cM), showed a significant epistatic interaction with D2Mit102, whereas the inner marker D2Mit369 (24.51 cM) did not, although both markers exhibited significant LOD scores of 3.6 and 4.4, respectively. On the other hand, D2Mit380 (40.89 cM) showed a significant epistatic interaction with D19Mit33, as did D2Mit66 (49.45 cM) and D2Mit102 (57.65 cM) (data not shown). These results suggest multiple QTLs for tubular index on Chr 2. The bootstrap analysis also supports this hypothesis (Fig. 2C). Next, to identify the candidate genes, we screened the QTLs on Chrs 2 and 13 for podocyte-related genes using XPodNet, and protein-protein interactions in the podocytes [19]. In total, 46 candidate genes are listed (Table 2 and 3).
Figure 4. Epistasis associated with QTLs.

Epistatic interactions were detected associated with the markers on chromosome 2 and 10. Open circles and closed circles represent homozygous and heterozygous variants for the tested markers, respectively. Asterisks signify significant differences between the two genotypes (**P<0.001; *P<0.01; Mann-Whitney’s U test).
Table 2. Podocyte-related genes on Chr 2 QTL.
| Candidate genes | Interacting proteins (XPodNet) |
| Enkur | Pak1, Pik3r2 |
| Apbb1ip | Enah, Vasp |
| Pax8 | Paxip1, Wt1 (Chr 2∶55 cM) |
| Grin1 | Ctnnb1, Src (Chr 2∶78 cM), Actb, Spnb2, Prkce, Camk2a, Camk2b, Dnm1 (Chr 2∶22 cM), Ppp3ca |
| Notch1 | Wwp1, Psen1, Psen2, Ep300, Rbpj, Wdr12, Fbxw7, Kat2a, Kat2b, Smarcd3 |
| Vav2 | Lcp2, Rac1, Nckipsd, Ptpn6, Src (Chr 2∶78 cM) |
| Rxra | Trim24, Rarb, Rara, Ppara, Thra, Thrb, Zfp423, Nsd1, Med25 |
| Olfm1 | N.D. |
| Tsc1 | Ezr, Ywhab (Chr 2∶85 cM) |
| Abl1 | Nck1, Pxn |
| Lamc3 | Nid1, Nid2, Lama5 (Chr 2∶103 cM) |
| Dnm1 | Atp9a (Chr 2∶89 cM), Grin1 (Chr 2∶17 cM), Grin2b, Sh3gl2, Itsn1, Pfn2, Prnp (Chr 2∶64 cM), Ctsl, Ap2a2, Kif2a, Kif5a, Tuba1a |
| Eng | Tgfbr1, Tgfbr2 |
| Cdk9 | Cdkn1a, Cdkn1b |
| Angptl2 | N.D. |
| Lmx1b | Ldb1, Tcf3, Bmpr2 |
| Mapkap1 | Akt1 |
| Crb2 | N.D. |
| Lrp1b | Pick1 |
| Zeb2 | Smad2 |
| Prpf40a | Wasl |
| Pkp4 | Ctnnb1 |
| Pla2r1 | N.D. |
| Itga6 | Lama5 (Chr.2∶103 cM), Itgb1, Itgb4, Nisch |
| Wipf1 | Cttn |
| Hoxd1 | Ldb1 (Chr.19∶38.75 cM) |
| Itgav | Itgb1, Itgb3, Itgb5, Spp1, Plaur, Col4a3 |
| Ctnnd1 | Yes, Cdh2, Cdh4 (Chr.2∶102 cM), Cdh22 (Chr.2∶85 cM) |
| Lrp4 | Pick1 |
| Syt13 | Nrxn1 |
| Wt1 | Pawr, Wtip, Fhl2, Fhl3, Pax2 (Chr.19∶38 cM), Pax8 (Chr.2∶16 cM) |
| Pax6 | Paxip1 |
N.D.: not detected.
Table 3. Podocyte-related genes on Chr 13 QTL.
| Candidate genes | Interacting proteins (XPodNet) |
| Net1 | Magi1 |
| Actn2 | Camk2a |
| Nid1 | Lama2, Lamc1, Lamc2, Lamc3, Hspg2, Eln |
| E2f3 | Ccnd1 |
| Agtr1a | Agtrap |
| Ripk1 | Atg5 |
| Nm1 | N.D. |
| Bmp6 | Bmpr1a |
| Nedd9 | Src, Nck2, Ptk2 |
| Gadd45g | Cdkn1a |
| Nsd1 | Rxra |
| Slc34a1 | Itgav |
| Dbn1 | Gja1, Lrrk2 |
| Ntrk2 | Cdk5, Cdk5r1 |
N.D.: not detected.
We also detected seven genetic loci associated with blood cell traits (Table S2). These loci (except for Chr 2) were not linked to the histopathological traits, so that the loci only influenced hematological parameters between the two mice, independent of CKD. Correspondingly, numerous genetic loci of this kind have been identified in recent genome-wide linkage analyses of humans and other mouse strains [26], [27], [28], [29].
Discussion
In this study, we report the mapping of genetic loci responsible for the resistance to congenital nephropathy caused by Tns2 deficiency in a B6 genetic background. Analysis of backcrossing between resistant B6 mice and susceptible ICGN mice identified significant QTLs on Chrs 2 and 13. Among the tested traits, tubulointerstitial injury (tubular index) and anemia (HGB and HCT) were strongly associated with the same locus on Chr 2. In patients with CKD, anemia develops due to decreased renal erythropoietin synthesis resulting from tubulointerstitial damage. A significant locus for tubular index, ranging approximately from 19.6 to 56.4 cM on Chr 2, which overlaps the QTLs of the other tested traits, appears to be separated into three QTLs with peaks at 22, 31 and 50 cM (Fig. 2). The proximal QTL (22 cM) has epistatic interactions with D2Mit102 and D2Mit282 (Fig. 4, data not shown). The region between the two markers exhibiting a peak LOD score of 3.8 in tubular index includes the syntenic locus for albuminuria previously reported in humans, rats and mice [30], [31]. The middle QTL (31 cM) maps to the same region that was found in a (B6×D2) F2 intercross and in a (B6×A/J) F2 intercross as a second QTL for albuminuria on Chr 2 [30], [31]. The distal QTL (50 cM), where the glomerular score and BUN showed a suggestive linkage, maps to the same region as the suggestive locus for proteinuria that was found in an HIV-associated nephropathy (HIVAN) model mouse on a mixed genetic background of B6 and FVB [32]. The syntenic human region (2q32.1) is associated with the early onset of end-stage renal disease in black families enriched for nondiabetic nephropathy [33]. Taken together, the resistance to CKD in Tns2-deficient B6 mice can be mainly exerted in combination with the multiple loci on Chr 2.
While Tns2 mRNA is broadly expressed in the kidney and other organs, significant abnormalities (i.e., podocytopenia) were only observed in the kidney of ICGN mice [1]. In the glomeruli, Tns2 expression is localized in podocytes [10], [34]. Therefore, the dysfunction of Tns2 in podocytes has been speculated to cause renal dysfunction. Tns is an actin-binding protein that is present in focal adhesions and has been suggested to play an important role in the actin cytoskeleton and integrin-mediated signaling [11]. Recently, it has been reported that Tns2 may bind to integrin β1, and regulate the activity of the integrin β1 downstream effector molecule, integrin-linked kinase (ILK), which is known to play an essential role in GBM assembly and podocyte function [22]. In addition, ILK expression is stimulated in podocytes by renal injury and induces podocyte epithelial-mesenchymal transition [35]. Interestingly, the podocyte-specific knockout of ILK induces progressive focal segmental GS with increased mesangial matrix deposition and downregulation of Nphs1 and Synpo with age, as observed in ICGN mice [12], [36], [37]. Activation of FAK, another effector molecule of the integrin β1 signaling pathway, is also observed in the glomeruli of Tns2 mutant mice and conditional Ilk knockout mice [22], [37]. This upregulation of FAK activity is considered to compensate for the ILK abnormality structurally, but not functionally [22], [37]. It is notable that the Ilk knockout genetic background is derived from B6 (NPHS2 promoter-Cre transgenic mice) and 129 (ILKflox /flox mice) [36], [37]. That is, the genetic backgrounds of B6 and 129 are resistant to Tns2-deficiency, but their resistance has no effect on Ilk-deficiency. Hence, it is assumed that the resistance effects are exhibited by functionally regulating the disorders in the integrin β1-ILK signaling pathway or structurally reinforcing the fragility of the actin cytoskeleton and slit diaphragm induced by aberrant ILK activity in Tns2-deficient podocytes.
Under this assumption, we screened QTLs mapped on Chr 2 for podocyte-related genes using XPodNet and identified 32 candidate genes (Table 2). Among these, we were especially interested in Notch1, Dnm1, Lmx1b, Zeb2 and Wt1. NOTCH1 (18.91 cM) is a transmembrane receptor protein whose signaling pathway correlates with GS and renal function [38]. DNM1 (22.09 cM), dynamin1, participates in endocytosis in the podocyte foot process and contributes to maintenance of the slit diaphragm [39]. WT1 (55.06 cM) and LMX1B (22.48 cM) are transcription factors that regulate podocyte-specific structural genes, Nphs1 and Podxl, and Nphs2 respectively, and play a key role in the development and maintenance of the actin cytoskeleton and slit diaphragm in podocytes [40], [41], [42]. ZEB2 (27.31 cM), a transcriptional repressor of E-cadherin, is upregulated by the integrin β1-ILK signaling pathway downstream target SNAIL and induces the epithelial–mesenchymal transition [43].
In the same way, 14 candidate genes of the Chr 13 QTL are listed in Table 3. This locus partially overlaps the HIVAN susceptibility locus, HIVAN2, that was found in an (HIV-1 transgenic FVB×B6) F2 intercross [32]. An expression quantitative trait locus (eQTL) analysis has revealed that HIVAN2 influences the transcript abundance of Nphs2, a membrane protein linking the slit diaphragm to the cytoskeleton [32]. However, HIVAN2 includes none of transcription factors known to regulate Nphs2 expression. In our cross, the locus was not linked to anemia, so that it is not considered to be essential for susceptibility to CKD induced by Tns2nph.
Our genomic analysis of CKD in a cross between B6 and ICGN assumes that the Chr 2 loci critically contribute to resistance in Tns2-deficient mice. The Chr 2 QTLs identified in crosses for proteinuria between resistant B6 and susceptible D2 or A/J or HIV-1 transgenic FVB are similar to ours [30], [31], [32]. This suggests that our QTL is not limited to resistance to the CKD induced by Tns2nph, but is generally associated with resistance to CKD. Although this locus has not been noted in previous reports due to the absence of a homologous human QTL, we believe that this locus also plays an important role in resistance to CKD. Identification of resistant genes on this locus would help reveal the mechanism by which Tns2 deficiency leads to CKD and provide a novel insight into susceptibility to CKD, leading to the development of treatment strategies for CKD.
Supporting Information
Representative examples of glomerular score.
(TIF)
Representative examples of tubular score.
(TIF)
Histopathological severity, resistant strain vs. susceptible strain.
(TIF)
Genotyping markers.
(XLSX)
QTLs identified for hematological phenotypes.
(PPTX)
Funding Statement
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Glant-in-Aid for Scientific Research, KAKENHI (20302614) to N.S. H.S. was a research assistant of the Program of Leading Graduate School supported by MEXT. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ogura A, Asano T, Matsuda J, Takano K, Nakagawa M, et al. (1989) Characteristics of mutant mice (ICGN) with spontaneous renal lesions: a new model for human nephrotic syndrome. Lab Anim 23: 169–174. [DOI] [PubMed] [Google Scholar]
- 2. Mizuno S, Matsumoto K, Kurosawa T, Mizuno-Horikawa Y, Nakamura T (2000) Reciprocal balance of hepatocyte growth factor and transforming growth factor-beta 1 in renal fibrosis in mice. Kidney Int 57: 937–948. [DOI] [PubMed] [Google Scholar]
- 3. Mizuno S, Mizuno-Horikawa Y, Yue BF, Okamoto M, Kurosawa T (1999) Nephrotic mice (ICGN strain): a model of diffuse mesangial sclerosis in infantile nephrotic syndrome. Am J Nephrol 19: 73–82. [DOI] [PubMed] [Google Scholar]
- 4. Ogura A, Fujimura H, Asano T, Koura M, Naito I, et al. (1995) Early ultrastructural glomerular alterations in neonatal nephrotic mice (ICGN strain). Vet Pathol 32: 321–323. [DOI] [PubMed] [Google Scholar]
- 5. Mizuno S, Yue BF, Okamoto M, Horikawa Y, Kurosawa T (1997) Diffuse glomerulosclerosis without tubular injury does not directly manifest renal dysfunction in nephrotic mice (ICGN strain). Exp Nephrol 5: 498–507. [PubMed] [Google Scholar]
- 6. Mundel P, Schwarz K, Reiser J (2001) Podocyte biology: a footstep further. Adv Nephrol Necker Hosp 31: 235–241. [PubMed] [Google Scholar]
- 7. Wiggins RC (2007) The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71: 1205–1214. [DOI] [PubMed] [Google Scholar]
- 8. Pavenstadt H, Kriz W, Kretzler M (2003) Cell biology of the glomerular podocyte. Physiol Rev 83: 253–307. [DOI] [PubMed] [Google Scholar]
- 9. Warsow G, Endlich N, Schordan E, Schordan S, Chilukoti RK, et al. (2013) PodNet, a protein-protein interaction network of the podocyte. Kidney Int 84: 104–115. [DOI] [PubMed] [Google Scholar]
- 10. Cho AR, Uchio-Yamada K, Torigai T, Miyamoto T, Miyoshi I, et al. (2006) Deficiency of the tensin 2 gene in the ICGN mouse: an animal model for congenital nephrotic syndrome. Mamm Genome 17: 407–416. [DOI] [PubMed] [Google Scholar]
- 11. Lo SH (2004) Tensin. Int J Biochem Cell Biol 36: 31–34. [DOI] [PubMed] [Google Scholar]
- 12. Kato T, Mizuno S, Taketo MM, Kurosawa TM (2008) The possible involvement of tensin 2 in the expression and extension of nephrin by glomerular podocytes in mice. Biomed Res 29: 279–287. [DOI] [PubMed] [Google Scholar]
- 13. Stevens LA, Claybon MA, Schmid CH, Chen J, Horio M, et al. (2011) Evaluation of the Chronic Kidney Disease Epidemiology Collaboration equation for estimating the glomerular filtration rate in multiple ethnicities. Kidney Int 79: 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kottgen A, Pattaro C, Boger CA, Fuchsberger C, Olden M, et al. (2010) New loci associated with kidney function and chronic kidney disease. Nat Genet 42: 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, et al. (2009) Finding the missing heritability of complex diseases. Nature 461: 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boger CA, Heid IM (2011) Chronic kidney disease: novel insights from genome-wide association studies. Kidney Blood Press Res 34: 225–234. [DOI] [PubMed] [Google Scholar]
- 17. Andrews KL, Mudd JL, Li C, Miner JH (2002) Quantitative trait loci influence renal disease progression in a mouse model of Alport syndrome. Am J Pathol 160: 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hartner A, Cordasic N, Klanke B, Veelken R, Hilgers KF (2003) Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol Dial Transplant 18: 1999–2004. [DOI] [PubMed] [Google Scholar]
- 19. Roselli S, Heidet L, Sich M, Henger A, Kretzler M, et al. (2004) Early glomerular filtration defect and severe renal disease in podocin-deficient mice. Mol Cell Biol 24: 550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nishino T, Sasaki N, Nagasaki K, Ahmad Z, Agui T (2010) Genetic background strongly influences the severity of glomerulosclerosis in mice. J Vet Med Sci 72: 1313–1318. [DOI] [PubMed] [Google Scholar]
- 21. Nishino T, Sasaki N, Nagasaki K, Ichii O, Kon Y, et al. (2012) The 129 genetic background affects susceptibility to glomerulosclerosis in tensin 2-deficient mice. Biomed Res 33: 53–56. [DOI] [PubMed] [Google Scholar]
- 22. Uchio-Yamada K, Sawada K, Tamura K, Katayama S, Monobe Y, et al. (2013) Tenc1-deficient mice develop glomerular disease in a strain-specific manner. Nephron Exp Nephrol 123: 22–33. [DOI] [PubMed] [Google Scholar]
- 23. Manly KF, Cudmore RH Jr, Meer JM (2001) Map Manager QTX, cross-platform software for genetic mapping. Mamm Genome 12: 930–932. [DOI] [PubMed] [Google Scholar]
- 24. Kruglyak L, Lander ES (1995) A nonparametric approach for mapping quantitative trait loci. Genetics 139: 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Elliot SJ, Karl M, Berho M, Potier M, Zheng F, et al. (2003) Estrogen deficiency accelerates progression of glomerulosclerosis in susceptible mice. Am J Pathol 162: 1441–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van der Harst P, Zhang W, Mateo Leach I, Rendon A, Verweij N, et al. (2012) Seventy-five genetic loci influencing the human red blood cell. Nature 492: 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peters LL, Shavit JA, Lambert AJ, Tsaih SW, Li Q, et al. (2010) Sequence variation at multiple loci influences red cell hemoglobin concentration. Blood 116: e139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Davis RC, van Nas A, Bennett B, Orozco L, Pan C, et al. (2013) Genome-wide association mapping of blood cell traits in mice. Mamm Genome 24: 105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kelada SN, Aylor DL, Peck BC, Ryan JF, Tavarez U, et al. (2012) Genetic analysis of hematological parameters in incipient lines of the collaborative cross. G3 (Bethesda) 2: 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sheehan S, Tsaih SW, King BL, Stanton C, Churchill GA, et al. (2007) Genetic analysis of albuminuria in a cross between C57BL/6J and DBA/2J mice. Am J Physiol Renal Physiol 293: F1649–1656. [DOI] [PubMed] [Google Scholar]
- 31. Doorenbos C, Tsaih SW, Sheehan S, Ishimori N, Navis G, et al. (2008) Quantitative trait loci for urinary albumin in crosses between C57BL/6J and A/J inbred mice in the presence and absence of Apoe. Genetics 179: 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Papeta N, Chan KT, Prakash S, Martino J, Kiryluk K, et al. (2009) Susceptibility loci for murine HIV-associated nephropathy encode trans-regulators of podocyte gene expression. J Clin Invest 119: 1178–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Freedman BI, Langefeld CD, Rich SS, Valis CJ, Sale MM, et al. (2004) A genome scan for ESRD in black families enriched for nondiabetic nephropathy. J Am Soc Nephrol 15: 2719–2727. [DOI] [PubMed] [Google Scholar]
- 34. Nishino T, Sasaki N, Chihara M, Nagasaki K, Torigoe D, et al. (2012) Distinct distribution of the tensin family in the mouse kidney and small intestine. Exp Anim 61: 525–532. [DOI] [PubMed] [Google Scholar]
- 35. Kang YS, Li Y, Dai C, Kiss LP, Wu C, et al. (2010) Inhibition of integrin-linked kinase blocks podocyte epithelial-mesenchymal transition and ameliorates proteinuria. Kidney Int 78: 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. El-Aouni C, Herbach N, Blattner SM, Henger A, Rastaldi MP, et al. (2006) Podocyte-specific deletion of integrin-linked kinase results in severe glomerular basement membrane alterations and progressive glomerulosclerosis. J Am Soc Nephrol 17: 1334–1344. [DOI] [PubMed] [Google Scholar]
- 37. Kanasaki K, Kanda Y, Palmsten K, Tanjore H, Lee SB, et al. (2008) Integrin beta1-mediated matrix assembly and signaling are critical for the normal development and function of the kidney glomerulus. Dev Biol 313: 584–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murea M, Park JK, Sharma S, Kato H, Gruenwald A, et al. (2010) Expression of Notch pathway proteins correlates with albuminuria, glomerulosclerosis, and renal function. Kidney Int 78: 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soda K, Balkin DM, Ferguson SM, Paradise S, Milosevic I, et al. (2012) Role of dynamin, synaptojanin, and endophilin in podocyte foot processes. J Clin Invest 122: 4401–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harendza S, Stahl RA, Schneider A (2009) The transcriptional regulation of podocin (NPHS2) by Lmx1b and a promoter single nucleotide polymorphism. Cell Mol Biol Lett 14: 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burghardt T, Kastner J, Suleiman H, Rivera-Milla E, Stepanova N, et al. (2013) LMX1B is essential for the maintenance of differentiated podocytes in adult kidneys. J Am Soc Nephrol 24: 1830–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo JK, Menke AL, Gubler MC, Clarke AR, Harrison D, et al. (2002) WT1 is a key regulator of podocyte function: reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Hum Mol Genet 11: 651–659. [DOI] [PubMed] [Google Scholar]
- 43. Beltran M, Puig I, Pena C, Garcia JM, Alvarez AB, et al. (2008) A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev 22: 756–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative examples of glomerular score.
(TIF)
Representative examples of tubular score.
(TIF)
Histopathological severity, resistant strain vs. susceptible strain.
(TIF)
Genotyping markers.
(XLSX)
QTLs identified for hematological phenotypes.
(PPTX)