

We recently reported picosecond time-resolved crystallographic investigations on photoactive yellow protein1. Now Schotte and colleagues2 challenge the structural interpretation of our results based on their work3. In particular, they disagree with structural details of our earliest intermediate IT and the next intermediate ICT based on their density functional theory (DFT) calculations. We stand by our results because, for both intermediates, the time-resolved X-ray data and the experimental electron densities favor the structures that we reported over the structures derived from DFT.
Our study1 and that by Schotte et al. 2 used the same experimental technique but differ in that (1) in our work we also studied the E46Q mutant as well as the wild-type (WT) protein, (2) the crystals were grown under quite different conditions, and (3) the X-ray data quality and crystallographic completeness differ.
Figure 1 shows the dependence of the R factor on three dihedral angles. Whereas IT is located at the minimum R factor, both the DFT structure (ITDFT) and their earliest structure of Schotte et al. 2 (pR0), which supposedly corresponds to IT, are far from the minimum. Notably, in `Structure Refinement' section of the Supporting Information of their paper, they report that pR0 also tends to adopt a C1'–C3'–C2'–C1 dihedral angle close to 90° as found in IT when the structure was not restrained to resemble ITDFT. Thus, the refined dihedral angle varies depending on whether the structure is restrained to mimic ITDFT (their approach) or is allowed to follow the experimental electron density (our approach). Our approach was to compare the qualitative features of ITDFT and IT instead of using ITDFT as the structural restraints. Although IT obtained without such restraints has a different dihedral angle than that of ITDFT, we were content with the fact that ITDFT also supports a non-planar structure consistent with IT. We believed that forcing the structural refinement of the experimental density to meet the restraints from the DFT structure removes the possibility that the experimental data could provide any new information other than the boundary given by DFT. Strictly speaking, no new information was obtained in their study, and such an approach would always yield only those structures compatible with DFT even when new experimental data with better temporal and spatial resolution become available from experiments at X-ray free electron lasers. The possibility of interactions that stabilize such a highly strained dihedral angle but are not fully accounted for by DFT cannot be ruled out. In addition, because DFT is a single-reference based method, DFT may not well describe such a highly distorted structure of the chromophore whose multiconfigurational character might be strong. It should also be noted that IT is observed in both E46Q and WT with a dihedral angle C1'–C3'–C2'–C1 that is smaller in E46Q by ~15°.
Figure 1.
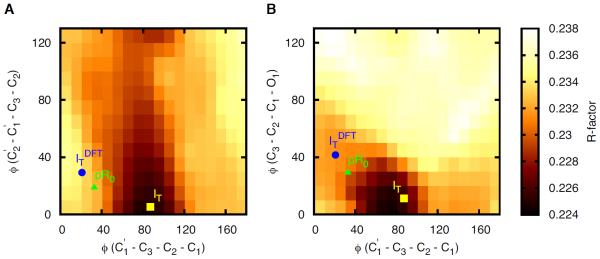
The dependence of the R factor on the three dihedral angles (A: C2'–C1'–C3'–C2 vs C1'–C3'–C2'–C1 and B: C3'–C2'–C1'–O1 vs C1'–C3'–C2'–C1). Whereas IT is located at the minimum R factor, ITDFT and pR0 are far from the minimum. The same situation is found also for ICT vs ICP (data not shown).
Regarding the choice between ICT and ICP: our maps in the present study are consistent with those Ihee et al. reported earlier4. In that earlier work, due to the limited time resolution, Ihee et al. used only a single structure, ICP, to fit the maps at the time delays on nanosecond time scales. This fit was only partly satisfactory and left some residual density. Further, the single ICP structure does not have the minimum R-factor. Our work in ref. 1 explains these observations: the maps in both the present and the earlier studies are structurally heterogeneous and contain ICT and pR1. Schotte et al.2 explain this residual, non-planar density differently: they assume an equilibrium between the first and second intermediates, but this kinetic scenario gives a worse fit to our experimental densities for both WT and E46Q. Moreover, such an equilibrium is highly unlikely because, at early times, the chromophore is highly strained and the reactions is likely to proceed strongly downhill.
Although Schotte et al. 2 direct their major attention to the detailed structural features of the early intermediates, a more serious discrepancy between us is found even on the well-established microsecond time range. They identify2 only one structural species (pR2) whereas our study11 and others4,5 reported that two species (pR1 and pR2) co-exist. It is not clear yet whether this discrepancy arises from the experimental conditions, or from data analysis and interpretation. Our conditions of lower salt (50 mM NaCl) and neutral pH (pH 7.0) have been extensively used for earlier time-resolved X-ray crystallographic investigations of PYP. In contrast, Schotte et al. used crystals grown in high salt (1.1 M NaCl) and D2O (pD 9.0), although their ammonium sulfate concentration (~2.5 M) was close to ours (~2.6 M). Because salt and pH may well affect the structure and dynamics6–8, the exact origin of these discrepancies remains to be studied.
Acknowledgements
This work was supported by Institute for Basic Science (IBS)[CA1301-01], and the National Science Foundation, NSF, USA, grants NSF-STC 1231306 and NSF 0952643.
References
- 1.Jung YO, et al. Volume-conserving trans–cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography. Nature Chem. 2013;5:212–220. doi: 10.1038/nchem.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schotte F, et al. Watching a signaling protein function in real time via 100-ps time-resolved Laue crystallography. Proc. Natl Acad. Sci. USA. 2012;109:19256–19261. doi: 10.1073/pnas.1210938109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaila VRI, Schotte F, Cho HS, Hummer G, Anfinrud PA. Contradictions in X-ray structures of intermediates in the photocycle of photoactive yellow protein. Nature Chem. 2014;6:258–259. doi: 10.1038/nchem.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ihee H, et al. Visualizing reaction pathways in photoactive yellow protein from nanoseconds to seconds. Proc. Natl Acad. Sci. USA. 2005;102:7145–7150. doi: 10.1073/pnas.0409035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim TW, et al. Protein structural dynamics of photoactive yellow protein in solution revealed by pump-probe X-ray solution scattering. J. Am. Chem. Soc. 2012;134:3145–3153. doi: 10.1021/ja210435n. [DOI] [PubMed] [Google Scholar]
- 6.Harigai M, Imamoto Y, Kamikubo H, Yamazaki Y, Kataoka M. Role of an N-terminal loop in the secondary structural change of photoactive yellow protein. Biochemistry. 2003;42:13893–13900. doi: 10.1021/bi034814e. [DOI] [PubMed] [Google Scholar]
- 7.Hoersch D, et al. Role of a conserved salt bridge between the PAS core and the N-terminal domain in the activation of the photoreceptor photoactive yellow protein. Biophys. J. 2007;93:1687–1699. doi: 10.1529/biophysj.107.106633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tripathi S, Srajer V, Purwar N, Henning R, Schmidt M. pH Dependence of the Photoactive Yellow Protein Photocycle Investigated by Time-Resolved Crystallography. Biophys. J. 2012;102:325–332. doi: 10.1016/j.bpj.2011.11.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]