

Abstract
Purpose
The aim of this study was to investigate the mutation spectrum and frequency of 34 known genes in 18 Chinese families with congenital cataracts.
Methods
Genomic DNA and clinical data was collected from 18 families with congenital cataracts. Variations in 34 cataract-associated genes were screened by whole exome sequencing and then validated by Sanger sequencing.
Results
Eleven candidate variants in seven of the 34 genes were detected by exome sequencing and then confirmed by Sanger sequencing, including two variants predicted to be benign and the other pathogenic mutations. The nine mutations were present in 9 of the 18 (50%) families with congenital cataracts. Of the four families with mutations in the X-linked NHS gene, no other abnormalities were recorded except for cataract, in which a pseudo-dominant inheritance form was suggested, as female carriers also had different forms of cataracts.
Conclusion
This study expands the mutation spectrum and frequency of genes responsible for congenital cataract. Mutation in NHS is a common cause of nonsyndromic congenital cataract with pseudo-autosomal dominant inheritance. Combined with our previous studies, a genetic basis could be identified in 67.6% of families with congenital cataracts in our case series, in which mutations in genes encoding crystallins, genes encoding connexins, and NHS are responsible for 29.4%, 14.7%, and 11.8% of families, respectively. Our results suggest that mutations in NHS are the common cause of congenital cataract, both syndromic and nonsyndromic.
Introduction
Congenital cataract is the most common cause of childhood blindness. Its prevalence is 1–6 per 10000 live births, but it can be as high as 5–15 per 10,000 in less developed areas of the world [1]–[3]. Congenital cataracts can occur independently or be accompanied by other ocular and/or systemic abnormalities, and are thus designated as “nonsyndromic” or “syndromic” forms [4]. Genetic defect is a common cause of congenital cataract and was estimated to account for about 25% of patients [5]. Autosomal dominant inheritance is the most common form, although other forms of inheritance have also been described. To date, at least 34 genes have been reported to cause congenital cataracts (Cat-Map; http://cat-map.wustl.edu/) [6] (Table 1). Identification of the genetic basis is a great challenge in congenital cataracts, as it is highly heterogeneous in term of genetic and clinical phenotypes. Some of the known genes were selected for analysis in a cohort of patients in few studies and the results showed that the mutation frequencies have great differences [7]–[12]. We previously conducted similar studies and were able to detect mutation in 14 of 34 (41%) families with congenital cataracts [13]–[15]. In order to identify the genetic cause of the remaining and newly recruited families with congenital cataracts, whole exome sequencing was used in this study to screen the mutations, and then the detected variants were confirmed by Sanger sequencing. Nine mutations were identified in the 18 families; of those NHS mutations were found in four families.
Table 1. Information of the 34 genes referred in this study.
Note: AD = autosomal dominant; AR = autosomal recessive; XL = X-linked.
Methods
Patients
Written informed consent conforming to the tenets of the Declaration of Helsinki and following the Guidance of Sample Collection of Human Genetic Diseases (863-plan) by the Ministry of Public Health of China were obtained from all participating individuals or their guardians of the 18 families enrolled in this study. This study was approved by the Institutional Review Board of the Zhongshan Ophthalmic Center. Of the 18 families, ten were from our previous studies with no mutations identified [13]–[15], while the other eight families had not been analyzed before. Thirteen families with family history showed autosomal dominant inheritance, two were sporadic patients, while the other three were not clear if they had family histories. None of them were recorded to have systemic abnormalities. Ocular examinations were performed by ophthalmologists in Zhongshan Ophthalmic Center. Congenital cataract means that cataracts were noticed at birth or in the first few months. Microcornea was defined as a cornea whose horizontal diameter was less than 10 mm. The procedures for obtaining written informed consent and for preparing of genomic DNA were the same as previously described [13],[14],[16].
Exome Sequencing
Exome sequencing was performed by Macrogen (http://www.macrogen.com/), a commercial service. The criteria to select samples for exome sequencing included: 1. The total amount of genome DNA can't be less than 5 ug; 2. There was no smear by running on an agarose gel. All the 18 samples of probands from 18 unrelated families were involved in exome sequencing. Exome capture was carried out using an Illumina TruSeq Exome Enrichment Kit (62 M) array. The kit included 340,427 probes (95 mer DNA probes) and could enrich about 201,121 exons and cover about 97.2% CCDS region. Exome-enriched DNA fragments were sequenced by an Illumina HiSeq2000; the average sequencing depth was 125-fold. Over 99% base call accuracy was up to Q20, which means that the probability of an incorrect base call is 0.01. After the low quality reads were filtered, the clean data will be aligned to the consensus sequence (UCSC hg19) to detect variants by SAMtools. Additional bioinformatics analysis of all the variants were provided from dbSNP (http://www.ncbi.nlm.nih.gov/), 1000 Genome (http://browser.1000genomes.org/index.html), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and GERP (http://mendel.stanford.edu/SidowLab/downloads/gerp/) online tools.
Variants Analysis
From the exome sequencing results of the 18 probands, detected variants in the 34 known causative genes were summarized. Then we excluded the variants which we don't considered pathogenic as the following criteria: 1. Minor allele frequency (MAF) ≥0.01 from 1000 Human Genome Project database; 2. Located in non-coding region without affecting splicing site; 3. Synonymous variants without affecting splicing site; 4. Only one single heterozygous variation detected in recessive genes. All the other variants were considered pathogenic and summarized for validation.
Sanger Sequencing
Sanger sequencing was used to confirm the potential pathogenic variants detected by exome sequencing, including missense, nonsense, indels, and splice site variants. Primers used to amplify the sequence with each variant were either referred to in previous studies [13],[15],[17] or designed by online tool Primer3 (http://frodo.wi.mit.edu/primer3/) (Table S1). The nucleotide sequences of amplicons were analyzed using an ABI BigDye Terminator cycle sequencing kit v3.1 (Applied Biosystems, Foster City, CA) on an ABI 3130 Genetic Analyzer (Applied Biosystems). Sequencing results were aligned with consensus sequences from the National Center for Biotechnology Information (NCBI) human genome database (http://www.ncbi.nlm.nih.gov/), using the SeqManII program of the Lasergene package (DNAStar Inc. Madison, WI). Confirmed variants were further sequenced in the available family members and 96 unrelated control individuals. The descriptions of the variants followed the nomenclature recommended by the Human Genomic Variation Society (HGVS; http://www.hgvs.org/mutnomen/). The effects of missense variations were evaluated by the PolyPhen-2 [18] and SIFT [19] online tools, and the effects of intronic variants on splicing site changes were predicted by the Berkeley Drosophila Genome Project (BDGP; http://www.fruitfly.org/seq_tools/splice.html [20].
Results
From the whole exome sequencing results of the 18 probands, a total of 1545 variants were identified in the 34 known genes. Twelve variants of them were considered potential pathogenic after we excluded all the variants which were not pathogenic. And 11 of them were confirmed while one was false positive by Sanger sequencing. The 11 variants were present in seven genes and were identified in 11 of the 18 families with congenital cataracts (Table 2, Table 3, Figure 1, and Figure S1). Two of the 11 variants were predicted to be benign (Table 3), while the other 9 were likely to be pathogenic (Table 2). None of the 11 variants was found in 96 normal controls. Four of the 11 variants were identified in NHS in four of the 18 (22.2%) families. The other seven mutations were identified in six genes, including two mutations in GJA8 (gap junction protein, alpha-8; MIM: 600897), and one mutation each in CRYBA4 (crystallin, beta-A4; MIM: 123631), CRYBB2 (crystallin, beta-B2; MIM: 123620), EPHA2 (ephrin receptor EphA2; MIM: 176946), MAF (v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog; MIM: 177075), and MIP (major intrinsic protein of lens fiber; MIM: 154050). The four mutations in NHS included two nonsense, one frameshift, and one splice site mutations. Analysis of available family members in three families demonstrated cosegregation of the mutation with the disease (Figure 1; families 4, 6, and 9). Conservation alignment analysis of the six missense mutations showed relatively conserved residues (Figure 2). None of the 11 variants was present in the 96 normal controls.
Table 2. The 9 mutations identified in 9 of 18 Chinese families with congenital cataract.
| Variations | Bioinformation prediction | Variations in | ||||||||
| Family ID | Gene | Nucleotide | Amino acid | Status | Polyphen-2 | SIFT | Splice | patients | controls | Note |
| Family 2 | CRYBB2 | c.326T>A | p.I109N | Hetero | PD | D | / | 1/18 | 0/96 | Novel |
| Family 3 | EPHA2 | c.1046C>T | p.T349M | Hetero | PD | D | / | 1/18 | 0/96 | rs200490325 |
| Family 4 | GJA8 | c.130G>A | p.V44M | Hetero | PD | D | / | 1/18 | 0/96 | Novel |
| Family 6 | MAF | c.880C>T | p.R294W | Hetero | PD | D | / | 1/18 | 0/96 | Novel |
| Family 7 | MIP | c.500delC | p.A169Pfs*15 | Hetero | / | / | / | 1/18 | 0/96 | Novel |
| Family 8 | NHS | c.556G>T | p.E186* | Hetero | / | / | / | 1/18 | 0/96 | Novel |
| Family 9 | NHS | c.853-1G>A | / | Hemi | / | / | DSA | 1/18 | 0/96 | Novel |
| Family 10 | NHS | c.1117C>T | p.R373* | Hemi | / | / | / | 1/18 | 0/96 | Reported (rs132630322) |
| Family 11 | NHS | c.2716_2719delTTAG | p.L906Mfs*24 | Hetero | / | / | / | 1/18 | 0/96 | Novel |
Note: Hetero = heterozygosity; Hemi = hemizygosity; D = damaging; PD = probably damaging; DSA = donor site abolished.
Table 3. The two variants that can't be excluded.
| Variations | Bioinformation prediction | Variations in | ||||||||
| Family ID | Gene | Nucleotide | Amino acid | Status | Polyphen-2 | SIFT | Splice | patients | controls | Note |
| Family 1 | CRYBA4 | c.26C>T | p.A9V | Hetero | Benign | Tolerate | / | 1/18 | 0/96 | Novel |
| Family 5 | GJA8 | c.367G>A | p.G123S | Hetero | Benign | Tolerate | / | 1/18 | 0/96 | COSM1333689 |
Note: Hetero = heterozygosity.
Figure 1. Pedigrees of the 9 available families with mutations.

The family number and their causative gene were just noted above the pedigree. The mutations of available members were noted beside or below the members. The – indicated that the mutation was not present in the chromosome while the 0 indicated that males had only one X chromosome. Family 8 was recorded to have a history of congenital cataracts but the details of the pedigree are not available. The
Figure 2. Conservation alignments for the five missense mutations.

The amoni acid position with p.I109N in CRYBB2, p.V44M in GJA8, and p.R294W in MAF are highly conserved, while the p.A9V in CRYBA4, the p.T349M inEPHA2, and the p. G123S in GJA8 are not highly conserved.
The clinical data of the probands with mutations are listed in Table 4. Of the four families with mutations in NHS, an X-linked gene, three (family 9, family 10, and family 11) showed congenital cataracts, as well as microcornea and nystagmus. The fourth one, family 8, with NHS c.556G>T (p.E186*) mutation, showed punctate cataract and high myopia (−7.5D for the right eye and −8.0D for the left eye). There were no records of other abnormalities in the patients with NHS mutations. Clinical re-examination was only possible for two affected individuals and an unaffected member (II:1, II∶2, and III∶1) of family 9, with the c.853-1G>A mutation in NHS. The proband (III∶1) harbored a hemizygous mutation and his affected mother (II∶2) harbored a heterozygous mutation, while his unaffected father (II∶1) did not carry the mutation. The proband (Figure 1; family 9-III∶1) had undergone cataract surgery nine years prior to the study, so that cataract was no longer observed (Figure 3A). His affected mother (Figure 1; family 9-II∶2) had posterior subcapsule opacification in her left eye; her right eye underwent cataract surgery when she was a teenager (Figure 3D). The horizontal corneal diameter of the proband (III∶1) was 10 mm in both eyes (Figure 3A) while those of his affected mother (II∶2) were 9 mm in the left eye (figure 3E) and 8 mm in the right eye (Figure 3F). Both the mother and son had bilateral nystagmus. In addition, the proband (III∶1) showed bilateral tigroid retinal change in the temporal region of the optic disc (Figure 3B). The proband (III∶1) had abnormal teeth (figure 3C), but atypical for Nance-Horan syndrome. His mother had lost all her teeth when she was forty.
Table 4. Clinical features of affected individuals with variants identified in this study.
| Variations | Sex | Age (yrs) at | Inheri- | Visual acuity | Cataract types | Cornea diameter | Nystagmus | Other abnormalities | |||
| Family ID | Gene | Nucleotide | Exam | Onset | tance | Right;Left | Right;Left | ||||
| Family 1 | CRYBA4 | c.26C>T | M | 5 | NA | S | 0.2;0.1 | Lamellar, punctate | NA | No | Myopia |
| Family 2 | CRYBB2 | c.326T>A | M | NA | NA | AD | 0.07;0.1 | membrane cataract | 10 mm;10 mm | Yes | No |
| Family 3 | EPHA2 | c.1046C>T | F | 6 | FMB | AD | 0.1;0.1 | Nuclear | NA | No | StrabismusOS |
| Family 4 | GJA8 | c.130G>A | F | 7 | FMB | AD | 0.1;0.1 | NA | NA | Yes | No |
| Family 5 | GJA8 | c.367G>A | M | 31 | NA | AD | FC;0.6 | Nuclear | NA | No | StrabismusOD |
| Family 6 | MAF | c.880C>T | M | 0.3 | FMB | AD | NA | Nuclear | NA | Yes | No |
| Family 7 | MIP | c.500delC | M | 30 | FMB | NA | 0.01;0.01 | Nuclear | 10 mm;10 mm | Yes | No |
| Family 8 | NHS | c.556G>T | F | 5 | NA | NA | 0.3;0.2 | Punctate | NA | No | Myopia |
| Family 9 | NHS | c.853-1G>A | M | 16 | FMB | XL | 0.05;0.05 | NA | 10 mm;10 mm | Yes | No |
| Family 10 | NHS | c.1117C>T | M | 19 | FMB | XL | FC;FC | Nuclear | 8 mm;8 mm | Yes | No |
| Family 11 | NHS | c.2716_2719delTTAG | F | 0.5 | FMB | XL | PL;PL | Nuclear | 9 mm;9 mm | Yes | No |
Note: NA = not available; S = sporadic; FMB = first few month; AD = autosomal dominant; XL = X-linked; FC = finger counting; PL = pursuit of light; OD = right eye; OS = left eye.
Figure 3. The clinical examination details of patients from the family 9.
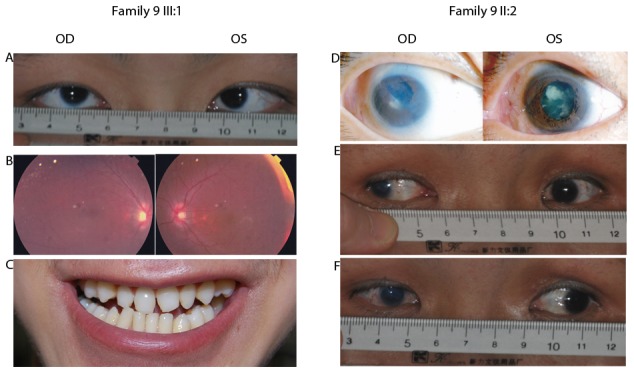
A–C showed that the member III∶1 had bilateral10 mm of the horizontal corneal diameter (A), bilateral mild tigroid retinal change in the temporal region of the optic disc (B), and dental condition without obvious screwdriver incisors or mesiodens (C). D–F showed that the member II∶2 had posterior capsule opacification in her left eye and postoperative pupil shift in her right eye (D), 10 mm of the horizontal corneal diameter in her left eye (E), and 8 mm in her right eye (F).
Discussion
In this study, we performed whole exome sequencing on probands from 18 families with congenital cataracts. Analysis of the sequencing information for 34 genes known to be associated with congenital cataract revealed 12 potential pathogenic mutations. Sanger sequencing confirmed the 11 of them and nine mutations in six genes were considered to be pathogenic in 9 (50.0%) of the 18 families.
To date, about 34 genes have been reported to be associated with congenital cataract (Cat-Map). About half of the mutations were from genes encoding crystallins and 15% of the mutations were from genes encoding connexins [21]. Some screening studies on a set of the two groups of genes in a cohort of patients showed that the frequencies were much lower than expected. Hansen et al. selected 28 Danish families with hereditary congenital cataracts to screen 17 genes and found that mutations in genes encoding crystallins and connexins accounted for 53.5% [7]. Other studies have screened only a few genes in different populations and all of their results showed that the mutation frequencies were no more than 20% [7]–[10],[12],[22]. In our previous study, we screened all of the 12 genes encoding crystallins and connexins in 25 Chinese families, and 40% of the families were found to carry mutations [13]. Regarding other genes, most reports were based on an individual gene in one family. Therefore, it is still not clear about the mutation frequencies of the known genes in a group of patients with congenital cataracts, especially it can be different in specific populations.
In the 9 potential variants identified in the current study, four were in NHS gene and were detected in four X-linked families, which we previously considered as autosomal dominant inheritance. Combining our previous studies with the current study, we identified mutations in 23 of 34 recruited families with congenital cataract, accounting for 67.6% (23/34) of the families [13]–[15]. Mutation frequencies in genes encoding crystallins, genes encoding connexins, and the NHS gene were 29.4% (10/34), 14.7% (5/34), and 11.8% (4/34), respectively. Our results indicated that the NHS gene is also a major causative gene besides the above two groups of genes, especially in some congenital cataracts with pseudo-autosomal dominant inheritance.
NHS gene is located in Xp22.13, and mutations in NHS can cause X-linked dominant Nance–Horan syndrome, which is also known as X-linked congenital cataract [23],[24] or X-linked cataract–dental syndrome [25]. Affected males with Nance–Horan syndrome typically show severe congenital cataracts and dental abnormalities, with occasional dysmorphic features and mental retardation, while females have milder symptoms[26]. While 100% of patients with Nance–Horan syndrome have bilateral congenital cataracts, only 65% have typical dental anomalies, including screwdriver incisors and mesiodens [26]. In some cases, Nance–Horan syndrome was diagnosed after mutations were identified in the NHS gene of affected males who were first noted to suffer from severe congenital cataracts [27]. In our study, all patients in the four families with NHS gene mutations were recruited as congenital cataract, and only three (family 9, family 10, and family 11) had microcornea and nystagmus, which are major manifestations of Nance–Horan syndrome. However, there were no records regarding abnormal dental features. Although it has been demonstrated in a study that Nance–Horan syndrome and X-linked cataract are allelic disorders, there have been no studies to date regarding mutation frequency in congenital cataracts [24]. Therefore, we strongly suggest that the NHS gene should be considered one of the major genes associated with congenital cataract.
In conclusion, combined with our previous studies [13]–[15], based on a total of 34 analyzed families, the results showed that mutations in the 34 known genes were responsible for about 67.6% of this set of Chinese families with congenital cataracts. And mutations in the NHS gene were identified in 11.8% of the families, in whom congenital cataract was the only recorded sign in their first visit. Atypical teeth abnormality was really detected in one patient, while there were no records about other abnormalities in the other three families with NHS mutations except for cataract. Therefore, we supposed that maybe it was not taken too much attention on the abnormal dental and face features caused by NHS mutations, especially in Chinese patients. We suggest that mutations in NHS are a common cause of congenital cataract, both syndromic and nonsyndromic.
Supporting Information
Sequence chromatography. The family number was shown in the left column. Sequences with mutations from patients and normal controls were shown in the middle and right column, respectively. Each mutation was noted under the corresponding sequence. For the Family 9, the proband and his affected mother showed the hemizygous mutant sequence (upper one in the middle column) and the heterozygous mutant sequence (the lower one in the middle column), respectively.
(TIF)
Primers used to amplify and sequence the variants regions in this study. This table listed 12 pairs of primers which were used to amplify the genomic fragments with variants detected by exome sequencing.
(XLSX)
Acknowledgments
The authors thank all patients and their family members for their participation.
Funding Statement
Supported by the National Natural Science Foundation of China (81170881), the National 973 plan (2010CB529904), the “985 project” of Sun Yat-sen University, and the Fundamental Research Funds of the State Key Laboratory of Ophthalmology. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Apple DJ, Ram J, Foster A, Peng Q (2000) Elimination of cataract blindness: a global perspective entering the new millenium. Surv Ophthalmol 45 Suppl 1S1–196. [PubMed] [Google Scholar]
- 2. Gilbert C, Foster A (2001) Childhood blindness in the context of VISION 2020–the right to sight. Bull World Health Organ 79: 227–232. [PMC free article] [PubMed] [Google Scholar]
- 3. Rahi JS, Sripathi S, Gilbert CE, Foster A (1995) Childhood blindness in India: causes in 1318 blind school students in nine states. Eye (Lond) 9 (Pt 5): 545–550. [DOI] [PubMed] [Google Scholar]
- 4. Hejtmancik JF (2008) Congenital cataracts and their molecular genetics. Semin Cell Dev Biol 19: 134–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haargaard B, Wohlfahrt J, Fledelius HC, Rosenberg T, Melbye M (2004) A nationwide Danish study of 1027 cases of congenital/infantile cataracts: etiological and clinical classifications. Ophthalmology 111: 2292–2298. [DOI] [PubMed] [Google Scholar]
- 6. Shiels A, Bennett TM, Hejtmancik JF (2010) Cat-Map: putting cataract on the map. Mol Vis 16: 2007–2015. [PMC free article] [PubMed] [Google Scholar]
- 7. Hansen L, Mikkelsen A, Nurnberg P, Nurnberg G, Anjum I, et al. (2009) Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci 50: 3291–3303. [DOI] [PubMed] [Google Scholar]
- 8. Devi RR, Yao W, Vijayalakshmi P, Sergeev YV, Sundaresan P, et al. (2008) Crystallin gene mutations in Indian families with inherited pediatric cataract. Mol Vis 14: 1157–1170. [PMC free article] [PubMed] [Google Scholar]
- 9. Burdon KP, Wirth MG, Mackey DA, Russell-Eggitt IM, Craig JE, et al. (2004) Investigation of crystallin genes in familial cataract, and report of two disease associated mutations. Br J Ophthalmol 88: 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang KJ, Wang BB, Zhang F, Zhao Y, Ma X, et al. (2011) Novel beta-crystallin gene mutations in Chinese families with nuclear cataracts. Arch Ophthalmol 129: 337–343. [DOI] [PubMed] [Google Scholar]
- 11. Dave A, Laurie K, Staffieri SE, Taranath D, Mackey DA, et al. (2013) Mutations in the EPHA2 Gene Are a Major Contributor to Inherited Cataracts in South-Eastern Australia. PLoS One 8: e72518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kumar M, Agarwal T, Khokhar S, Kaur P, Roy TS, et al. (2011) Mutation screening and genotype phenotype correlation of alpha-crystallin, gamma-crystallin and GJA8 gene in congenital cataract. Mol Vis 17: 693–707. [PMC free article] [PubMed] [Google Scholar]
- 13. Sun W, Xiao X, Li S, Guo X, Zhang Q (2011) Mutation analysis of 12 genes in Chinese families with congenital cataracts. Mol Vis 17: 2197–2206. [PMC free article] [PubMed] [Google Scholar]
- 14. Sun W, Xiao X, Li S, Guo X, Zhang Q (2011) Mutational screening of six genes in Chinese patients with congenital cataract and microcornea. Mol Vis 17: 1508–1513. [PMC free article] [PubMed] [Google Scholar]
- 15. Xiao X, Li W, Wang P, Li L, Li S, et al. (2011) Cerulean cataract mapped to 12q13 and associated with a novel initiation codon mutation in MIP. Mol Vis 17: 2049–2055. [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Q, Wang P, Li S, Xiao X, Jia X, et al. (2010) Mitochondrial DNA haplogroup distribution in Chaoshanese with and without myopia. Mol Vis 16: 303–309. [PMC free article] [PubMed] [Google Scholar]
- 17. Huang KM, Wu J, Brooks SP, Hardcastle AJ, Lewis RA, et al. (2007) Identification of three novel NHS mutations in families with Nance-Horan syndrome. Mol Vis 13: 470–474. [PMC free article] [PubMed] [Google Scholar]
- 18. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 20. Reese MG, Eeckman FH, Kulp D, Haussler D (1997) Improved splice site detection in Genie. J Comput Biol 4: 311–323. [DOI] [PubMed] [Google Scholar]
- 21. Shiels A, Hejtmancik JF (2013) Genetics of human cataract. Clin Genet 84: 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ponnam SP, Ramesha K, Matalia J, Tejwani S, Ramamurthy B, et al. (2013) Mutational screening of Indian families with hereditary congenital cataract. Mol Vis 19: 1141–1148. [PMC free article] [PubMed] [Google Scholar]
- 23. Francis PJ, Berry V, Hardcastle AJ, Maher ER, Moore AT, et al. (2002) A locus for isolated cataract on human Xp. J Med Genet 39: 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coccia M, Brooks SP, Webb TR, Christodoulou K, Wozniak IO, et al. (2009) X-linked cataract and Nance-Horan syndrome are allelic disorders. Hum Mol Genet 18: 2643–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burdon KP, McKay JD, Sale MM, Russell-Eggitt IM, Mackey DA, et al. (2003) Mutations in a novel gene, NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation. Am J Hum Genet 73: 1120–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tug E, Dilek NF, Javadiyan S, Burdon KP, Percin FE (2013) A Turkish family with Nance-Horan Syndrome due to a novel mutation. Gene 525: 141–145. [DOI] [PubMed] [Google Scholar]
- 27. Ramprasad VL, Thool A, Murugan S, Nancarrow D, Vyas P, et al. (2005) Truncating mutation in the NHS gene: phenotypic heterogeneity of Nance-Horan syndrome in an asian Indian family. Invest Ophthalmol Vis Sci 46: 17–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence chromatography. The family number was shown in the left column. Sequences with mutations from patients and normal controls were shown in the middle and right column, respectively. Each mutation was noted under the corresponding sequence. For the Family 9, the proband and his affected mother showed the hemizygous mutant sequence (upper one in the middle column) and the heterozygous mutant sequence (the lower one in the middle column), respectively.
(TIF)
Primers used to amplify and sequence the variants regions in this study. This table listed 12 pairs of primers which were used to amplify the genomic fragments with variants detected by exome sequencing.
(XLSX)