

Abstract
Phenol-soluble modulins (PSMs) are a recently discovered family of amphipathic, alpha-helical peptides that have multiple roles in staphylococcal pathogenesis and contribute to a large extent to the pathogenic success of virulent staphylococci, such as Staphylococcus aureus. PSMs may cause lysis of many human cell types including leukocytes and erythrocytes, stimulate inflammatory responses, and contribute to biofilm development. PSMs appear to have an original role in the commensal lifestyle of staphylococci, where they facilitate growth and spreading on epithelial surfaces. Aggressive, cytolytic PSMs seem to have evolved from that original role and are mainly expressed in highly virulent S. aureus. Here we will review the biochemistry, genetics and role of PSMs in the commensal and pathogenic lifestyles of staphylococci, discuss how diversification of PSMs defines the aggressiveness of staphylococcal species, and evaluate potential avenues to target PSMs for drug development against staphylococcal infections.
Keywords: Staphylococcus aureus, Staphylococcus epidermidis, phenol-soluble modulin, toxin, virulence, biofilm
Introduction
Staphylococcus aureus is a dangerous pathogen that is a frequent cause of hospital- and community-associated infections on a global scale (Lowy, 1998, Deleo et al., 2010). Many S. aureus hospital-associated infections present as infections of the respiratory tract, while skin and soft tissue infections dominate among community-associated infections (Lowy, 1998, Deleo et al., 2010). However, S. aureus can also cause a series of other diseases, including endocarditis or osteomyelitis. Often, S. aureus infection can proceed to septicemia and become life-threatening. Furthermore, S. aureus is a frequent cause of biofilm-associated infections, in particular those developing on indwelling medical devices (Otto, 2008). Finally, S. aureus can cause food poisoning.
S. aureus strains may produce a diverse and large repertoire of virulence factors, many of which are encoded on mobile genetic elements (MGE)s (Novick et al., 2001). MGE-encoded virulence factors can be transferred from strain to strain by horizontal gene transfer and are usually present only in a minority of strains. Some virulence factors appear to have been acquired from coagulase-negative staphylococci (CoNS) such as S. epidermidis (Otto, 2013). However, some S. aureus virulence factors are encoded on the core genome, such as alpha-toxin and phenol-soluble modulins (PSMs), which are thus produced by virtually all strains.
Toxins, secreted molecules that directly harm the human or animal host, are arguably the most important and dangerous virulence determinants of S. aureus. In particular, S. aureus produces a series of cytolytic toxins, which are proteins that cause lysis of host cells such as red and white blood cells. Those that lyse white blood cells (leukocytes) are called leukocidins. Leukocidins of S. aureus belong to the beta-barrel forming family, comprising alpha-toxin and the bicomponent leukocidins (Szmigielski et al., 1999). Of note, these toxins have recently been shown to exert their cytolytic activities dependent on recognition by specific receptors on the surface of the host cell (Wilke & Bubeck Wardenburg, 2010, Alonzo et al., 2011, Spaan et al., 2013). PSMs, the subject of the present review, are a family of peptides with multiple functions in pathogenesis. They gained recognition mostly due to their pronounced cytolytic activity toward leukocytes (Wang et al., 2007), which is why they can be regarded as a new class of staphylococcal leukocidins.
Antibiotic resistance is frequent among S. aureus and severely complicates treatment of S. aureus infections (Lowy, 2003). Penicillin resistance is common among S. aureus; and resistance to methicillin as the antibiotic of first choice against penicillin-resistant S. aureus is of great concern. Many countries report methicillin resistance rates among hospital isolates that exceed 50%. Community-associated methicillin-resistant S. aureus (CA-MRSA) strains, first reported in the late 1990s, represent an additional severe threat to public health systems (Deleo et al., 2010). Antibiotic resistance is also common among many other staphylococcal species, which are in general less aggressive than S. aureus, but are often involved with chronic infections. S. epidermidis in particular is the most frequent cause of infections on indwelling medical devices (Otto, 2009).
Staphylococci are colonizers of the human or animal skin and mucous surfaces. S. epidermidis is the most frequent skin colonizer in humans (Otto, 2009), while S. aureus colonizes the nares and ano-rectal areas of about one third of the population (Rimland & Roberson, 1986, Wertheim et al., 2005). Importantly, staphylococcal infections are believed to originate mainly from colonizing bacteria (von Eiff et al., 2001), making colonization a risk factor for infection. However, we know much less about the physiology of staphylococci during their commensal state as compared to our knowledge of the molecular underpinnings of staphylococcal infectivity. Of note, it has been argued that the physiological status of staphylococcal commensal life may resemble that seen in biofilms (Sugimoto et al., 2013), which are bacterial agglomerations embedded in an extracellular matrix commonly associated with device-related infections (Otto, 2008).
In this review article, we will present the current knowledge on PSMs, a recently discovered family of short, amphipathic peptides produced by S. aureus and other staphylococci, which have been recognized as premier determinants of staphylococcal virulence and physiology.
PSMs – history, definition, and structure
The term “phenol-soluble modulins” (PSMs) was coined by Seymour Klebanoff in 1999, when he and his coworkers discovered immune-modulatory peptides of S. epidermidis that partitioned into the phenol phase during hot phenol extraction (Mehlin et al., 1999). He described a pro-inflammatory “complex” of three peptides, PSMα, PSMβ, and PSMγ, with PSMγ being equivalent to the previously described δ-toxin of S. epidermidis. The Klebanoff group reported a series of pro-inflammatory activities of the PSM “complex” (Mehlin et al., 1999, Liles et al., 2001), many of which we believe today were due to impurities, as pure (synthetic) peptides and isogenic deletion mutants of psm genes were not used in those early studies. Furthermore, PSMs have now been found to signal through a specific receptor (FPR2, see below) (Kretschmer et al., 2010), which is different from the TLR2/TLR6 receptor reported to be activated by the PSM “complex” back then.
PSMs can be classified according to their length. PSMs of the α-type are ~ 20–25 amino acids in length, and those of the β-type are about double that size (43–45 amino acids). The PSMs of the β-type commonly have a negative net charge, while most, but not all, α-type PSMs have a neutral or positive net charge. All PSMs form an amphipathic α-helix, with the α-helical part stretching over the entire length of α-type PSMs and forming the C-terminal part of the β-type PSMs, as predicted by primary and secondary structure bioinformatics analysis (Fig. 1). The structure of S. aureus δ-toxin, which belongs to the α-type, was determined by nuclear magnetic resonance (NMR) analysis (Tappin et al., 1988), confirming amphipathy and α-helicity. These features explain why PSMs are phenol-soluble. PSMs are nowadays more easily identified by their behavior during reversed-phase high-pressure liquid chromatography (RP-HPLC) rather than by their partitioning into the phenol phase. Namely, they elute at exceptionally high concentration of organic solvent, much later than most other proteins or peptides (Wang et al., 2007). This feature is also likely due to the pronounced amphipathy of PSMs and their tendency to aggregate. Finally, it is important to stress that PSMs usually contain an N-terminal N-formyl methionine, indicative of the fact that they are secreted without a signal peptide (Wang et al., 2007). Accordingly, psm genes only encode the amino acids found in the secreted PSM peptides. Some portions of secreted PSMs may lack the N-terminal N-formyl group, which is due to the activity of the cytoplasmic enzyme N-deformylase (Somerville et al., 2003). Interestingly, this appears to happen at a much more pronounced extent in S. aureus than S. epidermidis (Cheung et al., 2010).
Fig. 1. Structure of PSMs.

Shown is a structural representation of PSMα3 modeled after NMR data available for δ-toxin (a) and an α-helical wheel representation of the same peptide (b) as examples. Hydrophobic amino acids are in yellow/brown, hydrophilic amino acids in blue (positive charge) or red (negative charge). The two C-terminal asparagine residues are in pink. Note that in the α-helix formed by PSMα3 hydrophilic and hydrophobic residues occupy opposite locations, making the α-helix distinctly amphipathic.
Purification and measurement of PSMs
As performed in the early studies by the Klebanoff group, a crude PSM preparation can be obtained as precipitate by hot phenol extraction followed by dialysis against distilled water (Mehlin et al., 1999). Precipitation with trichloroacetic acid (TCA) or extraction with 1-butanol can be used as alternatives to enrich PSMs from the culture supernatant (Otto et al., 2004, Joo et al., 2011). Crude PSM extract is further purified by RP-HPLC using a gradient of organic solvent such as acetonitrile or 1-propanol. Some PSMs (e.g., PSMα and PSMβ of S. epidermidis), which elute together at the first RPLC step, need one or more additional RP-HPLC steps for purification (Mehlin et al., 1999, Yao et al., 2005, Joo & Otto, 2014). As a special case of PSM purification, fibrous PSM complexes can be obtained from S. aureus biofilms cultured in drip bioreactors with a defined media, namely peptone-NaCl-glucose (Schwartz et al., 2012).
The amount of PSMs can be measured in complex bacterial culture supernatant by RP-HPLC with a proper column and eluent system (Fig. 2) (Joo & Otto, 2014). Due to the exceptionally late retention time of PSMs during RP-HPLC, PSMs are separated quite easily from other proteins in the culture supernatant. However, the fact that many PSMs do not separate from each other under those conditions necessitates coupling with mass spectrometry (MS) to quantify single PSMs accurately. The concentration of each PSM is determined by integration of the extracted ion chromatogram of the two major m/z ratios among multiply-charged ions of each PSM (Otto et al., 2004, Joo & Otto, 2014). For absolute quantification, purified or synthetic PSMs should be used to calibrate (Yao et al., 2005). As a useful tool for qualitative detection of PSMs, Nizet and colleagues have used an imaging mass spectrometry technique with MALDI-TOF mass spectrometry to discover PSMα derivatives (Gonzalez et al., 2012).
Fig. 2. Analysis of PSMs by RPLC/MS.
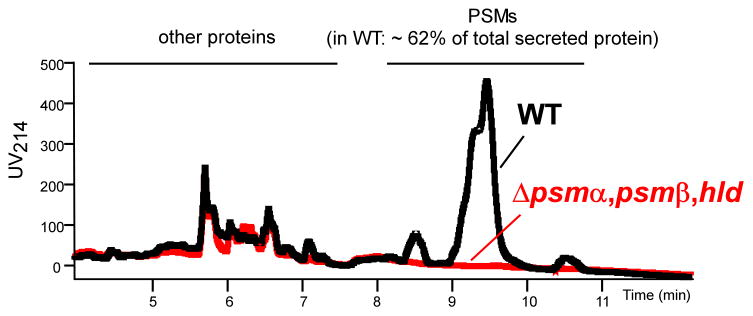
Shown is RPLC chromatography routinely used for RPLC/MS analysis of PSMs. Culture supernatant of an S. aureus strain (USA300) was directly applied to a 2.3 × 30 mm C8 reversed phase column and a brief gradient from 10 to 50% followed by an extended gradient from 50 to 90% water/0.1% triflouroacetic acid (TFA) to acetonitrile/0.1% TFA as described in Wang et al. (Wang et al., 2007) was run. Note that PSMs are separated from other proteins, but not all PSMs completely separate from other PSMs, necessitating coupling to MS for measurement of single PSM quantities. WT, wild-type USA300 (LAC); Δpsmα, psmβ, hld, isogenic mutant not expressing any PSM.
Occurrence of PSMs in staphylococci
PSMs occur in many staphylococcal strains, with the PSM profile apparently being specific for and conserved within a certain species (Vuong et al., 2004, Wang et al., 2007, Queck et al., 2009, Rautenberg et al., 2011). Although this would represent a possibly valid tool to distinguish staphylococcal species, PSM profiling has not yet been used for that purpose. It has been noted that PSM production is more frequent among pathogenic than non-pathogenic species (Rautenberg et al., 2011), but whether there is a clear correlation between pathogenicity and PSM production on the species level needs further verification.
For some PSMs, such as the δ-toxins and PSMβ peptides of S. aureus, S. epidermidis and many other staphylococci, amino acid sequences are fairly conserved between species. Some previously described peptides, such as the gonococcal growth inhibitor peptides from S. haemolyticus (Beaudet et al., 1982, Watson et al., 1988) or the slush peptides from S. lugdunensis (Donvito et al., 1997), are now recognized to belong to the β-type PSMs. Genes encoding δ-toxin and β-type PSMs can be found by similarity searches in many recently sequenced staphylococcal genomes (Tab. 1). In contrast, most α-type PSMs, such as the PSMα peptides of S. aureus, are too short to be routinely annotated in genome sequences and in general too short to yield meaningful results in similarity searches. Performing tblastn similarity searches with all S. aureus and S. epidermidis α-type PSMs, we detected some genes in staphylococci other than S. aureus and S. epidermidis that are similar to PSMε of S. epidermidis and were not previously described (Tab. 1). However, these searches did not yield any significant results for other α-type PSMs; and for the reasons outlined above, they are not likely to detect all α-type psm genes in a genome. For most staphylococcal species, we therefore do not have a complete list of PSM peptides, although we know from RP-HPLC/MS analyses that they produce PSMs (Rautenberg et al., 2011).
Table 1.
PSM homologues identified by similarity searches
| Species/strain | PSM name/annotated as (suggested name in brackets) | Amino acid sequence | Gene1 | Protein1 | Length (amino acids) | Net Charge |
|---|---|---|---|---|---|---|
| PSMβ homologues | ||||||
| S. aureus | PSMβ1 | MEGLFNAIKDTVTAAINNDGAKLGTSIVSIVENGVGLLGKLFGF | 44 | −2 | ||
| PSMβ2 | MTGLAEAIANTVQAAQQHDSVKLGTSIVDIVANGVGLLGKLFGF | 44 | −2 | |||
| S. epidermidis | PSMβ1 | MSKLAEAIANTVKAAQDQDWTKLGTSIVDIVESGVSVLGKIFGF | 44 | −2 | ||
| PSMβ2 | MEQLFDAIRSVVDAGINQDWSQLASGIAGIVENGISVISKLLGQ | 44 | −4 | |||
| S. warneri SG1 | PSMβ1 (PSMβ1) | MEGLIKAIKDTVEAGVNNDGAKLGTSIVGIVENGVGVLSKLFGF | A284_07765 | AGC90870.1 | 44 | −2 |
| PSMβ1 (PSMβ2a) | MTKLAEAIANTVQAAQGHDGAKLGTSIVSIVENGVSVLGKLFGF | A284_07760 | AGC90869.1 | 44 | −1 | |
| PSMβ1 (PSMβ3a) | MTKLAEAIANTVKAAQGHDGAKLGTSIVSIVENGVSVLGKLFGF | A284_07755 | AGC90868.1 | 44 | 0 | |
| PSMβ1 (PSMβ3b) | MTKLAEAIANTVKAAQGHDGAKLGTSIVSIVENGVSVLGKLFGF | A284_07750 | AGC90867.1 | 44 | 0 | |
| antibacterial protein (PSMβ2b) | MTKLAEAIANTVQAAQGHDGAKLGTSIVSIVENGVGVLSKLFGF | A284_07745 | AGC90866.1 | 44 | −1 | |
| S. haemolyticus JCSC1435 | unnamed protein (PSMβ1a) | MSKLVQAISDAVQAGQNQDWAKLGTSIVGIVENGVGILGKLFGF | SH1745 | BAE05054.1 | 44 | −1 |
| unnamed protein (PSMβ2a) | MEKIANAVKSAIEAGQNQDWTKLGTSILDIVSNGVTELSKIFGF | SH1744 | BAE05053.1 | 44 | −2 | |
| unnamed protein (PSMβ1b) | MSKLVQAISDAVQAGQNQDWAKLGTSIVGIVENGVGILGKLFGF | SH1743 | BAE05052.1 | 44 | −1 | |
| unnamed protein (PSMβ2b) | MEKIANAVKSAIEAGQNQDWTKLGTSILDIVSNGVTELSKIFGF | SH1742 | BAE05051.1 | 44 | −2 | |
| unnamed protein (PSMβ3) | MQKLAEAIAAAVQAGQDKDWGKMGTSIVGIVENGISVLGKIFGF | SH1741 | BAE05050.1 | 44 | −1 | |
| S. pseudinterme dius ED99 | putative hemolysin-H1C (PSMβ1)2 | MTELFDAIKNTIEAGINHDWVTLGTSIADIVANGINVISGFFG | SPSE_1635 | ADX76893.1 | 43 | −5 |
| putative hemolysin-H1C (PSMβ2)2 | MGDLFESITNIVTSAIQHDWVNMGAAIVATVQHGVELIAKLFGLG | SPSE_1546 | ADX76805.1 | 45 | −4 | |
| S. pseudinterme dius HKU10-03 | hypothetical protein (PSMβ2)2 | MGDLFESITNIVTSAIQHDWVHMGAAIVATVQHGVELIAKLFGLG | SPSINT_0962 | ADV05490.1 | 45 | −4 |
| hypothetical protein (PSMβ1)2 | MTELFDAIKNTIEAGINHDWVTLGTSIADIVANGINVISGFFG | SPSINT_0872 | ADV05400.1 | 43 | −5 | |
| S. saprophyticus subsp. saprophyticus ATCC 15305 | putative antibacterial protein (PSMβ1) | MEGLFEAIKNTVQAGIAADGAKIGSGVLDIISNGASLIGKALGL | SSP1598 | BAE18743.1 | 44 | −2 |
| putative antibacterial protein (PSMβ2) | MADLFNAIKETVQAGIAGDGAKLGTSIVSIVENGVGVLSKLFGF | SSP1597 | BAE18742.1 | 44 | −2 | |
| putative antibacterial peptide (PSMβ3)2 | MTGIVEAIGNAVNVGLAHDWATMGVSIADVLAKGVDSVLGFFK | SSP0130 | BAE17275.1 | 43 | −3 | |
| S. lugdunensis N920143 | SlushA | MSGIVDAISKAVQAGLDKDWATMATSIADAIAKGVDFIAGFFN | SLUSHA | CCB52870.1 | 43 | −3 |
| SlushB | MSGIIEAITKAVQAGLDKDWATMGTSIAEALAKGIDAISGLFG | SLUSHB | CCB52869.1 | 43 | −3 | |
| SlushC | MDGIFEAISKAVQAGLDKDWATMGTSIAEALAKGVDFIIGLFH | SLUSHC | CCB52868.1 | 43 | −3 | |
| S. carnosus TM300 | phenol soluble modulin (PSMβ1) | MEGLFNAIKGTVEAAINQDGGQLAVNIVDIVQNGIQIVSKFIGA | SCA_0784 | CAL27694.1 | 44 | −3 |
| phenol soluble modulin (PSMβ2) | MLTKLAQAIVDTVNAAQAQDPAKLGTSIVDIVQNGVGIVGKLFGF | SCA_0785 | CAL27695.1 | 45 | −1 | |
| S. simulans bv. staphylolyticus NRRL B-2628 plasmid pACK1 | unnamed protein product (PSMβ-pACK1) | MSGIVDAISQAVQSGLEQDWVTMGTNIAKALAQGIDVISGFFG | 43 | −4 | ||
| δ-toxin homologues | ||||||
| S. epidermidis | δ-toxin | MAADIISTIGDLVKWIIDTVNKFKK | 25 | 0 | ||
| S. aureus | δ-toxin | MAQDIISTIGDLVKWIIDTVNKFTKK | 26 | 0 | ||
| S. carnosus TM300 | (δ-toxin) | MAGDIVGTIGDFVKLIIETVNKFTNK | 26 | −1 | ||
| S. simulans | (δ-toxin) | MAGDIVGTIGEFVKLIIETVQKFTQK | AJ223775.1 | CAA11542.1 | 26 | −1 |
| S. intermedius | (δ-toxin) | MAGDIISTIVDFIKLIAETVKKFTK | AY860843.1 | AAW55662.1 | 25 | 0 |
| S. pseudinterme dius HKU10-03 | (δ-toxin) | MAADIISTIVEFVKLIAETVEKFIK | 25 | −2 | ||
| S. pseudinterme dius ED99 | (δ-toxin) | MAADIISTIVEFVKLIAETVAKFIK | 25 | −1 | ||
| S. warneri SG1 | (δ-toxin 1) | MAADIISTIGDLVKLIINTVKKFQK | A284_04270 | AGC90171.1 | 25 | 1 |
| (δ-toxin 2) | MTADIISTIGDFVKWILDTVKKFTK | A284_04275 | AGC90172.1 | 25 | 0 | |
| PSMε(S. epidermidis) homologues | ||||||
| S. epidermidis | PSM ε | MFIINLVKKVISFIKGLFGNNENE | 24 | 0 | ||
| S. pseudinterme dius ED99 | (PSM ε) | MFIIDLIKKVIEFLKGLFGNK | 21 | 1 | ||
| S. pseudinterme dius HKU10-03 | (PSM ε) | MFIIDLIKKVIEFLKGLFGNK | 21 | 1 | ||
| S. lugdunensis N920143 | (PSM ε) | MFIVNLIKKVIEFIKKLFGNK | 21 | 3 | ||
| S. lugdunensis HKU09-01 | (PSM ε) | MFIVNLIKKVIEFIKKLFGNK | 21 | 3 | ||
| S. warneri SG1 | (PSM ε) | MQFITDLIKKAVDFFKGLFGNK | 22 | 1 | ||
if annotated. The psm genes in S. aureus and S. epidermidis are found and annotated in many strains. See Fig. 3 for gene numbers in strains USA300 and RP62A.
encoded separately. Other psmβ genes are commonly grouped in one genetic locus.
S. aureus and S. epidermidis are the only species for which peptide purification approaches led to a complete list of produced PSMs and PSM-encoding genes (Mehlin et al., 1999, Vuong et al., 2004, Yao et al., 2005, Wang et al., 2007) (Fig. 3). S. aureus produces four PSMα peptides, encoded in the psmα locus, two PSMβ peptides, encoded in the psmβ locus, and the δ-toxin, which is encoded within RNAIII, the effector molecule of the Agr system (Wang et al., 2007). S. epidermidis produces the α-type PSMs PSMα, PSMδ, PSMε, and the δ-toxin (Mehlin et al., 1999, Vuong et al., 2004, Yao et al., 2005). PSMα and PSMδ are somewhat similar to the S. aureus PSMα peptides and encoded together in a locus that resembles the S. aureus psmα locus, indicating that they are evolutionarily related. Furthermore, S. epidermidis produces two PSMβ peptides. The S. epidermidis psmβ locus contains another gene, psmβ3, but we have never found a secreted peptide corresponding to that gene (Vuong et al., 2004, Queck et al., 2009). In some strains including the genome-sequence strains RP62A and ATCC12228, the psmβ1 gene is duplicated, leading to enhanced production of PSMβ1 (Wang et al., 2011).
Fig. 3. PSMs in S. aureus and S. epidermidis.

Amino acid sequences are shown at the top. Numbers at the right show the net charge of the peptides at pH 7.0, rounded to whole numbers, and considering N-formylation. Genes are shown at the bottom. Gene numbering is according to strains S. aureus USA300 FPR3757 (Diep et al., 2006) and S. epidermidis RP62A (Gill et al., 2005), respectively.
Notably, all known psm genes are encoded on the core genome or on MGEs that are present in virtually all strains of the species. As a noticeable exception, the PSM-mec peptide is encoded in staphylococcal cassette chromosome mec (SCCmec) types II, III and VIII, representing a rare example of combined presence of antibiotic resistance and virulence factors on a staphylococcal MGE (Queck et al., 2009, Chatterjee et al., 2011). In addition to the MRSA strains harboring these SCCmec elements, PSM-mec is frequently produced by methicillin-resistant S. epidermidis (Queck et al., 2009); and psm-mec genes are also found in other methicillin-resistant staphylococci (Monecke et al., 2012). The presence of a psmβ homologue on a plasmid of S. simulans ssp. staphylolyticus is a further exception to the common encoding of psm genes on the genome. Of note, to clearly distinguish PSM peptides carrying the same name, a PSM peptide should always be described together with the species, such as in “S. epidermidis PSMβ1”.
Interestingly, the PSM production pattern reflects the clearly distinct “lifestyles” of S. aureus and S. epidermidis. Despite the capacity of S. epidermidis to produce strongly aggressive and cytolytic PSMs such as PSMδ (Cheung et al., 2010), the organism shifts the production of PSMs to less aggressive forms, such as the PSMβ peptides (Otto, 2009, Wang et al., 2011). In contrast, many virulent S. aureus strains produce high amounts of the strongly cytolytic PSMα peptides and only low amounts of PSMβ peptides (Wang et al., 2007).
Regulation of PSMs
Similar to several other major staphylococcal exotoxins, PSMs are positively regulated by the global regulatory quorum sensing system, accessory gene regulator (Agr) (Wang et al., 2007, Queck et al., 2008) (Fig. 4). Notably, Agr thus connects the response to cell density (quorum-sensing) with virulence (Novick, 2003). The agr locus is comprised of a divergent promoter system that transcribes two important mRNAs. RNAIII is a regulatory RNA that controls the transcription of several major exotoxins and surface proteins, and also contains the gene coding for the PSM δ-toxin, named hld for hemolysin delta. RNAII codes for two genes belonging to a classical two-component signal transduction system, a histidine kinase (agrC), and a response regulator (agrA), and two genes that code for an auto-inducing peptide (AIP) precursor (agrD) and an AIP maturation and export protein (agrB). AgrA becomes activated when AgrC detects a critical concentration of secreted AIP. In turn, activated AgrA induces the transcription of rnaiii and its own operon (rnaii), constituting a positive feedback loop. A functional Agr regulatory system is essential for S. aureus virulence as demonstrated by severe attenuation of pathogenicity in isogenic agr mutants (Abdelnour et al., 1993, Cheung et al., 1994, Gillaspy et al., 1995, Cheung et al., 2011).
Fig. 4. Regulation of PSMs by Agr.

The Agr quorum-sensing circuit, as described in the text, is shown at the top. Direct regulation of the psmα and psmβ operons by AgrA is shown left (blue shadowing) and RNAIII-dependent Agr control of other target genes at the right (yellow shadowing). The likely evolution of the connection of quorum-sensing, RNAIII-dependent and –independent control is apparent in the encoding of the PSM δ-toxin (hld) by RNAIII. Agr also controls the psm-mec gene in an RNAIII-independent manner, but binding of AgrA to the psm-mec promoter has not yet been demonstrated.
In contrast to the regulation of virtually all Agr target genes via RNAIII, psm genes are regulated by direct binding of AgrA to their promoters (Queck et al., 2008). This represents an exceptionally direct control by the Agr system, indicating that cell density-dependent regulation of PSMs preceded a later connection to further virulence determinants. The fact that the PSM δ-toxin is encoded within rnaiii, which thus probably evolved around the δ-toxin gene hld, further supports this notion. Although the regulation of PSM transcription is dominated by AgrA (Queck et al., 2008, Chatterjee et al., 2011), additional levels of PSM regulation exist. For example, the biofilm mode of growth appears to lead to enhanced relative production of β-type PSMs over α-type PSMs, as shown in S. epidermidis (Wang et al., 2011), but how differential production of α- versus β-type PSMs is achieved mechanistically is not known. Recent studies show that the PSMs, in addition to other toxins, are degraded by aureolysin, a protease which is regulated by several global regulators, which include Agr, staphylococcal accessory regulator (SarA), and SaeRS (Zielinska et al., 2011, Cassat et al., 2013, Rasigade et al., 2013). Accordingly, SarA impacts PSM production by a general impact on protease activity (Zielinska et al., 2011).
The psm-mec gene is embedded in a gene encoding a regulatory RNA (Kaito et al., 2011), in a way very similar to the connection of hld with RNAIII (Fig. 5). The psm-mec regulatory RNA decreases agrA activity (Kaito et al., 2013), and thus has an indirect effect on PSM production. However, the effect of the psm-mec regulatory RNA on PSM production is quite low in extent, reaching a maximal factor of ~ 3, and is only seen in some strains (Kaito et al., 2013). Lastly, Agr also regulates transcription of psm-mec itself in an RNAIII-independent manner (Chatterjee et al., 2011).
Fig. 5. PSM-mec.
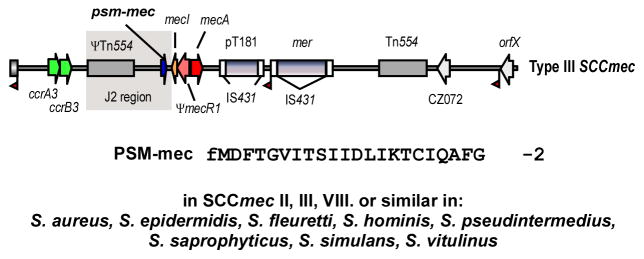
The PSM-mec peptide and the psm-mec RNA surrounding the PSM-mec-encoding gene are encoded on SCCmec elements of types II, II, and VIII, of which SCCmec type III is shown here as example.
S. aureus can survive within a variety of host cells to hide from the immune system and disseminate to other sites in the host (Gresham et al., 2000, Kubica et al., 2008, Giese et al., 2011, Thwaites & Gant, 2011, Cassat et al., 2013). Importantly, intracellular environments were found to activate the expression of PSMs (Geiger et al., 2012, Surewaard et al., 2012). When phagocytosed by polymorphonuclear leukocytes (PMNs, or neutrophils), S. aureus quickly produces high levels of the intracellular signaling molecule, (p)ppGpp, which activates the production of PSMs by an unknown mechanism (Geiger et al., 2012). Unlike the Agr quorum sensing system, which requires time for the accumulation of enough AIP for activation of AgrA (Carnes et al., 2010, Pang et al., 2010), activation of the stringent response is independent of AgrA. It is likely that both the stringent response (Geiger et al., 2012) and Agr (Surewaard et al., 2012) are induced by S. aureus to ensure that PSMs are produced immediately and continually in intracellular environments, maximizing the capacity of S. aureus to trigger phagosomal escape and subsequent cell lysis.
Finally, it was reported that PSM transcription is up-regulated, in addition to other important virulence factors, in the presence of calf serum (Oogai et al., 2011). It is tempting to speculate that S. aureus may up-regulate PSM production as a means to compensate for the observed sequestration of PSMs by lipoproteins in human serum (Surewaard et al., 2012). Taken together, while the Agr global regulatory system is by far the main effector of PSM production, other factors, which include the regulatory psm-mec mRNA, proteolytic degradation, and various responses to the intracellular and extracellular environments, all play important roles affecting PSM concentrations in vitro and in vivo.
Export of PSMs
The fact that PSMs carry an N-terminal N-formyl methionine indicates that they are secreted without a signal peptide using a non-canonical protein export system. Recently, the PSM export system was identified to be an ABC transporter, which was named phenol soluble modulin transporter (Pmt) (Chatterjee et al., 2013) (Fig. 6). The pmt locus encodes four genes, pmtA-D, of which pmtA and pmtC code for ATPases, whereas pmtB and pmtD encode membrane-associated proteins. In S. aureus, Pmt was shown to be able to export all types of PSMs into the surrounding milieu. Pmt is present in all Staphylococcus species and absent from non-staphylococcal bacterial genomes, suggesting that Pmt is specific for staphylococci, as is PSM production. Notably, Pmt is essential for bacterial survival, because in its absence PSMs accumulate in the cytoplasm, causing acute growth deficiency and cellular defects. As expected, Pmt is required for mediating PSM-associated virulence phenotypes. Due to its key impact on virulence and additional essentiality for bacterial survival, and because it is highly conserved among the staphylococci, Pmt appears to be a exceptionally promising target for interference with staphylococcal pathogenesis (Chatterjee & Otto, 2013).
Fig. 6. PSM export.

The genetic locus encoding the four Pmt ABC transporter components is shown at the top (a). Numbers refer to gene numbers in the USA300 FPR3757 genome (Diep et al., 2006). (b) Pmt functions. (c) Consequences of Pmt absence/blocking.
PSMs in the natural habitat of staphylococci
PSMs have been attributed at least two main functions that are related to their detergent-like features and may facilitate the life of staphylococci in their natural habitat, that is, on the epithelial surfaces of humans and other mammals. First, the observed PSM-dependent spreading of S. aureus on agar plates most likely translates to PSMs having a key function in the spreading of staphylococci on epithelial surfaces (Tsompanidou et al., 2013). Second, PSMs may assist in the emulsification of nutrients in those environments, although this remains to be demonstrated. Of note, PSMs are produced at the high levels that these functions presumably require. In fact, ~ 60% of the secreted protein mass in S. aureus in-vitro culture was shown to be PSMs (Chatterjee et al., 2013). Finally, PSMs are responsible for biofilm structuring (Wang et al., 2011, Periasamy et al., 2012), which is discussed further below. As biofilm-like bacterial agglomerations are believed to exist on epithelial surfaces, this PSM function likely also contributes to the physiology of staphylococci in their commensal state. However, in general we still lack detailed information on the role of PSMs in the non-infectious lifestyle of staphylococci.
PSMs as cytolysins
Several PSMs have an extraordinary ability to lyse many eukaryotic cell types, such as bone cells (osteoblasts) (Cassat et al., 2013, Rasigade et al., 2013), endothelial and epithelial cells (Giese et al., 2011), monocytes (Wang et al., 2007), erythrocytes (Cheung & Otto, 2011), and most importantly, PMNs (Wang et al., 2007, Cheung et al., 2010, Forsman et al., 2012). The wide variety of cell types that are subject to PSM-mediated lysis is in accordance with the notion that PSM cytolytic activity is due to membrane perturbation in a receptor-independent fashion (Kretschmer et al., 2010) (Fig. 7). However, not all PSMs show this pronounced cytolysis of eukaryotic cells. For instance, the PSM β-type peptides are generally much less cytolytic than the α-type peptides. Whether this is due to their commonly negative net charge is not known. Notably, S. aureus PSMα3 and S. epidermidis PSMδ are by far the most potent cytolytic PSMs described to date, with pronounced cytolytic potencies observed in the low micromolar range (Wang et al., 2007, Cheung et al., 2010). Interestingly, the cytolytic activities of the PSMs are enhanced in the presence of S. aureus β-toxin (Cheung & Otto, 2011) and PVL (Hongo et al., 2009) in addition to antimicrobial peptides (Cogen et al., 2010), signifying that the PSMs may not always exert their cytolytic effects independently.
Fig. 7. PSM-mediated cytolysis.

Likely mechanism of receptor-independent membrane attachment and disintegration, and subsequent pore formation by PSMs. This model is based on experimental evidence obtained with S. aureus δ-toxin, which forms receptor-independent short-lived pores in artificial membranes (Verdon et al., 2009). (b) Mechanism of intracellular neutrophil killing by PSMα peptides of S. aureus (Surewaard et al., 2013).
Much of our understanding about the lytic nature of the PSMs stems from extensive research performed with S. aureus δ-toxin, which forms small transient pores in artificial lipid vesicles (Pokorny et al., 2002). Phosphatidylcholine (POPC), which is a major phospholipid constituent of eukaryotic membranes (Devaux & Morris, 2004), is generally used to study membrane perturbation. A more detailed study of the interaction of the PSMs of S. aureus and S. epidermidis with POPC vesicles revealed that POPC-lytic activities of PSMs did not correlate with those previously described for leukocytes and erythrocytes (Duong et al., 2012). In particular the PSMβ peptides showed strongly lytic behavior toward POPC phospholipid vesicles, implying that the pore-forming activities of PSMs are more complex than originally thought and possibly dependent on the membrane lipid composition. Some PSMs can lyse bacterial protoplasts (Kreger & Bernheimer, 1971) and even whole bacteria, such as Streptococcus pyogenes and Legionella pneumophilla (Verdon et al., 2008, Cogen et al., 2010, Joo et al., 2011). This is discussed further below.
A hallmark of a typical subcutaneous S. aureus infection is the rapid infiltration of PMNs. PMNs are important host innate immune cells that have a key role in controlling local infections by taking up bacteria by phagocytosis (Amulic et al., 2012). Phagocytosed bacteria, compartmentalized into phagosomes, are exposed to a battery of antimicrobial agents. However, virulent S. aureus strains such as CA-MRSA strains have developed strategies to evade killing by cells of the host innate immune system (Voyich et al., 2005, Kobayashi et al., 2010, Rigby & DeLeo, 2012). These strategies include the ability to survive within the PMNs, which provides protection from detection by the immune system and also represents a means of transportation to disseminate to other sites of the body (Rogers, 1956, Gresham et al., 2000, Thwaites & Gant, 2011). Several recent studies have shown that PSMα peptides are responsible for the lysis of neutrophils after ingestion (Geiger et al., 2012, Chatterjee et al., 2013, Surewaard et al., 2013) (Fig. 7). Furthermore, expression levels of PSMs correlate with cytotoxicity within PMNs (Rasigade et al., 2013). Among the classical bicomponent S. aureus leukocidins, only LukAB (also known as LukGH) (Ventura et al., 2010, Dumont et al., 2011) contributes to PMN lysis after phagocytosis (Ventura et al., 2010, DuMont et al., 2013). PSMα expression is also important for the phagosomal escape or survival of S. aureus within osteoblasts (Rasigade et al., 2013), professional phagocytes other than neutrophils, and non-professional phagocytes such as endothelial cells (Grosz et al., 2013), indicating a general crucial role in intracellular survival of S. aureus.
Recognition of PSMs by the host
The PSMs are potent pro-inflammatory agents that mediate their activities primarily through the interaction with human formyl peptide receptor 2 (FPR2) (Kretschmer et al., 2010) (Fig. 8). FPR2 belongs to a family of G protein-coupled receptors that aid cells of the innate immune system to recognize pathogen-associated molecular patterns (PAMPs) produced by bacteria. In humans, three FPR paralogs exist; FPR1, FPR2/ALX (previously known as FPR-like 1), and FPR3. FPR1 and FPR2 are expressed on monocytes, macrophages, PMNs, immature dendritic cells and some other cell types, while FPR3 is only present on monocytes and macrophages (Migeotte et al., 2006).
Fig. 8. PSM receptor interaction.
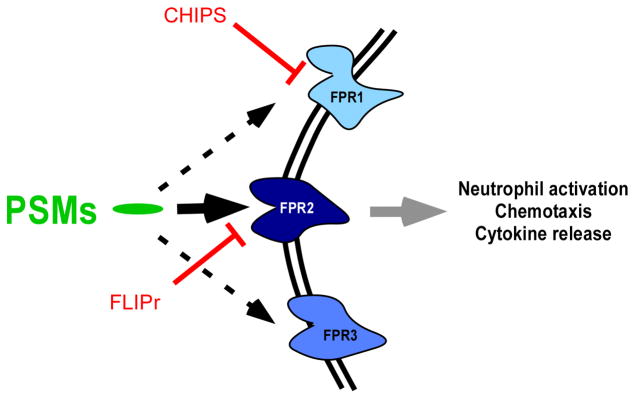
In the nanomolar range, PSMs bind to and activate FPR receptors, with by far the strongest activation occurring with FPR2. Pro-inflammatory, FPR2-mediated activities of PSMs include neutrophil activation, chemotaxis, and release of specific cytokines such as IL-8. N-formylated and N-deformylated PSMs activate FPR2 receptors, with activation by formylated PSMs being somewhat stronger. The S. aureus virulence factors CHIPS and FLIPr block recognition by FPR1 and FPR2, respectively.
The FPRs detect N-formyl peptides, such as the N-formylated PSMs. However, they were shown to also recognize non-formylated forms of PSMs (Kretschmer et al., 2011), which are secreted under certain conditions (Somerville et al., 2003). At nanomolar concentrations, the PSMs activate many FPR2 receptor-dependent effector functions, which include the induction of chemotaxis, exocytosis and superoxide generation, pro-inflammatory cytokine production, up-regulation of CD11b expression, and the induction of intracellular calcium flux (Fu et al., 2006, Wang et al., 2007, Queck et al., 2009, Kretschmer et al., 2010, Chaffin et al., 2012, Forsman et al., 2012). These pro-inflammatory activities are essential for an effective immune response to invading pathogens. The activation of FPR2-dependent pro-inflammatory activities by non-formylated PSMs from S. aureus and S. epidermidis is generally less pronounced than that by PSMs with an N-formyl methionine residue (Kretschmer et al., 2011).
The specificity of the PSMs for FPR2 was discovered with the help of two small protein inhibitors (the chemotaxis-inhibitory protein of S. aureus (CHIPS) (de Haas et al., 2004) and FPR2/ALX-inhibitory protein (FLIPr) (Prat et al., 2006)). These inhibitors, secreted by S. aureus, block the ability of FPR1 and FPR2 to recognize formylated peptides, respectively. Based on the specificity of FPR2 for the detection of PSMs, it is thought that the innate immune system may discriminate between virulent S. aureus strains, such as CA-MRSA, and commensal staphylococci, which generally have a reduced PSM output (Kretschmer et al., 2010). Notably, FPR2-specific inhibition by FLIPr dramatically reduces chemotaxis and IL-8 release in neutrophils as stimulated by culture filtrate of the CA-MRSA strain USA300 (Kretschmer et al., 2010), emphasizing the importance of PSM recognition in the interaction of the innate immune system with S. aureus. Evidently, PSMs and FLIPr have an opposite impact on FPR2 activation. Most likely, production of FLIPr and PSMs is controlled by the bacteria in a way that allows establishment of an infection without immune recognition. This is achieved by the strict control of PSM production by the Agr quorum-sensing system (Queck et al., 2008), delaying PSM production until the bacterial density is high. Then, strong PSM production likely outcompetes FLIPr to promote inflammation via FPR2. However, the in-vivo kinetics of FLIPr and PSM production still need to be investigated. Similarly, it is yet unknown whether PSM-dependent induction of inflammatory responses is part of the bacterial pathogenesis program or PSMs represent pathogen-associated molecular patterns that the immune system recognizes to launch a defensive response.
While the PMNs are critical for innate immune defense, macrophages and dendritic cells (DCs) play an essential role in linking innate and adaptive immunity by serving as antigen presenting cells (APCs). In particular, APCs process and present antigens to T-lymphocytes, which then initiate the stimulation of an adaptive immune response. Interestingly, a recent study demonstrated that PSMs modulate murine DC function; PSM-stimulated DCs increased in vitro priming of regulatory T cells, which led to a suppression of Th1 immune responses (Schreiner et al., 2013). Murine macrophages stimulated with isogenic psmα and hld CA-MRSA mutants showed decreased production of IL-6, IL-12 and TNFα cytokines as compared to stimulation with the corresponding wild-type strain (Spentzas et al., 2011) confirming that both PSMα peptides and δ-toxin are important mediators of inflammatory cytokine production. Taken together, the PSMs appear to be capable of manipulating the host immune system in various ways to promote survival of S. aureus during infection.
PSMs in different infection types
PSMs in skin and soft-tissue infections and sepsis
In humans, skin and soft tissue infections represent ~ 90% of all clinical manifestations of community-associated S. aureus disease (Deleo et al., 2010). However, S. aureus can cause severe invasive disease resulting in bacteremia (sepsis). Even under antibiotic therapy, invasive S. aureus disease is associated with high mortality (Klevens et al., 2007). Notably, on average, greater levels of PSMs are detected in the supernatants of CA-MRSA strains as compared to HA-MRSA strains, suggesting a correlation with the virulence potential of those strains (Wang et al., 2007, Li et al., 2010). Accordingly, isogenic psmα CA-MRSA mutants were severely attenuated in their ability to cause subcutaneous abscesses in the skin of mice compared to the wild-type strain (Wang et al., 2007) (Fig. 9a). Furthermore, mice infected with isogenic psmα CA-MRSA mutants were significantly delayed in reaching moribund status compared to mice infected with the wild-type strain in a bacteremia model of infection (Wang et al., 2007), confirming a link between high expression of PSMα peptides and the virulence potential of CA-MRSA. In contrast, psmβ deletion mutants had none and hld mutants only a slight impact on disease progression in these mouse models. Similarly, in a rabbit model of skin infection, a psmα deletion mutant was strongly impaired in forming abscesses, to an extent similar to that caused by an agr deletion mutant and clearly exceeding that of a lukSF (PVL) deletion mutant, which did not show a significantly different ability to form abscesses compared to the wild-type strain in that model (Kobayashi et al., 2011) (Fig. 9b).
Fig. 9. PSMα peptides and skin infection.

Representative pictures of abscesses formed by the USA300 CA-MRSA strain and its isogenic psmα deletion mutant in mice (Wang et al., 2007). (b) Comparative analysis of virulence determinants in a rabbit skin infection model (Kobayashi et al., 2011). The abscess volume is shown on the y-axis. Significantly smaller abscesses compared to the wild-type strain were found with the hla, psmα, and agr, but not lukSF mutants.
As for the impact of the psm-mec locus on virulence, remarkable differences were reported in murine models of skin and bacteremia infections (Queck et al., 2009, Kaito et al., 2013). In S. aureus strains that produce relatively high levels of PSM-mec, a significant attenuation in virulence was observed in isogenic S. aureus psm-mec mutants, suggesting that the PSM-mec peptide contributes to virulence similarly to the α-class PSMs (Queck et al., 2009). In contrast, deletion of the psm-mec locus caused an increase in virulence in some HA-MRSA isolates (Kaito et al., 2013) as a result of the negative effect of the psm-mec RNA on Agr, but not in others (Chatterjee et al., 2011, Kaito et al., 2013). The impact that the psm-mec locus has on virulence thus is highly strain-dependent and appears to result from a combination of the PSM-mec peptide, which has a positive effect, and the psm-mec RNA, which has a negative effect on virulence.
PSMs in biofilm-associated infection
Biofilms are surface-attached agglomerations with a distinct physiology and pronounced capacity to withstand attacks by host defenses and antibiotics (Costerton et al., 1999). Biofilm-associated infections often develop on indwelling medical devices; staphylococci are the most frequent pathogens involved with such infections (Otto, 2008). The factors that determine aggregation of cells in a biofilm, such as the production of an extracellular matrix, are fairly well understood. This includes staphylococci, in which the matrix consists of proteins, extracellular DNA, teichoic acids, and a specific exopolysaccharide called polysaccharide intercellular adhesin (PIA) or poly-N-acetylglucosamine (PNAG), according to its chemical composition. In contrast, the factors that contribute to the structuring of biofilms and the detachment of cells from biofilms, a process vital for the systemic dissemination of a biofilm infection, have remained poorly understood until recently (Otto, 2013).
S. epidermidis in particular, often regarded as an opportunistic pathogen, is the leading cause of chronic catheter-related infections in hospital settings (Otto, 2009, Rogers et al., 2009). S. epidermidis has a strong aptitude to form biofilms on inert surfaces (Kogan et al., 2006, Rohde et al., 2007). PSMβ peptides are strongly produced in S. epidermidis, and in a relatively higher proportion compared to other PSMs during biofilm as compared to planktonic growth (Wang et al., 2011). PSMβ peptides were shown to structure S. epidermidis biofilms, leading to the characteristic biofilm structure with channels deemed important for nutrient delivery to all biofilm layers (Wang et al., 2011) (Fig. 10). Furthermore, these peptides were found to be crucial for the detachment of biofilm clusters in vitro and dissemination from biofilms on indwelling devices in vivo, as demonstrated in a mouse model of indwelling device-associated infection (Wang et al., 2011). In S. aureus, all PSM peptides were shown later to contribute to these phenotypes in vitro and in vivo (Periasamy et al., 2012). The extent to which other PSMs of S. epidermidis contribute to biofilm structuring and detachment has not yet been analyzed using gene deletion mutants. The finding that the PSM surfactant peptides are responsible for biofilm structuring and detachment are in line with similar findings in other bacteria (Branda et al., 2001, Boles et al., 2005, Angelini et al., 2009), indicating a common mechanism involved in these phenotypes that is dependent on surfactant molecules. However, the chemical nature of the surfactants underlying biofilms structuring is different in different bacteria. Notably, the experiments on biofilm dissemination in S. epidermidis and S. aureus biofilm-associated infection were the first to show the impact of biofilm-structuring surfactants on dissemination from in-vivo biofilms.
Fig. 10. PSMs and biofilm structuring.
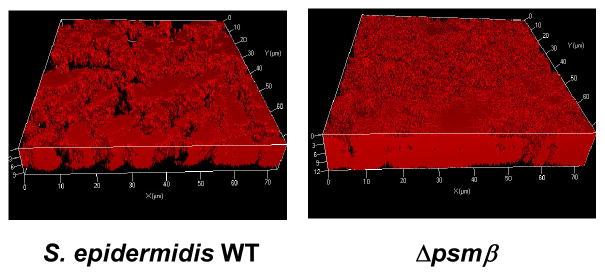
Confocal laser scanning microscopy pictures of biofilms formed by an S. epidermidis wild-type strain (strain 1457) and an isogenic deletion mutant of the psmβ operon (Wang et al., 2011). Note the thicker biofilm formed by the mutant and the absence of channels in the biofilm.
A recent report demonstrated that PSMs can polymerize to form fibrillar, amyloid structures that help stabilize S. aureus biofilms in vitro (Schwartz et al., 2012). Interestingly, the amyloid structures isolated from S. aureus cultures consist of only deformylated PSMs. However, further research is needed to investigate whether these structures exist in vivo and how they fit into the pathogenic lifestyle of S. aureus and S. epidermidis during biofilm infection.
PSMs in bone infection (osteomyelitis)
S. aureus is the leading cause of bone infections (osteomyelitis) in adults and infants (Arnold et al., 2006, Vander Have et al., 2009). CA-MRSA-related osteomyelitis in infants is often associated with increased interval and severity compared to HA-MRSA-related osteomyelitis (Martinez-Aguilar et al., 2004, Arnold et al., 2006, Bocchini et al., 2006). Intracellular residence within osteoblasts appears to be essential for the chronic presence and dissemination of S. aureus (Tuchscherr et al., 2010, Tuchscherr et al., 2011). Recently, the global S. aureus regulators Agr, SarA, and SaeRS were shown to regulate the expression of aureolysin, a protease responsible for degrading important S. aureus exoproteins (Zielinska et al., 2011, Gonzalez et al., 2012, Cassat et al., 2013, Rasigade et al., 2013). Although there is conflicting evidence supporting the roles SaeRS and SarA in aureolysin-mediated degradation of exoproteins (Cassat et al., 2013, Rasigade et al., 2013), it is clear that PSMα peptides are crucial for cytotoxicity toward murine and human osteoblasts in vitro (Cassat et al., 2013, Rasigade et al., 2013) and that they are subject to aureolysin-mediated degradation during bone infection (Cassat et al., 2013). Furthermore, PSMα peptides were essential for bone remodeling and destruction in a mouse model of osteomyelitis infection as visualized by high-resolution microcomputed tomography (Cassat et al., 2013). Taken together, these results implicate the PSMα peptides as important virulence determinants in the pathogenesis of osteomyelitis.
Delta-toxin and atopic dermatitis
Atopic dermatitis (AD) is a chronic inflammatory skin disease that commonly affects infants (Williams & Flohr, 2006). Mast cells are believed to be key contributors to the pathology of AD and other allergic diseases through the release of inflammatory mediators from intracellular granules activated by antigen cross-linked to Fc3RI receptor-bound IgE (Galli et al., 2011, Galli & Tsai, 2012). Although more than 90% of AD patients are tested positive for S. aureus (Rudikoff & Lebwohl, 1998), the link between S. aureus and AD pathogenesis has remained unclear. However, a recent study implicates a link between S. aureus colonization and allergic skin disease and correlates these observations with the potent mast cell-degranulating activity of δ-toxin (Nakamura et al., 2013). Notably, other S. aureus PSMs lacked such activity. Importantly, non-cytolytic concentrations of both formylated and non-formylated forms of δ-toxin are equally potent in stimulating mast cell degranulation, these activities are not dependent on FPR2, and the intracellular signaling pathways stimulated by δ-toxin leading to mast cell degranulation are different from those induced by antigen cross-linked to IgE. The ability of δ-toxin to induce mast cell degranulation was also confirmed in vivo using in murine models of inflammatory skin disease. Furthermore, skin colonization with an isogenic S. aureus hld mutant promoted less IgE and IL-4 production and inflammatory skin disease when compared to mice colonized with the wild-type δ-toxin expressing S. aureus strain. Moreover, enhancement of IgE production and skin inflammation by δ-toxin was abolished in mast cell-deficient mice and restored by adoptive transfer of mast cells (Nakamura et al., 2013).
Commensal staphylococci such as S. epidermidis, which are not linked to AD, also produce δ-toxin peptides that are highly similar to that produced by S. aureus (Scheifele et al., 1987). S. epidermidis δ-toxin showed some, but reduced activity compared to S. aureus δ-toxin in causing mast cell degranulation (Nakamura et al., 2013). Whether δ-toxin producing S. epidermidis isolates contribute to allergic skin disease requires further investigation.
Relationship of PSMs with other S. aureus toxins
Clearly, the PSMs are not the only toxins that contribute to skin and soft tissue and many of the other infections mentioned above. For instance, α-toxin (Hla), an important pore-forming and pro-inflammatory (Thelestam et al., 1973, Bhakdi & Tranum-Jensen, 1991, Walev et al., 1993, Ezepchuk et al., 1996, Valeva et al., 1997, Hocke et al., 2006, Liang & Ji, 2006, Bartlett et al., 2008) toxin, contributes to abscess formation in mice (Inoshima et al., 2011) and rabbits (Kobayashi et al., 2011). Furthermore, bi-component pore-forming leukotoxins are increasingly recognized as important virulence determinants involved in S. aureus pathogenesis. These leukotoxins include LukED, LukAB (LukGH) and PVL (Diep et al., 2010, Alonzo et al., 2011, Dumont et al., 2011). They are cytolytic towards cells of myeloid origin of different animal species. Notably, cytolysis triggered by those toxins has recently been found to be receptor-dependent (Alonzo et al., 2013, DuMont et al., 2013, Spaan et al., 2013), as has α-toxin-dependent cell lysis and pro-inflammatory activities (Wilke & Bubeck Wardenburg, 2010, Inoshima et al., 2011). Studies using isogenic S. aureus deletion mutants showed that LukED and LukAB contribute to the virulence of S. aureus in murine sepsis (Alonzo et al., 2011) and renal abscess models (Dumont et al., 2011), respectively. The contribution of PVL to the pathogenesis especially of CA-MRSA-induced disease is controversial (Otto, 2010, Otto, 2011).
While PSMs play a key role as virulence determinants during many types of S. aureus infection, the pathogenesis of S. aureus disease is multi-factorial. The PSMs may work in concert with other toxins, possibly in a synergistic fashion as reported for PVL and PSMs (Hongo et al., 2009), to exert maximal pathogenesis as in the case of skin and soft tissue infections. Notably, unlike α-toxin, LukAB, LukED and PVL, the cytolytic activity of the PSMs very likely does not require a receptor (Kretschmer et al., 2010). Thus, the non-specific lytic nature of the PSMs may provide a distinct advantage over the aforementioned toxins, inasmuch as they may attack a wide variety of cell types, and offer a possible explanation as to why the PSMs contribute to pathogenicity in a variety of S. aureus diseases.
PSMs as antimicrobials
Although periodically, antimicrobial activities of δ-toxin analogs and other PSM-like peptides of staphylococci have been reported (Beaudet et al., 1982, Dhople & Nagaraj, 1995, Dhople & Nagaraj, 2005), more intensive studies on such activities of PSMs have only been performed more recently (Tab. 2). In these studies, PSMδ and δ-toxin of S. epidermidis were found to be active predominantly against Streptococcus pyogenes (Cogen et al., 2010). Furthermore, S. epidermidis δ-toxin recruited host antimicrobial peptides (AMPs), such as LL-37, CRAMP, and hBDs, to reduce survival of S. pyogenes in a synergistic fashion (Cogen et al., 2010). In another study, most intact PSMs exhibited very low or no antimicrobial activity on S. pyogenes, but proteolytically processed PSMα1 and PSMα2 showed significantly increased activities (Joo et al., 2011). The N-terminal two amino acids, formyl-methionine and glycine, of these PSMs are missing in the antimicrobially active derivatives. Whether these processed PSMs contribute to bacterial interference in vivo is unclear. Also, which proteases are involved in the processing is not known, but recent findings on additional proteolytically processed derivatives of PSMα1 and PSMα4 implicates aureolysin. Overall, the biological significance of the antimicrobial activities found in some PSMs is unclear. The facts that these activities are rare and the antimicrobially active processed PSMα peptide derivatives are only produced in low amounts support the notion that they are a side effect of the general membrane-damaging effect of PSMs. The features that most PSMs exhibit suggest that they evolved predominantly to harm eukaryotic rather than prokaryotic membranes. Nevertheless, that staphylococci are protected from the antimicrobial effects of PSMs by the Pmt export system (Chatterjee et al., 2013) indicates that they may at least theoretically contribute to bacterial interference with other genera, which remains to be clearly demonstrated in real-life bacterial communities.
Table 2.
PSMs with antimicrobial activities.
| Strain | PSM type | Derivatization | Target | MBC (μM) | Note | Reference |
|---|---|---|---|---|---|---|
| CoNS isolate | unknown | - | Neisseria gonorrhoeae G-10 | 16 kD protein dissociated to 1.4 kD | (Beaudet, et al., 1982) | |
| S. epidermidis | δ-toxin | intact | S. aureus 113 | 4–8 | KAM2 used except for S. pyogenes (25% Todd-Hewitt broth, 75% PBS) | (Cogen, et al., 2010) |
| S. pyogenes, NZ131 | 16 | |||||
| Escherichia coli | 8–16 | |||||
| PSMδ | intact | MRSA, Sanger 252 | 16–32 | |||
| MRSA, USA300 | 32 | |||||
| S. aureus,113 | 8 | |||||
| S. pyogenes NZ131 | 16 | |||||
| Escherichia coli | 8 | |||||
| S. aureus | δ-toxin | synthesized and modified 16 to 26 residues-derivatives | Escherichia coli | 2.1–3.91 | intact S. aureus δ-toxin is not antimicrobial | (Dhople & Nagaraj, 1995, Dhople & Nagaraj, 2005) |
| S. aureus | 3.7–221 | |||||
| PSMα1 | N-terminal two residues are absent | S. pyogenes | 8 | THY2 used | (Joo, et al., 2011) | |
| N-terminal five residues are absent | S. pyogenes | 11 | (Gonzalez, et al., 2012) | |||
| S. epidermidis, ATCC35984 | 42 | |||||
| PSMα2 | N-terminal two residues are absent | S. pyogenes | 8 | THY used | (Joo, et al., 2011) | |
| Staphylococcus epidermidis 12228 | 16 | TSB2 used | ||||
| PSMα3 | Synthesized alanine exchange derivatives: F3A, V4A, F8A, F10A, F11A, L14A, F18A, L19A | S. pyogenes, Micrococcus luteus | 3 | (Cheung, et al., 2013) | ||
| PSMα4 | N-terminal three residues are absent | S. pyogenes | 10 | aureolysin-dependent | (Gonzalez, et al., 2012) |
MIC (μM)
KAM, killing assay media (Dorschner, et al., 2006); THY, Todd-Hewitt broth containing 0.2% yeast extract; TSB, tryptic soy broth
determined by growth inhibition zones
Structure-function analysis of PSMs and role of cytolytic versus pro-inflammatory capacity during infection
Recently, an alanine substitution peptide library of PSMα3 was used to investigate which structural determinants are responsible for the diverse biological activities of PSMs (Cheung et al., 2013) (Fig. 11). PSMα3 was chosen because it is the most cytolytic PSM and significantly contributes to all phenotypes associated with PSMs, including hemolysis, biofilm structuring and pro-inflammatory activities (Wang et al., 2007, Kretschmer et al., 2010, Cheung & Otto, 2011, Periasamy et al., 2012). The results obtained with the alanine substitution peptide library of PSMα3 identified several important variants that have unique phenotypes. Namely, PSMα3 L7A and K17A showed reductions in cytolytic but retained pro-inflammatory activities, whereas PSMα3 N21A and N22A showed the reverse phenotype. PSMα3 K6A and PSMα3 K12A showed marked reductions in both cytolytic and pro-inflammatory activities. To investigate whether the in-vitro pro-inflammatory phenotypes of those derivatives were also reflected in vivo, a murine peritonitis model was used to measure the numbers of infiltrating leukocytes after exposure to select PSMα3 alanine variants. Interestingly, only PSMα3 L7A and K17A induced levels of leukocyte influx similar to those induced by wild-type PSMα3. S. aureus constructs expressing these PSMα3 derivatives from the genome will be valuable to investigate the biological role of PSM-receptor interaction in S. aureus disease in vivo. Furthermore, the identified non-toxic PSMα3 derivatives may serve to develop a toxoid vaccine.
Fig. 11. PSM structure-function analysis.

An alanine exchange library of the PSMα3 peptide was created and used to determine changes in PSM function caused by structural changes (Cheung, et al., 2013). Overall, large hydrophobic side chains impacted biofilm-structuring capacities (such as some phenylalanine residues shown in regular font) and predominantly cationic amino acids impacted cytolytic and pro-inflammatory functions. In addition, the C-terminus exclusively affected receptor-mediated but not cytolytic activities. Finally, removal of large hydrophobic side chains such as all phenylalanine residues led to derivatives that showed antimicrobial activities, indicating that these large hydrophobic side chains are involved in preventing cytolysis of bacterial versus eukaryotic membranes in PSMs.
Changes at the hydrophobic side of PSMα3, especially the exchange of large side chains such as phenylalanine, led to a reduction in biofilm-structuring activities and sometimes to antimicrobial activity toward S. pyogenes not observed in native PSMα3 (Cheung et al., 2013). The latter suggests that large hydrophobic side chains in PSMs prevent the lysis of bacterial versus that of eukaryotic cells. Accordingly, PSMα3, which has only strong capacity to lyse eukaryotic cells, has five phenylalanine residues, whereas PSMδ of S. epidermidis, which lyses both eukaryotic and prokaryotic cells, has only one.
Diversification of PSMs and PSM production
Based on recent findings, it has become evident that PSMs have undergone divergent evolution to develop different functions. For example, many α-type, but not β-type, PSMs are cytolytic and only the δ-toxin leads to mast cell degranulation (Wang et al., 2007, Cheung et al., 2010, Nakamura et al., 2013). The PSMβ peptides do not appear to have a distinct role, unless this role has not yet been identified. However, their lack of cytolytic function may be beneficial for commensal bacteria such as S. epidermidis in their effort to use PSM peptides in a “non-aggressive” fashion primarily to structure biofilms and facilitate life on epithelial surfaces, without simultaneously harming host cells.
While the capacity to produce strongly cytolytic PSMs is not limited to S. aureus, as we know from the cytolytic PSMδ of S. epidermidis (Cheung et al., 2010), only S. aureus appears to produce high amounts of cytolytic, “aggressive” PSMs to lyse host cells and thereby evade elimination by host defenses. Additionally, S. aureus uses increased production of PSMs to augment virulence, as seen with highly virulent S. aureus strains such as CA-MRSA as compared to HA-MRSA (Wang et al., 2007, Li et al., 2009). Whether S. epidermidis or other staphylococcal species up-regulate production of “aggressive” PSMs under conditions of systemic invasion or specific disease conditions is not known.
Targeting PSMs for anti-staphylococcal drug development
Historically, inactivated bacterial toxins proved to be successful candidates for active immunization to protect against bacterial disease, of which, the diphtheria, tetanus, acellular pertussis (DTaP) vaccine is a classic example (Adkins et al., 2012). However, unlike in S. aureus, the toxins produced by Corynebacterium diphtheria, Clostridium tetani, and Bordetella pertussis are, for the most part, the only and primary toxins that are responsible for disease development. In contrast, S. aureus produces a plethora of toxins that have different contributions to different S. aureus diseases. As an additional complication, the toxins produced by S. aureus are not uniformly expressed among all isolates, making the identification of a toxin target for immunotherapy against S. aureus disease challenging (Proctor, 2012). With the exception of α-toxin (Kennedy et al., 2010) and PVL (Brown et al., 2009), few other S. aureus toxins have been thoroughly investigated as candidates for active vaccination against a variety of pathogenic staphylococci (Spellberg & Daum, 2011).
Targeting the PSMs for anti-staphylococcal therapy and drug development would be advantageous for several reasons: the PSMs form a major constituent of the S. aureus exoprotein repertoire; they have an integral role in different facets of S. aureus disease; they show little variation in amino acid sequences between S. aureus strains; and the genome-encoded psm genes are present in all sequenced S. aureus strains (Wang et al., 2007). Therefore, neutralizing the cytolytic and pro-inflammatory activity of all PSMs would reduce their potency against host cells and possibly their overall contribution towards S. aureus disease progression.
Although protective humoral immune responses to S. aureus infection are generally not observed (Spellberg & Daum, 2011), a recent serological study showed a correlation between increased antibody levels to S. aureus exotoxins (α-toxin, PVL, S. aureus enterotoxin C1, δ-toxin, and PSMα3) and a lower incidence of sepsis in hospitalized adults with invasive S. aureus infections (Adhikari et al., 2012). These data indicate that the PSMs are immunogenic, since naturally circulating anti-PSM antibodies are detected in human adults (Adhikari et al., 2012). Furthermore, this observation correlates with studies that show robust anti-PSM antibody responses in mice and rabbits after active immunization with PSMs (Nolte & Kapral, 1981, Wang et al., 2011, Spaulding et al., 2012). In addition, rabbits immunized with a combination of PSMs show weak protection in a bacteremia infection model (Spaulding et al., 2012). The protective effect may be improved with the discovery of non-toxic PSMα3 derivatives (Cheung 2013), as high concentrations of PSMs are known to be cytotoxic in vitro (Wang et al., 2007).
The benefits of passive administration of anti-PSM antibodies were demonstrated in a study, in which antibodies against the S. epidermidis PSMβ peptides effectively blocked the dissemination of S. epidermidis from bacteria-coated catheters in mice (Wang et al., 2011). However, passive immunization with anti-PSM antibodies may not be generally beneficial in biofilm-related S. aureus and S. epidermidis infections, because – while they may inhibit dissemination - they may also lead to more compact and thicker biofilms (Wang et al., 2011). Accordingly, Agr-dysfunctional, and therefore PSM-negative, S. epidermidis and S. aureus strains are frequently isolated from biofilm-associated infections (Vuong et al., 2004, Traber et al., 2008). Thus, the benefits of passive vaccination with anti-PSM antibodies in the case of biofilm-associated staphylococcal disease need to be thoroughly investigated further. In contrast, passive immunization with antibodies directed against cytolytic PSMs may be more adequate in acute forms of staphylococcal disease. Problems with this approach are the following. First, PSMs are produced at high levels; likely requiring a high concentration of antibodies, at least if the primary mode of function is sequestration rather than promotion of opsonophagocytosis, whose efficacy in the case of S. aureus vaccines is debatable (DeLeo & Otto, 2008). Second, PSMs may exert much of their contributions to pathogenesis in the intracellular environment (Surewaard et al., 2012), which may be difficult to reach for antibodies in sufficient amounts. Third, the variety of PSMs produced by S. aureus would require an antibody cocktail, further complicating such an approach.
Keeping these difficulties in mind, developing drugs or mAbs against Pmt, the dedicated PSM ABC transporter (Chatterjee et al., 2013), has several key advantages over targeting single PSMs or other S. aureus toxins (Chatterjee & Otto, 2013). Inhibiting PSM transport would reduce the overall pathogenicity of S. aureus in vivo because of the interference of the secretion of all PSMs. Furthermore, because the fact that the pmt genes are found genome-encoded among all genome-sequenced staphylococci, it is probable that other pathogenic staphylococci may also be targeted by a potential drug inhibiting Pmt. Additionally, deletion of the pmt genes is harmful to S. aureus viability (Chatterjee et al., 2013) suggesting that blocking Pmt may have a direct bactericidal in addition to a virulence-diminishing effect.
Lastly, while lysis of immune cells very likely is of key importance for the strong impact of α-type PSMs on staphylococcal virulence, the biological role of PSM pro-inflammatory capacity and receptor-mediated recognition of PSMs by the human innate immune system is not understood. If indeed FPR2-dependent activation turns out to be crucial for the pathogenesis of staphylococcal disease, FPR antagonistic molecules, such as CHIPS and FLIPr, may be evaluated as potential drugs against staphylococcal infections.
In conclusion, the PSMs, FPR2-receptor mediated signaling, and especially Pmt are all possible targets for anti-staphylococcal drug development. However, as attractive as these single targets may be, successful protection against S. aureus may depend on disarming multiple targets that are involved in S. aureus virulence (Cheung & Otto, 2012).
Conclusion/open questions
PSMs are a fairly recently discovered group of peptides. Naturally, therefore, many aspects of PSM biology and biochemistry remain to be analyzed. First, our knowledge on psm genes and PSM peptides is restricted to S. aureus, S. epidermidis, and the psmβ-like genes that can be found by similarity searches in other staphylococci. A comprehensive analysis of PSMs of most staphylococcal species is still missing. Second, how the physico-chemical features of PSMs determine their function in surface spreading, emulsification, biofilm formation, and interaction with membranes during cytolysis remains to be more clearly analyzed. Most of this unknown and the only information we have on PSM structure is derived from experiments on S. aureus δ-toxin. One important question to be answered is the role of PSM aggregation, in particular in the reported form of ordered amyloid fibers, during biofilm formation and in-vivo biofilm infection. Third, the structural determinants of FPR2 activation by PSMs are not understood, as is the role of PSM-receptor interaction in pathogenesis, especially in comparison with the non-receptor-mediated cytolytic function of PSMs. Regarding cytolysis, further details of the interaction of PSMs with different cell types, including professional and non-professional phagocytes before and after ingestion, and the role of these interactions during infection and in possible synergy with other toxins need to be elucidated in more detail. Finally, PSMs may be used as vaccine targets, while small molecule inhibitors that target PSM export could induce killing bacteria during infection. However, while this newly recognized family of virulence determinants is clearly a promising therapeutic target, drug development efforts are still in their infancy.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), U.S. National Institutes of Health (NIH).
Bibliography
- Abdelnour A, Arvidson S, Bremell T, Ryden C, Tarkowski A. The accessory gene regulator (agr) controls Staphylococcus aureus virulence in a murine arthritis model. Infect Immun. 1993;61:3879–3885. doi: 10.1128/iai.61.9.3879-3885.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari RP, Ajao AO, Aman MJ, Karauzum H, Sarwar J, Lydecker AD, Johnson JK, Nguyen C, Chen WH, Roghmann MC. Lower antibody levels to Staphylococcus aureus exotoxins are associated with sepsis in hospitalized adults with invasive S. aureus infections. J Infect Dis. 2012;206:915–923. doi: 10.1093/infdis/jis462. [DOI] [PubMed] [Google Scholar]
- Adkins I, Holubova J, Kosova M, Sadilkova L. Bacteria and their toxins tamed for immunotherapy. Curr Pharm Biotech. 2012;13:1446–1473. doi: 10.2174/138920112800784835. [DOI] [PubMed] [Google Scholar]
- Alonzo F, 3rd, Benson MA, Chen J, Novick RP, Shopsin B, Torres VJ. Staphylococcus aureus leukocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol Microbiol. 2011 doi: 10.1111/j.1365-2958.2011.07942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonzo F, 3rd, Kozhaya L, Rawlings SA, Reyes-Robles T, DuMont AL, Myszka DG, Landau NR, Unutmaz D, Torres VJ. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature. 2013;493:51–55. doi: 10.1038/nature11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459–489. doi: 10.1146/annurev-immunol-020711-074942. [DOI] [PubMed] [Google Scholar]
- Angelini TE, Roper M, Kolter R, Weitz DA, Brenner MP. Bacillus subtilis spreads by surfing on waves of surfactant. Proc Natl Acad Sci U S A. 2009;106:18109–18113. doi: 10.1073/pnas.0905890106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SR, Elias D, Buckingham SC, Thomas ED, Novais E, Arkader A, Howard C. Changing patterns of acute hematogenous osteomyelitis and septic arthritis: emergence of community-associated methicillin-resistant Staphylococcus aureus. J Pediatr Orthop. 2006;26:703–708. doi: 10.1097/01.bpo.0000242431.91489.b4. [DOI] [PubMed] [Google Scholar]
- Bartlett AH, Foster TJ, Hayashida A, Park PW. Alpha-toxin facilitates the generation of CXC chemokine gradients and stimulates neutrophil homing in Staphylococcus aureus pneumonia. J Infect Dis. 2008;198:1529–1535. doi: 10.1086/592758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudet R, Bisaillon JG, Saheb SA, Sylvestre M. Production, purification, and preliminary characterization of a gonococcal growth inhibitor produced by a coagulase-negative staphylococcus isolated from the urogenital flora. Antimicrob Agents Chemother. 1982;22:277–283. doi: 10.1128/aac.22.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakdi S, Tranum-Jensen J. Alpha-toxin of Staphylococcus aureus. Microbiol Rev. 1991;55:733–751. doi: 10.1128/mr.55.4.733-751.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchini CE, Hulten KG, Mason EO, Jr, Gonzalez BE, Hammerman WA, Kaplan SL. Panton-Valentine leukocidin genes are associated with enhanced inflammatory response and local disease in acute hematogenous Staphylococcus aureus osteomyelitis in children. Pediatrics. 2006;117:433–440. doi: 10.1542/peds.2005-0566. [DOI] [PubMed] [Google Scholar]
- Boles BR, Thoendel M, Singh PK. Rhamnolipids mediate detachment of Pseudomonas aeruginosa from biofilms. Mol Microbiol. 2005;57:1210–1223. doi: 10.1111/j.1365-2958.2005.04743.x. [DOI] [PubMed] [Google Scholar]
- Branda SS, Gonzalez-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. Fruiting body formation by Bacillus subtilis. Proc Natl Acad Sci U S A. 2001;98:11621–11626. doi: 10.1073/pnas.191384198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EL, Dumitrescu O, Thomas D, et al. The Panton-Valentine leukocidin vaccine protects mice against lung and skin infections caused by Staphylococcus aureus USA300. Clin Microbiol Infect. 2009;15:156–164. doi: 10.1111/j.1469-0691.2008.02648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes EC, Lopez DM, Donegan NP, Cheung A, Gresham H, Timmins GS, Brinker CJ. Confinement-induced quorum sensing of individual Staphylococcus aureus bacteria. Nat Chem Biol. 2010;6:41–45. doi: 10.1038/nchembio.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassat JE, Hammer ND, Campbell JP, Benson MA, Perrien DS, Mrak LN, Smeltzer MS, Torres VJ, Skaar EP. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe. 2013;13:759–772. doi: 10.1016/j.chom.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffin DO, Taylor D, Skerrett SJ, Rubens CE. Changes in the Staphylococcus aureus transcriptome during early adaptation to the lung. PLoS One. 2012;7:e41329. doi: 10.1371/journal.pone.0041329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee SS, Otto M. How can Staphylococcus aureus phenol-soluble modulins be targeted to inhibit infection? Fut Microbiol. 2013;8:693–696. doi: 10.2217/fmb.13.37. [DOI] [PubMed] [Google Scholar]
- Chatterjee SS, Chen L, Joo HS, Cheung GY, Kreiswirth BN, Otto M. Distribution and regulation of the mobile genetic element-encoded phenol-soluble modulin PSM-mec in methicillin-resistant Staphylococcus aureus. PLoS One. 2011;6:e28781. doi: 10.1371/journal.pone.0028781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee SS, Joo HS, Duong AC, Dieringer TD, Tan VY, Song Y, Fischer ER, Cheung GY, Li M, Otto M. Essential Staphylococcus aureus toxin export system. Nat Med. 2013;19:364–367. doi: 10.1038/nm.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung AL, Eberhardt KJ, Chung E, Yeaman MR, Sullam PM, Ramos M, Bayer AS. Diminished virulence of a sar-/agr- mutant of Staphylococcus aureus in the rabbit model of endocarditis. J Clin Invest. 1994;94:1815–1822. doi: 10.1172/JCI117530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GY, Otto M. Direct and synergistic hemolysis caused by Staphylococcus phenol-soluble modulins: Implications for diagnosis and pathogenesis. Microbes Infect. 2011;14:380–386. doi: 10.1016/j.micinf.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GY, Otto M. The potential use of toxin antibodies as a strategy for controlling acute Staphylococcus aureus infections. Expert Opin Ther Targets. 2012;16:601–612. doi: 10.1517/14728222.2012.682573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GY, Wang R, Khan BA, Sturdevant DE, Otto M. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect Immun. 2011;79:1927–1935. doi: 10.1128/IAI.00046-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GY, Rigby K, Wang R, Queck SY, Braughton KR, Whitney AR, Teintze M, DeLeo FR, Otto M. Staphylococcus epidermidis strategies to avoid killing by human neutrophils. PLoS Pathog. 2010;6:e1001133. doi: 10.1371/journal.ppat.1001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GY, Kretschmer D, Queck SY, et al. Insight into structure-function relationship in phenol-soluble modulins using an alanine screen of the phenol-soluble modulin (PSM) alpha3 peptide. Faseb J. 2013 doi: 10.1096/fj.13-232041. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogen AL, Yamasaki K, Muto J, Sanchez KM, Crotty Alexander L, Tanios J, Lai Y, Kim JE, Nizet V, Gallo RL. Staphylococcus epidermidis antimicrobial delta-toxin (phenol-soluble modulin-gamma) cooperates with host antimicrobial peptides to kill group A Streptococcus. PLoS One. 2010;5:e8557. doi: 10.1371/journal.pone.0008557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogen AL, Yamasaki K, Sanchez KM, et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J Invest Dermatol. 2010;130:192–200. doi: 10.1038/jid.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- de Haas CJ, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, Poppelier MJ, Van Kessel KP, van Strijp JA. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med. 2004;199:687–695. doi: 10.1084/jem.20031636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo FR, Otto M. An antidote for Staphylococcus aureus pneumonia? J Exp Med. 2008;205:271–274. doi: 10.1084/jem.20080167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet. 2010;375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux PF, Morris R. Transmembrane asymmetry and lateral domains in biological membranes. Traffic. 2004;5:241–246. doi: 10.1111/j.1600-0854.2004.0170.x. [DOI] [PubMed] [Google Scholar]
- Dhople VM, Nagaraj R. Generation of analogs having potent antimicrobial and hemolytic activities with minimal changes from an inactive 16-residue peptide corresponding to the helical region of Staphylococcus aureus delta-toxin. Protein Eng. 1995;8:315–318. doi: 10.1093/protein/8.3.315. [DOI] [PubMed] [Google Scholar]
- Dhople VM, Nagaraj R. Conformation and activity of delta-lysin and its analogs. Peptides. 2005;26:217–225. doi: 10.1016/j.peptides.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Diep BA, Gill SR, Chang RF, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367:731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- Diep BA, Chan L, Tattevin P, et al. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc Natl Acad Sci U S A. 2010;107:5587–5592. doi: 10.1073/pnas.0912403107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donvito B, Etienne J, Denoroy L, Greenland T, Benito Y, Vandenesch F. Synergistic hemolytic activity of Staphylococcus lugdunensis is mediated by three peptides encoded by a non-agr genetic locus. Infect Immun. 1997;65:95–100. doi: 10.1128/iai.65.1.95-100.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuMont AL, Yoong P, Day CJ, Alonzo F, 3rd, McDonald WH, Jennings MP, Torres VJ. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci U S A. 2013;110:10794–10799. doi: 10.1073/pnas.1305121110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, Shopsin B, Unutmaz D, Voyich JM, Torres VJ. Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol Microbiol. 2011;79:814–825. doi: 10.1111/j.1365-2958.2010.07490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong AC, Cheung GY, Otto M. Interaction of phenol-soluble modulins with phosphatidylcholine vesicles. Pathogens. 2012;1:3–11. doi: 10.3390/pathogens1010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezepchuk YV, Leung DY, Middleton MH, Bina P, Reiser R, Norris DA. Staphylococcal toxins and protein A differentially induce cytotoxicity and release of tumor necrosis factor-alpha from human keratinocytes. J Invest Dermatol. 1996;107:603–609. doi: 10.1111/1523-1747.ep12583377. [DOI] [PubMed] [Google Scholar]
- Forsman H, Christenson K, Bylund J, Dahlgren C. Receptor-dependent and -independent immunomodulatory effects of phenol-soluble modulin peptides from Staphylococcus aureus on human neutrophils are abrogated through peptide inactivation by reactive oxygen species. Infect Immun. 2012;80:1987–1995. doi: 10.1128/IAI.05906-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Karlsson J, Bylund J, Movitz C, Karlsson A, Dahlgren C. Ligand recognition and activation of formyl peptide receptors in neutrophils. J Leukoc Biol. 2006;79:247–256. doi: 10.1189/jlb.0905498. [DOI] [PubMed] [Google Scholar]
- Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18:693–704. doi: 10.1038/nm.2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12:1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T, Francois P, Liebeke M, Fraunholz M, Goerke C, Krismer B, Schrenzel J, Lalk M, Wolz C. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog. 2012;8:e1003016. doi: 10.1371/journal.ppat.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese B, Glowinski F, Paprotka K, Dittmann S, Steiner T, Sinha B, Fraunholz MJ. Expression of delta-toxin by Staphylococcus aureus mediates escape from phago-endosomes of human epithelial and endothelial cells in the presence of beta-toxin. Cell Microbiol. 2011;13:316–329. doi: 10.1111/j.1462-5822.2010.01538.x. [DOI] [PubMed] [Google Scholar]
- Gill SR, Fouts DE, Archer GL, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187:2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun. 1995;63:3373–3380. doi: 10.1128/iai.63.9.3373-3380.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez DJ, Okumura CY, Hollands A, et al. Novel phenol-soluble modulin derivatives in community-associated methicillin-resistant Staphylococcus aureus identified through imaging mass spectrometry. J Biol Chem. 2012;287:13889–13898. doi: 10.1074/jbc.M112.349860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol. 2000;164:3713–3722. doi: 10.4049/jimmunol.164.7.3713. [DOI] [PubMed] [Google Scholar]
- Grosz M, Kolter J, Paprotka K, et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell Microbiol. 2013 doi: 10.1111/cmi.12233. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocke AC, Temmesfeld-Wollbrueck B, Schmeck B, Berger K, Frisch EM, Witzenrath M, Brell B, Suttorp N, Hippenstiel S. Perturbation of endothelial junction proteins by Staphylococcus aureus alpha-toxin: inhibition of endothelial gap formation by adrenomedullin. Histochem Cell Biol. 2006;126:305–316. doi: 10.1007/s00418-006-0174-5. [DOI] [PubMed] [Google Scholar]
- Hongo I, Baba T, Oishi K, Morimoto Y, Ito T, Hiramatsu K. Phenol-soluble modulin alpha 3 enhances the human neutrophil lysis mediated by Panton-Valentine leukocidin. J Infect Dis. 2009;200:715–723. doi: 10.1086/605332. [DOI] [PubMed] [Google Scholar]
- Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, Bubeck Wardenburg J. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. 2011;17:1310–1314. doi: 10.1038/nm.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo HS, Otto M. The isolation and analysis of phenol-soluble modulins of Staphylococcus epidermidis. Methods Mol Biol. 2014;1106:93–100. doi: 10.1007/978-1-62703-736-5_7. [DOI] [PubMed] [Google Scholar]
- Joo HS, Cheung GY, Otto M. Antimicrobial activity of community-associated methicillin-resistant Staphylococcus aureus is caused by phenol-soluble modulin derivatives. J Biol Chem. 2011;286:8933–8940. doi: 10.1074/jbc.M111.221382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaito C, Saito Y, Nagano G, et al. Transcription and translation products of the cytolysin gene psm-mec on the mobile genetic element SCCmec regulate Staphylococcus aureus virulence. PLoS Pathog. 2011;7:e1001267. doi: 10.1371/journal.ppat.1001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaito C, Saito Y, Ikuo M, et al. Mobile genetic element SCCmec-encoded psm-mec RNA suppresses translation of agrA and attenuates MRSA virulence. PLoS Pathog. 2013;9:e1003269. doi: 10.1371/journal.ppat.1003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis. 2010;202:1050–1058. doi: 10.1086/656043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klevens RM, Morrison MA, Nadle J, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Jama. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- Kobayashi SD, Malachowa N, Whitney AR, Braughton KR, Gardner DJ, Long D, Bubeck Wardenburg J, Schneewind O, Otto M, DeLeo FR. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J Infect Dis. 2011;204:937–941. doi: 10.1093/infdis/jir441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi SD, Braughton KR, Palazzolo-Ballance AM, et al. Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J Innate Immun. 2010;2:560–575. doi: 10.1159/000317134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan G, Sadovskaya I, Chaignon P, Chokr A, Jabbouri S. Biofilms of clinical strains of Staphylococcus that do not contain polysaccharide intercellular adhesin. FEMS Microbiol Lett. 2006;255:11–16. doi: 10.1111/j.1574-6968.2005.00043.x. [DOI] [PubMed] [Google Scholar]
- Kreger AS, Bernheimer AW. Disruption of bacterial protoplasts and spheroplasts by staphylococcal delta hemolysin. Infect Immun. 1971;3:603–605. doi: 10.1128/iai.3.4.603-605.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer D, Nikola N, Durr M, Otto M, Peschel A. The virulence regulator Agr controls the staphylococcal capacity to activate human neutrophils via the formyl peptide receptor 2. J Innate Immun. 2011;4:201–212. doi: 10.1159/000332142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer D, Gleske AK, Rautenberg M, et al. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe. 2010;7:463–473. doi: 10.1016/j.chom.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubica M, Guzik K, Koziel J, et al. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One. 2008;3:e1409. doi: 10.1371/journal.pone.0001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Cheung GY, Hu J, Wang D, Joo HS, DeLeo FR, Otto M. Comparative analysis of virulence and toxin expression of global community-associated methicillin-resistant Staphylococcus aureus strains. J Infect Dis. 2010;202:1866–1876. doi: 10.1086/657419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Diep BA, Villaruz AE, Braughton KR, Jiang X, DeLeo FR, Chambers HF, Lu Y, Otto M. Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci U S A. 2009;106:5883–5888. doi: 10.1073/pnas.0900743106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Ji Y. Alpha-toxin interferes with integrin-mediated adhesion and internalization of Staphylococcus aureus by epithelial cells. Cell Microbiol. 2006;8:1656–1668. doi: 10.1111/j.1462-5822.2006.00740.x. [DOI] [PubMed] [Google Scholar]
- Liles WC, Thomsen AR, O’Mahony DS, Klebanoff SJ. Stimulation of human neutrophils and monocytes by staphylococcal phenol-soluble modulin. J Leukoc Biol. 2001;70:96–102. [PubMed] [Google Scholar]
- Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- Lowy FD. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest. 2003;111:1265–1273. doi: 10.1172/JCI18535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Aguilar G, Avalos-Mishaan A, Hulten K, Hammerman W, Mason EO, Jr, Kaplan SL. Community-acquired, methicillin-resistant and methicillin-susceptible Staphylococcus aureus musculoskeletal infections in children. Pediatr Infect Dis. 2004;23:701–706. doi: 10.1097/01.inf.0000133044.79130.2a. [DOI] [PubMed] [Google Scholar]
- Mehlin C, Headley CM, Klebanoff SJ. An inflammatory polypeptide complex from Staphylococcus epidermidis: isolation and characterization. J Exp Med. 1999;189:907–918. doi: 10.1084/jem.189.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17:501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Monecke S, Engelmann I, Archambault M, et al. Distribution of SCCmec-associated phenol-soluble modulin in staphylococci. Mol Cell Probes. 2012;26:99–103. doi: 10.1016/j.mcp.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Oscherwitz J, Cease KB, et al. Staphylococcus δ-toxin promotes allergic skin disease by inducing mast cell degranulation. Nature. 2013;503:397–401. doi: 10.1038/nature12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte FS, Kapral FA. Immunogenicity of Staphylococcus aureus delta-toxin. Infect Immun. 1981;31:1251–1260. doi: 10.1128/iai.31.3.1251-1260.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol. 2003;48:1429–1449. doi: 10.1046/j.1365-2958.2003.03526.x. [DOI] [PubMed] [Google Scholar]
- Novick RP, Schlievert P, Ruzin A. Pathogenicity and resistance islands of staphylococci. Microbes Infect. 2001;3:585–594. doi: 10.1016/s1286-4579(01)01414-9. [DOI] [PubMed] [Google Scholar]
- Oogai Y, Matsuo M, Hashimoto M, Kato F, Sugai M, Komatsuzawa H. Expression of virulence factors by Staphylococcus aureus grown in serum. Appl Environ Microbiol. 2011;77:8097–8105. doi: 10.1128/AEM.05316-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcal biofilms. Curr Top Microbiol Immunol. 2008;322:207–228. doi: 10.1007/978-3-540-75418-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcus epidermidis--the ‘accidental’ pathogen. Nat Rev Microbiol. 2009;7:555–567. doi: 10.1038/nrmicro2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu Rev Microbiol. 2010;64:143–162. doi: 10.1146/annurev.micro.112408.134309. [DOI] [PubMed] [Google Scholar]
- Otto M. A MRSA-terious enemy among us: end of the PVL controversy? Nat Med. 2011;17:169–170. doi: 10.1038/nm0211-169. [DOI] [PubMed] [Google Scholar]
- Otto M. Coagulase-negative staphylococci as reservoirs of genes facilitating MRSA infection: Staphylococcal commensal species such as Staphylococcus epidermidis are being recognized as important sources of genes promoting MRSA colonization and virulence. BioEssays. 2013;35:4–11. doi: 10.1002/bies.201200112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu Rev Med. 2013;64:175–188. doi: 10.1146/annurev-med-042711-140023. [DOI] [PubMed] [Google Scholar]
- Otto M, O’Mahoney DS, Guina T, Klebanoff SJ. Activity of Staphylococcus epidermidis phenol-soluble modulin peptides expressed in Staphylococcus carnosus. J Infect Dis. 2004;190:748–755. doi: 10.1086/422157. [DOI] [PubMed] [Google Scholar]
- Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. agr-Dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun. 2010;2:546–559. doi: 10.1159/000319855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periasamy S, Joo HS, Duong AC, Bach TH, Tan VY, Chatterjee SS, Cheung GY, Otto M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc Natl Acad Sci U S A. 2012;109:1281–1286. doi: 10.1073/pnas.1115006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokorny A, Birkbeck TH, Almeida PF. Mechanism and kinetics of delta-lysin interaction with phospholipid vesicles. Biochemistry. 2002;41:11044–11056. doi: 10.1021/bi020244r. [DOI] [PubMed] [Google Scholar]
- Prat C, Bestebroer J, de Haas CJ, van Strijp JA, van Kessel KP. A new staphylococcal anti-inflammatory protein that antagonizes the formyl peptide receptor-like 1. J Immunol. 2006;177:8017–8026. doi: 10.4049/jimmunol.177.11.8017. [DOI] [PubMed] [Google Scholar]
- Proctor RA. Challenges for a universal Staphylococcus aureus vaccine. Clin Infect Dis. 2012;54:1179–1186. doi: 10.1093/cid/cis033. [DOI] [PubMed] [Google Scholar]
- Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, Otto M. RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol Cell. 2008;32:150–158. doi: 10.1016/j.molcel.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queck SY, Khan BA, Wang R, Bach TH, Kretschmer D, Chen L, Kreiswirth BN, Peschel A, Deleo FR, Otto M. Mobile genetic element-encoded cytolysin connects virulence to methicillin resistance in MRSA. PLoS Pathog. 2009;5:e1000533. doi: 10.1371/journal.ppat.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasigade JP, Trouillet-Assant S, Ferry T, et al. PSMs of hypervirulent Staphylococcus aureus act as intracellular toxins that kill infected osteoblasts. PLoS One. 2013;8:e63176. doi: 10.1371/journal.pone.0063176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautenberg M, Joo HS, Otto M, Peschel A. Neutrophil responses to staphylococcal pathogens and commensals via the formyl peptide receptor 2 relates to phenol-soluble modulin release and virulence. Faseb J. 2011;25:1254–1263. doi: 10.1096/fj.10-175208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby KM, DeLeo FR. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin Immunopathol. 2012;34:237–259. doi: 10.1007/s00281-011-0295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimland D, Roberson B. Gastrointestinal carriage of methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 1986;24:137–138. doi: 10.1128/jcm.24.1.137-138.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DE. Studies on bacteriemia. I. Mechanisms relating to the persistence of bacteriemia in rabbits following the intravenous injection of staphylococci. J Exp Med. 1956;103:713–742. doi: 10.1084/jem.103.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers KL, Fey PD, Rupp ME. Coagulase-negative staphylococcal infections. Infect Dis Clin North Am. 2009;23:73–98. doi: 10.1016/j.idc.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Rohde H, Burandt EC, Siemssen N, et al. Polysaccharide intercellular adhesin or protein factors in biofilm accumulation of Staphylococcus epidermidis and Staphylococcus aureus isolated from prosthetic hip and knee joint infections. Biomaterials. 2007;28:1711–1720. doi: 10.1016/j.biomaterials.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Rudikoff D, Lebwohl M. Atopic dermatitis. Lancet. 1998;351:1715–1721. doi: 10.1016/S0140-6736(97)12082-7. [DOI] [PubMed] [Google Scholar]
- Scheifele DW, Bjornson GL, Dyer RA, Dimmick JE. Delta-like toxin produced by coagulase-negative staphylococci is associated with neonatal necrotizing enterocolitis. Infect Immun. 1987;55:2268–2273. doi: 10.1128/iai.55.9.2268-2273.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner J, Kretschmer D, Klenk J, Otto M, Buhring HJ, Stevanovic S, Wang JM, Beer-Hammer S, Peschel A, Autenrieth SE. Staphylococcus aureus phenol-soluble modulin peptides modulate dendritic cell functions and increase in vitro priming of regulatory T cells. J Immunol. 2013;190:3417–3426. doi: 10.4049/jimmunol.1202563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012;8:e1002744. doi: 10.1371/journal.ppat.1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville GA, Cockayne A, Durr M, Peschel A, Otto M, Musser JM. Synthesis and deformylation of Staphylococcus aureus delta-toxin are linked to tricarboxylic acid cycle activity. J Bacteriol. 2003;185:6686–6694. doi: 10.1128/JB.185.22.6686-6694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan AN, Henry T, van Rooijen WJ, et al. The staphylococcal toxin Panton-Valentine Leukocidin targets human C5a receptors. Cell Host Microbe. 2013;13:584–594. doi: 10.1016/j.chom.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Spaulding AR, Lin YC, Merriman JA, Brosnahan AJ, Peterson ML, Schlievert PM. Immunity to Staphylococcus aureus secreted proteins protects rabbits from serious illnesses. Vaccine. 2012;30:5099–5109. doi: 10.1016/j.vaccine.2012.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellberg B, Daum R. Development of a vaccine against Staphylococcus aureus. Semin Immunopathol. 2011;34:335–348. doi: 10.1007/s00281-011-0293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spentzas T, Kudumula R, Acuna C, Talati AJ, Ingram KC, Savorgnan F, Meals EA, English BK. Role of bacterial components in macrophage activation by the LAC and MW2 strains of community-associated, methicillin-resistant Staphylococcus aureus. Cell Immunol. 2011;269:46–53. doi: 10.1016/j.cellimm.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Sugimoto S, Iwamoto T, Takada K, Okuda K, Tajima A, Iwase T, Mizunoe Y. Staphylococcus epidermidis Esp degrades specific proteins associated with Staphylococcus aureus biofilm formation and host-pathogen interaction. J Bacteriol. 2013;195:1645–1655. doi: 10.1128/JB.01672-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surewaard BG, Nijland R, Spaan AN, Kruijtzer JA, de Haas CJ, van Strijp JA. Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog. 2012;8:e1002606. doi: 10.1371/journal.ppat.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surewaard BG, de Haas CJ, Vervoort F, Rigby KM, DeLeo FR, Otto M, van Strijp JA, Nijland R. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell Microbiol. 2013;15:1427–1437. doi: 10.1111/cmi.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szmigielski S, Prevost G, Monteil H, Colin DA, Jeljaszewicz J. Leukocidal toxins of staphylococci. Zentralbl Bakteriol. 1999;289:185–201. doi: 10.1016/s0934-8840(99)80105-4. [DOI] [PubMed] [Google Scholar]
- Tappin MJ, Pastore A, Norton RS, Freer JH, Campbell ID. High-resolution 1H NMR study of the solution structure of delta-hemolysin. Biochemistry. 1988;27:1643–1647. doi: 10.1021/bi00405a038. [DOI] [PubMed] [Google Scholar]
- Thelestam M, Mollby R, Wadstrom T. Effects of staphylococcal alpha-, beta-, delta-, and gamma-hemolysins on human diploid fibroblasts and HeLa cells: evaluation of a new quantitative as say for measuring cell damage. Infect Immun. 1973;8:938–946. doi: 10.1128/iai.8.6.938-946.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thwaites GE, Gant V. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol. 2011;9:215–222. doi: 10.1038/nrmicro2508. [DOI] [PubMed] [Google Scholar]
- Traber KE, Lee E, Benson S, Corrigan R, Cantera M, Shopsin B, Novick RP. Agr function in clinical Staphylococcus aureus isolates. Microbiology. 2008;154:2265–2274. doi: 10.1099/mic.0.2007/011874-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsompanidou E, Denham EL, Becher D, de Jong A, Buist G, van Oosten M, Manson WL, Back JW, van Dijl JM, Dreisbach A. Distinct roles of phenol-soluble modulins in spreading of Staphylococcus aureus on wet surfaces. Appl Environ Microbiol. 2013;79:886–895. doi: 10.1128/AEM.03157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchscherr L, Heitmann V, Hussain M, Viemann D, Roth J, von Eiff C, Peters G, Becker K, Loffler B. Staphylococcus aureus small-colony variants are adapted phenotypes for intracellular persistence. J Infect Dis. 2010;202:1031–1040. doi: 10.1086/656047. [DOI] [PubMed] [Google Scholar]
- Tuchscherr L, Medina E, Hussain M, et al. Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol Med. 2011;3:129–141. doi: 10.1002/emmm.201000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeva A, Walev I, Pinkernell M, Walker B, Bayley H, Palmer M, Bhakdi S. Transmembrane beta-barrel of staphylococcal alpha-toxin forms in sensitive but not in resistant cells. Proc Natl Acad Sci U S A. 1997;94:11607–11611. doi: 10.1073/pnas.94.21.11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Have KL, Karmazyn B, Verma M, Caird MS, Hensinger RN, Farley FA, Lubicky JP. Community-associated methicillin-resistant Staphylococcus aureus in acute musculoskeletal infection in children: a game changer. J Pediatr Orthop. 2009;29:927–931. doi: 10.1097/BPO.0b013e3181bd1e0c. [DOI] [PubMed] [Google Scholar]
- Ventura CL, Malachowa N, Hammer CH, Nardone GA, Robinson MA, Kobayashi SD, DeLeo FR. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS One. 2010;5:e11634. doi: 10.1371/journal.pone.0011634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdon J, Berjeaud JM, Lacombe C, Hechard Y. Characterization of anti-Legionella activity of warnericin RK and delta-lysin I from Staphylococcus warneri. Peptides. 2008;29:978–984. doi: 10.1016/j.peptides.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Verdon J, Girardin N, Lacombe C, Berjeaud JM, Hechard Y. delta-hemolysin, an update on a membrane-interacting peptide. Peptides. 2009;30:817–823. doi: 10.1016/j.peptides.2008.12.017. [DOI] [PubMed] [Google Scholar]
- von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. New Engl J Med. 2001;344:11–16. doi: 10.1056/NEJM200101043440102. [DOI] [PubMed] [Google Scholar]
- Voyich JM, Braughton KR, Sturdevant DE, et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol. 2005;175:3907–3919. doi: 10.4049/jimmunol.175.6.3907. [DOI] [PubMed] [Google Scholar]
- Vuong C, Kocianova S, Yao Y, Carmody AB, Otto M. Increased colonization of indwelling medical devices by quorum-sensing mutants of Staphylococcus epidermidis in vivo. J Infect Dis. 2004;190:1498–1505. doi: 10.1086/424487. [DOI] [PubMed] [Google Scholar]
- Vuong C, Durr M, Carmody AB, Peschel A, Klebanoff SJ, Otto M. Regulated expression of pathogen-associated molecular pattern molecules in Staphylococcus epidermidis: quorum-sensing determines pro-inflammatory capacity and production of phenol-soluble modulins. Cell Microbiol. 2004;6:753–759. doi: 10.1111/j.1462-5822.2004.00401.x. [DOI] [PubMed] [Google Scholar]
- Walev I, Martin E, Jonas D, Mohamadzadeh M, Muller-Klieser W, Kunz L, Bhakdi S. Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect Immun. 1993;61:4972–4979. doi: 10.1128/iai.61.12.4972-4979.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Khan BA, Cheung GY, Bach TH, Jameson-Lee M, Kong KF, Queck SY, Otto M. Staphylococcus epidermidis surfactant peptides promote biofilm maturation and dissemination of biofilm-associated infection in mice. J Clin Invest. 2011;121:238–248. doi: 10.1172/JCI42520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Braughton KR, Kretschmer D, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- Watson DC, Yaguchi M, Bisaillon JG, Beaudet R, Morosoli R. The amino acid sequence of a gonococcal growth inhibitor from Staphylococcus haemolyticus. Biochem J. 1988;252:87–93. doi: 10.1042/bj2520087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005;5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci U S A. 2010;107:13473–13478. doi: 10.1073/pnas.1001815107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams H, Flohr C. How epidemiology has challenged 3 prevailing concepts about atopic dermatitis. J Allergy Clin Immunol. 2006;118:209–213. doi: 10.1016/j.jaci.2006.04.043. [DOI] [PubMed] [Google Scholar]
- Yao Y, Sturdevant DE, Otto M. Genomewide analysis of gene expression in Staphylococcus epidermidis biofilms: insights into the pathophysiology of S. epidermidis biofilms and the role of phenol-soluble modulins in formation of biofilms. J Infect Dis. 2005;191:289–298. doi: 10.1086/426945. [DOI] [PubMed] [Google Scholar]
- Zielinska AK, Beenken KE, Joo HS, Mrak LN, Griffin LM, Luong TT, Lee CY, Otto M, Shaw LN, Smeltzer MS. Defining the strain-dependent impact of the Staphylococcal accessory regulator (sarA) on the alpha-toxin phenotype of Staphylococcus aureus. J Bacteriol. 2011;193:2948–2958. doi: 10.1128/JB.01517-10. [DOI] [PMC free article] [PubMed] [Google Scholar]