

Abstract
Coronary heart disease (CHD) continues to be the greatest mortality risk factor in the developed world. Estrogens are recognized to have great therapeutic potential to treat CHD and other cardiovascular diseases; however, a significant array of potentially debilitating side effects continues to limit their use. Moreover, recent clinical trials have indicated that long-term postmenopausal estrogen therapy may actually be detrimental to cardiovascular health. An exciting new development is the finding that the more recently discovered G-protein-coupled estrogen receptor (GPER) is expressed in coronary arteries-both in coronary endothelium and in smooth muscle within the vascular wall. Accumulating evidence indicates that GPER activation dilates coronary arteries and can also inhibit the proliferation and migration of coronary smooth muscle cells. Thus, selective GPER activation has the potential to increase coronary blood flow and possibly limit the debilitating consequences of coronary atherosclerotic disease. This review will highlight what is currently known regarding the impact of GPER activation on coronary arteries and the potential signaling mechanisms stimulated by GPER agonists in these vessels. A thorough understanding of GPER function in coronary arteries may promote the development of new therapies that would help alleviate CHD, while limiting the potentially dangerous side effects of estrogen therapy.
Keywords: G-protein-coupled estrogen receptor, Coronary arteries, G-1, Atherosclerosis, Estrogen
Core tip: A continuing controversy in cardiology is the impact of estrogen on coronary arteries. This review provides the latest information on the discovery of a novel estrogen receptor in these vessels: the G-protein-coupled estrogen receptor (GPER). Recent findings demonstrate that GPER activation induces coronary artery relaxation and attenuates the proliferation and migration of coronary smooth muscle cells. Thus, GPER appears to be a promising, novel pharmacological target that could increase coronary blood flow in diseased arteries and prevent or reverse the progression of coronary atherosclerotic disease, and do so with potentially fewer dangerous side effects associated with traditional estrogen therapy.
INTRODUCTION
Evidence that ovarian hormones can influence blood flow in other vascular beds was provided well over a century ago[1]; yet the overall impact of estrogens on cardiovascular function is still quite controversial. For example, there are conflicting reports regarding the potential therapeutic uses of estrogens to alleviate or prevent cardiovascular disease, as clinical trials have found both preventative[2] and deleterious[3] effects of conjugated equine estrogens (a mixture of at least 10 different estrogens) on coronary heart disease (CHD) in menopausal women. More directly, 17β-estradiol (E2), the predominant and most potent circulating estrogen in premenopausal women, has clearly defined effects on blood vessel function. For example, numerous studies have demonstrated that estradiol dilates coronary arteries from humans or other species, and does so by specifically targeting both vascular smooth muscle (VSM) and endothelial cells. Estradiol increases coronary blood flow in intact hearts[4,5], dilates coronary arteries in situ[6-8], and relaxes coronary arteries isolated in vitro[9-12]. Such studies strongly suggest an inherent therapeutic potential of E2 for alleviating or possibly even preventing coronary insufficiency. As a caution, however, it should be noted that we have demonstrated that E2 can also constrict coronary arteries under experimental[13] or pathological[14] conditions via a non-genomic mechanism. Thus, E2, a “well-known vasodilator”, is actually a powerful, multifunctional vasoactive hormone whose signaling mechanisms are heterogeneous and complicated, and whose overall physiological effect on coronary arteries apparently depends upon the biochemical disposition of cells located in the vascular wall.
In addition to directly modulating coronary artery function, estrogens may also slow the progression of coronary atherosclerotic disease. Young women are normally protected from significant atherosclerotic plaque accumulation in coronary arteries. Menopause, however, brings unhealthy changes in plasma lipoproteins [i.e., increased low-density lipoproteins (LDLs), and decreased high-density lipoproteins (HDLs), respectively], whereas postmenopausal estrogen replacement therapy (ERT) reverses these potentially harmful changes by decreasing LDL and increasing HDL[15-17]. More specifically, the phenolic ring structure of estrogens appears to exert an antioxidant effect that may attenuate oxidation of lipids[18,19] and LDLs[20-22]; and it is oxidized LDLs are accumulated by macrophages in the early stages of atherosclerosis leading to foam cell formation and atherogenesis. Further, E2 can lower the activity and expression of vascular NADPH oxidase, a potent source of reactive oxygen species[23-25]. Thus, there is increasing evidence that an antioxidant effect of estrogen may attenuate the development of coronary atherosclerosis to help preserve optimal cardiac function throughout a woman’s reproductive years[26].
One might expect oxidant stress to increase during menopause as the antioxidant influence of estrogen begins to wane. Indeed, menopausal women do exhibit greater levels of oxidative stress compared to women of childbearing age[27]. In light of the demonstrated beneficial effects of E2 on plasma lipoproteins, it is puzzling that recent clinical trials continue to indicate a mostly deleterious effect of long-term postmenopausal hormone therapy on cardiovascular health. A potential reason for this apparent contradiction is that ERT, while reducing total LDL cholesterol level, does not reduce the number of LDL particles-which, become smaller, therefore more atherogenic[28]. In addition, we have demonstrated that E2 can actually promote oxidative stress in coronary arteries by enhancing the activity of uncoupled nitric oxide synthase expressed in VSM cells[13]. Thus, it seems likely that the influence of estrogens on cardiovascular health cannot be explained solely by an antioxidant effect on plasma lipids or blood vessels.
In summary, there is convincing evidence that estrogens can exert powerful effects on both the structure and function of coronary arteries, and that E2 can produce both beneficial and harmful effects on cardiovascular function. As our appreciation of estrogen grows, so should the potential to develop estrogen-like compounds as novel therapeutic agents to prevent or treat CHD. The current challenge remains to fully elucidate the cellular/molecular basis of estrogen action on coronary arteries. At present, however, our understanding of these mechanisms is far from complete, and is at times quite controversial. An important first step is to understand the nature of the specific estrogen receptor molecules that bind E2 and thereby initiate the complicated process of estrogen signaling in coronary arteries.
G-PROTEIN-COUPLED ESTROGEN RECEPTOR AND CORONARY ARTERY TONE
For years it was believed that E2 action on coronary arteries was mediated only via activation of one or both of the “classic” estrogen receptors (ER), ERα or ERβ. For example, both ERs are expressed in human coronary artery smooth muscle cells (CASMC); but it is ERα that appears to play a great role in mediating acute responses to E2 in these cells via increased nitric oxide (NO) generation[29]. Similarly, porcine coronary arteries were relaxed via a NO-dependent mechanism in vitro by an ERα-selective agonist, whereas an ERβ-selective agonist appeared to induce an NO-independent relaxation response[30]. It is the ERα subtype that helps protect against ischemia/reperfusion injury in rabbit hearts[31], whereas ERβ did not impact ischemic tolerance significantly[32]. In addition, optimal functioning of coronary artery endothelial cells is abrogated in mice lacking expression of ERα[33]. On the other hand, studies of coronary arteries obtained from human females indicated that it was ERβ, which was associated with coronary calcification and atherosclerosis, not ERα[34]. These studies suggest that although ERα and ERβ are both expressed in coronary arteries, it is ERα that may play the greater role in an acute vasodilatory response to E2 and possibly cardioprotection as well.
It is the dependency of estrogen on ERα and ERβ activation that has limited the potential use of estrogen as a therapeutic agent to alleviate coronary artery dysfunction. Estrogens are powerful endocrine hormones, and these endocrine effects (e.g., secondary sex characteristics) are mediated primarily by classic ERs which are expressed in most cell types. In terms of specifically treating cardiovascular disease the need is to pharmacologically mimic the beneficial effects of estrogens on coronary arteries while minimizing the oftentimes unwanted endocrine side effects in other tissues. Selective ER modulators (SERMs)-agents which act as ER agonists in some target tissues but not in others-have had some success in meeting this therapeutic ideal, but their use still falls short of providing protection against coronary artery disease without endocrine and other side effects. An exciting new development in this important field of investigation is the discovery of a novel G-protein-coupled estrogen receptor (GPR30, now GPER). GPER activation constitutes an acute, non-genomic mechanism of estrogen action that may avoid many, if not most, potential effects of estrogen on endocrine target tissues. Discovery of GPER expression and function in blood vessels has opened the potential for this receptor to serve as a novel therapeutic target.
What was often noticed, yet seldom appreciated, was that some commonly employed ER “antagonists” did not always attenuate the vasodilatory effect of E2 on coronary arteries, and sometimes actually exhibited a direct vascular action themselves. For example, ICI182,780 (fulvestrant) has long been considered a “pure” ERα/ERβ antagonist, an estrogen receptor down regulator (SERD); however, this ER blocker did not significantly affect E2-induced coronary dilation or blood flow in canine hearts[8]. These studies suggested a vasodilatory effect of E2 on coronary arteries that was not mediated by either of the classic ERs. Moreover, we[35] and others[36] have demonstrated that ICI182,780 can itself relax porcine coronary arteries in vitro (i.e., an effect independent of ERα or ERβ activation). In addition, ICI182,780 does not inhibit coronary artery relaxation induced by the SERM raloxifene, which acts directly on CASMC[37]. Taken together, these findings strongly suggest that E2 (and possibly ICI182,780 and raloxifene as well) can relax coronary arteries via a mechanism that does not involve activation of ERα or ERβ. Interestingly, it is now known that ICI182,780 and raloxifene are agonists for GPER[36,38-40].
GPER was originally cloned in 1997 from breast cancer[41] and other cells, including endothelial cells[42,43], and was found to exhibit the canonical seven transmembrane spanning regions common to all G-protein-coupled receptors. GPER is expressed in the heart and in a variety of blood vessels[44,45], and GPER mRNA and protein have been detected in porcine and human CASMC[46-48]. Functionally, studies employing the selective GPER agonist, G-1, have indicated a vasodilatory response to G-1 stimulation. Acute treatment with G-1 relaxes rat aorta[49], rat mesenteric arteries[50], human internal mammary arteries[50], and rat carotid arteries[40,51], whereas infusion of G-1 induces an acute decrease in arterial pressure[50]. In addition, recent studies have demonstrated that G-1 induces an acute relaxation response in porcine coronary arteries[36,46]. These pharmacological studies have been bolstered by use of G15, a selective GPER antagonist, which attenuates vascular relaxation induced by E2[52] or G-1[46,52]. Thus, there is increasingly consistent evidence obtained from both protein and pharmacological studies that activation of GPER exerts a vasodilatory effect, particularly in coronary arteries. These findings also substantiate the putative role of GPER in mediating E2-induced coronary artery relaxation.
Relatively few studies have investigated the signaling mechanisms mediating GPER-induced coronary artery relaxation, and these reveal that GPER can relax coronary arteries acutely via diverse mechanisms. There is good agreement that G-1 induces a maximal 30%-40% relaxation of porcine epicardial coronary arteries in vitro (after taking into account the effect of ethanol or dimethylsulfoxide vehicle on these vessels); however, this relaxant effect of G-1 may be endothelium-dependent or -independent. Meyer et al[36] report that activation of GPER induces a rapid relaxation of coronary arteries, and that this process is mediated by NO release from endothelial cells. In contrast, Yu et al[46] provide evidence for a direct relaxant effect of G-1 on CASMC, which is bolstered by patch-clamp experiments demonstrating that G-1 opens calcium-activated potassium (BKCa) channels in isolated porcine and human CASMC (i.e., in the absence of endothelium). In this later study G-1-induced relaxation was inhibited by blocking these same potassium channels, but was not attenuated by inhibiting NO or cyclic guanosine monophosphate synthesis. Thus, there appears to be a redundancy of mechanisms mediating GPER-induced coronary artery relaxation: indirect (endothelial cells) and direct (CASMC), and may or may not involve production of NO. Because E2 is a very lipophilic hormone that easily traverses biological membrane, it is likely that both cell types mediate GPER-induced coronary artery relaxation in vivo (unless there is significant endothelial dysfunction in which case the direct action on CASMC should predominate). In addition, there is significant expression of aromatase in both endothelial and VSM cells allowing estrogen to be synthesized directly within the coronary artery wall (i.e., independent of plasma E2 levels)[53].
As mentioned above, raloxifene is also an agonist for GPER and stimulates an endothelium-independent relaxation of porcine coronary arteries[37]. In this study the presence of high extracellular potassium (i.e., 30 or 60 mmol) significantly reduced raloxifene-induced relaxation, as did iberiotoxin, a highly specific inhibitor BKCa channels. In contrast, inhibiting ERα or ERβ with ICI182,780 had no effect on the response to raloxifene. Further patch-clamp studies demonstrated that raloxifene elevates iberiotoxin-sensitive outward currents in isolated CASMC. Thus, the endothelium-independent relaxation effect of raloxifene on porcine coronary arteries appears very similar to that of G-1 on CASMC in that stimulation of BKCa channels is likely an important effector of GPER-induced coronary artery relaxation. In contrast to these findings, however, are previous studies indicating that ICI182,780 can inhibit raloxifene-induced, endothelium-dependent relaxation of rabbit coronary arteries[54], also attenuates raloxifene-induced NO production from human endothelial cells[55]. Thus, it seems likely that endothelium-dependent effects of raloxifene are mediated primarily by classic ERs, whereas the direct effects on CASMC are via GPER.
It is interesting that the diverse mechanisms mediating acute, non-genomic estrogen-induced coronary artery relaxation often converge upon a common cellular effector-the BKCa channel. Nearly 20 years ago we first demonstrated that E2 could activate this powerful hyperpolarizing mechanism in CASMC[11], and a number of studies have since confirmed and extended the role this protein plays in mediating acute estrogen signaling in coronary arteries. An exciting new development is that there is now increasing evidence that coronary artery relaxation induced by GPER activation appears to involve BKCa activity as well. In addition to E2, single-channel and whole-cell patch-clamp studies have demonstrated that agents known to stimulate GPER, i.e., G-1[46], raloxifene[37], tamoxifen[10], also exert significant stimulatory effects on BKCa channel activity in isolated CASMC. Although not tested directly, it is also quite likely that the endothelium-dependent coronary artery relaxation effect of G-1[36] may also indirectly open BKCa channels in CASMC via release of NO[10,11,56]. These cellular studies are bolstered by the fact that iberiotoxin attenuates coronary relaxation induced by either G-1[46] or raloxifene[37]. At present, mechanisms coupling GPER activation to BKCa channel activity remain undefined; however, there is likely to be significant therapeutic potential here. For example, identification of the BKCa channel as a molecular effector of rapid estrogen signaling in CASMC could lead to the development of new agents which could specifically target these proteins in coronary arteries to provide the beneficial vasodilatory effect of E2 without the substantial endocrine side effects of hormone treatment.
GPER AND CORONARY ARTERY CELL PROLIFERATION
In healthy arteries CASMC retain a contractile phenotype and are localized in the medial layer; however, intimal injury (e.g., atherosclerosis, angioplasty) causes CASMC to dedifferentiate, lose their contractile phenotype, and proliferate[57,58]. Dedifferentiated CASMC can then migrate into the intimal region and contribute to the narrowing of the coronary artery lumen. Estrogen is known to inhibit injury-induced VSM proliferation[46,59-64], but, interestingly, genetic deletion of classic ERs does not abolish this anti-proliferative effect of E2[65,66]. Thus, it is likely that the protective, anti-proliferative effect of estrogens is due, at least in part, to activation of another ER-quite possibly GPER-in coronary arteries.
We recently reported that stimulation of GPER by G-1 inhibits proliferation of human and porcine CASMC[47]. In this study we found that 24-h exposure of primary porcine CASMC to G-1 inhibited serum-induced cell growth via repression of cell cycle progression. Further, we found that G-1 completely inhibited CASMC migration, and this inhibitory effect was attenuated by G-15. Similarly, Haas et al[50] found that G-1 decreased proliferation of human umbilical vein VSM. Further, VSM proliferation, assessed by measuring the media-to-lumen ratio, in murine resistance vessels was significantly increased in animals lacking the GPER gene[67]. Thus, it is very likely that GPER helps maintain VSM cells in a differentiated, contractile phenotype, and may thereby help retard the development of atherosclerotic buildup in the vascular intima.
Estrogen is also known to regulate the proliferation of vascular endothelial cells, and, specifically, can influence endothelial cell growth and re-endothelialization[68]. For example, direct delivery of E2 promotes reendothelialization and endothelial nitric oxide synthase expression in coronary arteries after damage due to coronary angioplasty[69]. In addition to this protective effect that may promote healing of endothelial damage, there is also evidence that GPER may prevent excessive proliferation of endothelial cells. G-1 reduces proliferation, DNA synthesis, and number of microvascular endothelial cells[70]. These studies suggest that an important role of GPER may be to provide an optimal balance for the effects of E2 on endothelial cell proliferation, and thereby prevent excessive endothelial cell proliferation; for instance, as occurs in tumor-associated angiogenesis.
The mechanism(s) of how GPER attenuates vascular cell growth remain to be elucidated, although several lines of evidence point to specific alterations in mitogenic signaling pathway such as extracellular signal-regulated protein kinases (ERKs) and protein kinase B (Akt). For example, E2 has been shown to phosphorylate ERK-1 and ERK-2 in breast cancer cells expressing GPER[71], thus enhancing cell proliferation. In contrast, we recently reported that GPER activation decreases phosphorylation of ERK1/2 and Akt activity in human and porcine CASMC[47], thus suppressing proliferation. This decreased kinase activity was consistent with a similar inhibitory effect of GPER stimulation on ERK1/2 activity in breast cancer cells[72]. Further, Gros et al[45] reported that E2 enhanced apoptosis in rat aortic VSM cells in which GPER was overexpressed, and did so in an ERK1/2-dependent manner. GPER overexpression altered downstream signaling from protein kinase A to a pertussis toxin-sensitive pathway which increased Akt phosphorylation and ERK1/2 activation, resulting in VSM cell apoptosis. In these VSM cells, G-1 stimulated ERK1/2 phosphorylation; however, other GPER agonists (i.e., tamoxifen, ICI182,780) failed to do so. These studies indicate that E2 can induce cell apoptosis via GPER signaling; however, the signaling mechanisms underlying this effect are complicated and require further study. A summary of the currently known effects of GPER activation is presented in Table 1 and the proposed mechanisms mediating the effects of GPER activation in coronary arteries is summarized in Figure 1.
Table 1.
Primary effects of G-protein-coupled estrogen receptor activation on coronary arteries
| Effect | Species | Drug |
| Relaxation | Porcine | G-1[36,46] |
| ICI182,780[35,36] | ||
| Raloxifene[37] | ||
| Tamoxifen[73] | ||
| Rabbit | Raloxifene[54] | |
| Tamoxifen[74] | ||
| Endothelial NO production | Porcine | G-1[36] |
| Raloxifene[54] | ||
| Tamoxifen[74] | ||
| CASMC BKCa channel opening | Porcine | G-1[46] |
| Raloxifene[37] | ||
| Tamoxifen[10] | ||
| Human | G-1[46] | |
| Inhibition of CASMC proliferation | Porcine | G-1[47] |
| Human | G-1[47] | |
| Inhibition of CASMC migration | Porcine | G-1[47] |
CASMC: Coronary artery smooth muscle cells; BKCa: G-1 opens calcium-activated potassium; NO: Nitric oxide.
Figure 1.
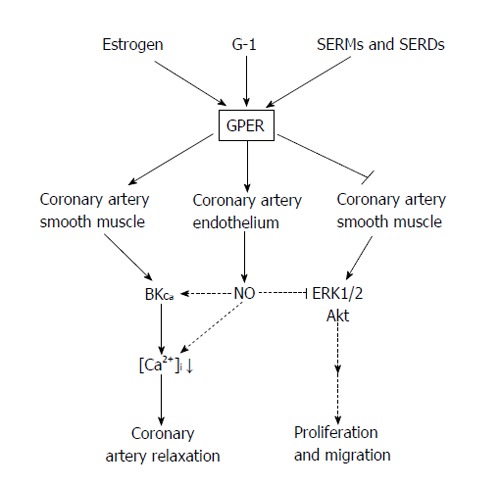
Summary of proposed mechanisms mediating the effects of G-protein-coupled estrogen receptor activation in coronary arteries. GPER is activated by 17β-estradiol and the selective agonist, G-1. In addition, selective estrogen receptor modulators (e.g., raloxifene, tamoxifen) and selective estrogen receptor down regulators (e.g., ICI182,780) also appear to be agonists for GPER. GPER activation induces an endothelium-independent relaxation of coronary artery smooth muscle mediated by the large-conductance, calcium-activated potassium channel. In addition, GPER activation can stimulated release of NO from coronary endothelial cells to relax these arteries. Besides this vasodilatory effect, GPER activation can attenuate proliferation and migration of coronary artery smooth muscle cells by inhibiting signaling via the ERK1/2 and Akt pathways. GPER: G-protein-coupled estrogen receptor; SERMs: Selective estrogen receptors modulators; SERD: Estrogen receptor down regulator; ERK: Extracellular signal-regulated protein kinase.
GPER: A NOVEL THERAPEUTIC TARGET FOR CORONARY ARTERY DISEASE
The first rule of medical practice is to “do no harm”. Despite the considerable therapeutic potential of estrogen and estrogen-like compounds, fear of potentially dangerous ancillary effects continues to limit their usage. A primary obstacle to overcome is the pleotropic effects of E2 on a wide diversity of tissues, most of which express one or more classes of ERs. For several years an important goal was to define the levels of ER expression in target tissues, and then deliver a receptor- or tissue-specific agonist that would produce the desired therapeutic response with limited side effects on non-target sites. However, because of the ubiquitous expression of ERs and the fact that multiple ERs are often expressed in the same cells, this goal has not been realized to any significant extent. With the more recent discovery of GPER, pursuit of this goal has been reinvigorated. In particular, the findings that GPER is highly expressed in both coronary endothelial cells and CASMC has opened the search to understand the importance of this non-genomic estrogen signaling mechanism and explore pharmacological means whereby GPER activity could be modulated for therapeutic benefit. A summary of experimental evidence suggesting possible therapeutic benefits of GPER activation is presented in Table 2.
Table 2.
Evidence for possible therapeutic effects of G-protein-coupled estrogen receptor agonists and selective estrogen receptor modulators
| Species | Drug | |
| Coronary artery relaxation | ||
| Endothelium-dependent, in vitro | Porcine | G-1[36] |
| Rabbit | Raloxifen[54] | |
| Tamoxifen[73,74] | ||
| Endothelium-independent, in vitro | Porcine | G-1[46] |
| Raloxifen[37] | ||
| Reduced cardiac ischemic injury/infarct | Rat | G-1[75,76] |
| Reduced cerebral ischemic injury/infarct | Mice | G-1[77] |
| Middle cerebral artery relaxation, in vitro | Rat | G-1[78] |
| Systemic artery relaxation, in vitro | Mice | G-1[40] |
| G-1[49] | ||
| G-1[50] | ||
| G-1[79] | ||
| Rat | G-1[51] | |
| G-1[52] | ||
| Human | G-1[50] | |
| Reduced systemic blood pressure, infusion | Rats | G-1[49] |
| G-1[50] | ||
| Inhibit VSM cell proliferation | Porcine | G-1[47] |
| Human | G-1[50] | |
| Inhibit endothelial cell proliferation | Mice | G-1[70] |
| Prevents calcium-induced increases in plasma cholesterol | Rats | G-1[80] |
VSM: Vascular smooth muscle.
CHD continues to be the greatest mortality risk factor in the developed world. Although our understanding of the causes of CHD continues to increase, therapeutic measures to prevent and treat this serious health problem have not improved dramatically over the past several decades. Invasive procedures such as bypass grafting or balloon angioplasty have been refined, but are still routinely practiced. Pharmacological measures (e.g., nitrates, calcium channel antagonists, beta blockers) can be effective at treating the symptoms of CHD (e.g., angina pectoris), but are seldom a viable long-term option, much less a cure. Ideally, what is needed is a widely-available therapeutic agent which might slow or reverse the progression of atherosclerosis, restore endothelial function, and induce coronary vasodilation in cases where blood flow was compromised significantly; and this agent would produce these beneficial cardiac effects with few side effects on other organs. In light of current research, it is possible that activation of GPER might be a promising new approach to achieving this desired therapeutic end.
Evidence is clear that activation of GPER produces an acute (i.e., in minutes) dilation of coronary arteries due to relaxation of CASMC. This action appears to be both direct (acting on and relaxing CASMC) and indirect (via NO release from endothelial cells), and this dual action could prove to be very important as many CHD patients have dysfunctional or damaged coronary endothelium. Thus, stimulation of GPER has the potential to induce a direct coronary artery dilation, as well as lowering afterload due to its ability to decrease peripheral vascular resistance. As a consequence of GPER activation myocardial oxygen supply should increase with increased coronary blood flow as metabolic oxygen demand declines in face of lower peripheral vascular resistance. In addition, relaxation of venous smooth muscle could lower venous return and preload, thus further lowering myocardial oxygen demand. Thus, the vasodilatory potential of GPER activation could influence a number of favorable hemodynamic parameters to alleviate the pain and risk of CHD, and could be used acutely or prophylactically. In addition, there is similar evidence that GPER activation may also reduce the risk of ischemic stroke due to dilation of cerebral arteries[77,78], and that GPER exerts a tonic suppression of arterial tone[79].
Although we are only beginning to understand the mechanisms whereby GPER activation influences cell proliferation, there is accumulating evidence that GPER agonists exert an anti-proliferative and anti-migratory effect on CASMC-as it does for human urothelial cells[81] and endothelial cells[70]. Because CASMC dedifferentiation, proliferation, intimal migration, and secretion are important steps in the process of atherogenesis, these studies strongly suggest a potentially important protective effect of GPER activation on coronary atherosclerotic disease. Further, it appears that GPER activation can also help heal intimal damage and quite possibly help restore normal function to dysfunctional coronary endothelial cells-particularly because of its ability to enhance NO synthesis and release from endothelium. These intimal effects involving NO release would likely prevent coronary vasospasm and also help to further limit CASMC proliferation/migration, as well as attenuate the formation of coronary thrombi that could precipitate an acute ischemic attack or infarction. Although the potential effects of GPER stimulation of plasma lipoproteins are as yet unknown, a recent study has reported that GPER activation prevents increases in plasma total cholesterol levels in postmenopausal women taking calcium supplements[80]. Thus, a new and promising effect of GPER activation may be outside the vascular system to help promote optimal cardiovascular health. Clearly then, there are potentially multiple sites of action for agents that would selectively stimulate GPER and produce beneficial effects on cardiovascular function-particularly treatment of CHD.
As always, potential side effects of GPER action must be considered. Initially, however, it could be predicted that GPER stimulation might produce significantly less risk of limiting side effects compared to E2 therapy or currently prescribed estrogenic agents (e.g., breast or uterine cancer, venothromboembolism). For example, raloxifene has been demonstrated to lower overall risk of cardiovascular disease or breast cancer and strengthen bones in younger postmenopausal women[82]; however, raloxifene does not lower blood pressure in these women, and its anti-estrogen side effects (e.g., hot flushes, vaginal dryness) continue to limit its use somewhat. Tamoxifen has been widely employed as a treatment for estrogen-sensitive breast tumors. As a SERM, tamoxifen can increase bone density and produce beneficial changes in plasma lipids; however, its anti-estrogenic effects can increase the risk of uterine cancer and produce many negative symptoms of menopausal[83]. It is likely that action on classic ERs (sometimes agonistic; other times antagonistic) mediates many of the undesirable side effects of SERM action.
At present, we are unaware of any reports from clinical trials evaluating the potential of G-1 (or another GPER agonist) as a therapeutic agent. As noted above, there appears to be great potential for GPER activation to enhance cardiovascular health. These effects, particularly those on coronary arteries, appear to be mediated almost exclusively via GPER with little or no concomitant activation of ERα or ERβ. If so, then this more specific pharmacodynamic profile should do much to help limit the potential side effects of GPER activation on targets outside the cardiovascular system. A caveat, however, is that we are only beginning to understand the impact of GPER activation and its signaling mechanisms in a diversity of cell types. Thus, caution must be exercised in promoting GPER as a therapeutic target. Nonetheless, there is a substantial cautious optimism that pharmacological targeting of this novel non-genomic estrogen signaling mechanism may finally provide a means of producing the many beneficial effects of estrogen on the cardiovascular system while eliciting fewer side effects on the reproductive and other non-cardiovascular systems that continue to limit the use of other less specific estrogenic compounds.
Footnotes
Supported by The American Heart Association, Texas Affiliate, No. 7370061; and the Center for Chronic Disorders of Aging, PCOM
P- Reviewers: Luthra S, Petretta M S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
References
- 1.Mackenzie J. Irritation of the sexual apparatus as an etiological factor in the production of nasal disease. Am J Med Sci. 1884;88:360–365. [Google Scholar]
- 2.Grodstein F, Stampfer MJ, Manson JE, Colditz GA, Willett WC, Rosner B, Speizer FE, Hennekens CH. Postmenopausal estrogen and progestin use and the risk of cardiovascular disease. N Engl J Med. 1996;335:453–461. doi: 10.1056/NEJM199608153350701. [DOI] [PubMed] [Google Scholar]
- 3.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 4.Raddino R, Manca C, Poli E, Bolognesi R, Visioli O. Effects of 17 beta-estradiol on the isolated rabbit heart. Arch Int Pharmacodyn Ther. 1986;281:57–65. [PubMed] [Google Scholar]
- 5.Santos RL, Marin EB, Gonçalves WL, Bissoli NS, Abreu GR, Moysés MR. Sex differences in the coronary vasodilation induced by 17 β-oestradiol in the isolated perfused heart from spontaneously hypertensive rats. Acta Physiol (Oxf) 2010;200:203–210. doi: 10.1111/j.1748-1716.2010.02140.x. [DOI] [PubMed] [Google Scholar]
- 6.Node K, Kitakaze M, Kosaka H, Minamino T, Sato H, Kuzuya T, Hori M. Roles of NO and Ca2+-activated K+ channels in coronary vasodilation induced by 17beta-estradiol in ischemic heart failure. FASEB J. 1997;11:793–799. doi: 10.1096/fasebj.11.10.9271364. [DOI] [PubMed] [Google Scholar]
- 7.Reis SE, Gloth ST, Blumenthal RS, Resar JR, Zacur HA, Gerstenblith G, Brinker JA. Ethinyl estradiol acutely attenuates abnormal coronary vasomotor responses to acetylcholine in postmenopausal women. Circulation. 1994;89:52–60. doi: 10.1161/01.cir.89.1.52. [DOI] [PubMed] [Google Scholar]
- 8.Sudhir K, Chou TM, Mullen WL, Hausmann D, Collins P, Yock PG, Chatterjee K. Mechanisms of estrogen-induced vasodilation: in vivo studies in canine coronary conductance and resistance arteries. J Am Coll Cardiol. 1995;26:807–814. doi: 10.1016/0735-1097(95)00248-3. [DOI] [PubMed] [Google Scholar]
- 9.Chester AH, Jiang C, Borland JA, Yacoub MH, Collins P. Oestrogen relaxes human epicardial coronary arteries through non-endothelium-dependent mechanisms. Coron Artery Dis. 1995;6:417–422. doi: 10.1097/00019501-199505000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Darkow DJ, Lu L, White RE. Estrogen relaxation of coronary artery smooth muscle is mediated by nitric oxide and cGMP. Am J Physiol. 1997;272:H2765–H2773. doi: 10.1152/ajpheart.1997.272.6.H2765. [DOI] [PubMed] [Google Scholar]
- 11.White RE, Darkow DJ, Lang JL. Estrogen relaxes coronary arteries by opening BKCa channels through a cGMP-dependent mechanism. Circ Res. 1995;77:936–942. doi: 10.1161/01.res.77.5.936. [DOI] [PubMed] [Google Scholar]
- 12.Mügge A, Riedel M, Barton M, Kuhn M, Lichtlen PR. Endothelium independent relaxation of human coronary arteries by 17 beta-oestradiol in vitro. Cardiovasc Res. 1993;27:1939–1942. doi: 10.1093/cvr/27.11.1939. [DOI] [PubMed] [Google Scholar]
- 13.White RE, Han G, Dimitropoulou C, Zhu S, Miyake K, Fulton D, Dave S, Barman SA. Estrogen-induced contraction of coronary arteries is mediated by superoxide generated in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2005;289:H1468–H1475. doi: 10.1152/ajpheart.01173.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White RE, Gerrity R, Barman SA, Han G. Estrogen and oxidative stress: A novel mechanism that may increase the risk for cardiovascular disease in women. Steroids. 2010;75:788–793. doi: 10.1016/j.steroids.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stampfer MJ, Sacks FM, Salvini S, Willett WC, Hennekens CH. A prospective study of cholesterol, apolipoproteins, and the risk of myocardial infarction. N Engl J Med. 1991;325:373–381. doi: 10.1056/NEJM199108083250601. [DOI] [PubMed] [Google Scholar]
- 16.Paganini-Hill A, Dworsky R, Krauss RM. Hormone replacement therapy, hormone levels, and lipoprotein cholesterol concentrations in elderly women. Am J Obstet Gynecol. 1996;174:897–902. doi: 10.1016/s0002-9378(96)70322-8. [DOI] [PubMed] [Google Scholar]
- 17.Bhavnani BR, Stanczyk FZ. Pharmacology of conjugated equine estrogens: efficacy, safety and mechanism of action. J Steroid Biochem Mol Biol. 2014;142:16–29. doi: 10.1016/j.jsbmb.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Subbiah MT, Kessel B, Agrawal M, Rajan R, Abplanalp W, Rymaszewski Z. Antioxidant potential of specific estrogens on lipid peroxidation. J Clin Endocrinol Metab. 1993;77:1095–1097. doi: 10.1210/jcem.77.4.8408459. [DOI] [PubMed] [Google Scholar]
- 19.Sugioka K, Shimosegawa Y, Nakano M. Estrogens as natural antioxidants of membrane phospholipid peroxidation. FEBS Lett. 1987;210:37–39. doi: 10.1016/0014-5793(87)81293-0. [DOI] [PubMed] [Google Scholar]
- 20.Sack MN, Rader DJ, Cannon RO. Oestrogen and inhibition of oxidation of low-density lipoproteins in postmenopausal women. Lancet. 1994;343:269–270. doi: 10.1016/s0140-6736(94)91117-7. [DOI] [PubMed] [Google Scholar]
- 21.Wilcox JG, Hwang J, Hodis HN, Sevanian A, Stanczyk FZ, Lobo RA. Cardioprotective effects of individual conjugated equine estrogens through their possible modulation of insulin resistance and oxidation of low-density lipoprotein. Fertil Steril. 1997;67:57–62. doi: 10.1016/s0015-0282(97)81856-0. [DOI] [PubMed] [Google Scholar]
- 22.Knopp RH, Paramsothy P, Retzlaff BM, Fish B, Walden C, Dowdy A, Tsunehara C, Aikawa K, Cheung MC. Sex differences in lipoprotein metabolism and dietary response: basis in hormonal differences and implications for cardiovascular disease. Curr Cardiol Rep. 2006;8:452–459. doi: 10.1007/s11886-006-0104-0. [DOI] [PubMed] [Google Scholar]
- 23.Miller AA, Drummond GR, Mast AE, Schmidt HH, Sobey CG. Effect of gender on NADPH-oxidase activity, expression, and function in the cerebral circulation: role of estrogen. Stroke. 2007;38:2142–2149. doi: 10.1161/STROKEAHA.106.477406. [DOI] [PubMed] [Google Scholar]
- 24.Wagner AH, Schroeter MR, Hecker M. 17beta-estradiol inhibition of NADPH oxidase expression in human endothelial cells. FASEB J. 2001;15:2121–2130. doi: 10.1096/fj.01-0123com. [DOI] [PubMed] [Google Scholar]
- 25.Laufs U, Adam O, Strehlow K, Wassmann S, Konkol C, Laufs K, Schmidt W, Böhm M, Nickenig G. Down-regulation of Rac-1 GTPase by Estrogen. J Biol Chem. 2003;278:5956–5962. doi: 10.1074/jbc.M209813200. [DOI] [PubMed] [Google Scholar]
- 26.Barton M. Cholesterol and atherosclerosis: modulation by oestrogen. Curr Opin Lipidol. 2013;24:214–220. doi: 10.1097/MOL.0b013e3283613a94. [DOI] [PubMed] [Google Scholar]
- 27.Vassalle C, Mercuri A, Maffei S. Oxidative status and cardiovascular risk in women: Keeping pink at heart. World J Cardiol. 2009;1:26–30. doi: 10.4330/wjc.v1.i1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howard BV, Rossouw JE. Estrogens and cardiovascular disease risk revisited: the Women’s Health Initiative. Curr Opin Lipidol. 2013;24:493–499. doi: 10.1097/MOL.0000000000000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han G, Yu X, Lu L, Li S, Ma H, Zhu S, Cui X, White RE. Estrogen receptor alpha mediates acute potassium channel stimulation in human coronary artery smooth muscle cells. J Pharmacol Exp Ther. 2006;316:1025–1030. doi: 10.1124/jpet.105.093542. [DOI] [PubMed] [Google Scholar]
- 30.Traupe T, Stettler CD, Li H, Haas E, Bhattacharya I, Minotti R, Barton M. Distinct roles of estrogen receptors alpha and beta mediating acute vasodilation of epicardial coronary arteries. Hypertension. 2007;49:1364–1370. doi: 10.1161/HYPERTENSIONAHA.106.081554. [DOI] [PubMed] [Google Scholar]
- 31.Booth EA, Obeid NR, Lucchesi BR. Activation of estrogen receptor-alpha protects the in vivo rabbit heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2005;289:H2039–H2047. doi: 10.1152/ajpheart.00479.2005. [DOI] [PubMed] [Google Scholar]
- 32.Tomicek NJ, Miller-Lee JL, Hunter JC, Korzick DH. Estrogen receptor beta does not influence ischemic tolerance in the aged female rat heart. Cardiovasc Ther. 2013;31:32–37. doi: 10.1111/j.1755-5922.2011.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Favre J, Gao J, Henry JP, Remy-Jouet I, Fourquaux I, Billon-Gales A, Thuillez C, Arnal JF, Lenfant F, Richard V. Endothelial estrogen receptor {alpha} plays an essential role in the coronary and myocardial protective effects of estradiol in ischemia/reperfusion. Arterioscler Thromb Vasc Biol. 2010;30:2562–2567. doi: 10.1161/ATVBAHA.110.213637. [DOI] [PubMed] [Google Scholar]
- 34.Christian RC, Liu PY, Harrington S, Ruan M, Miller VM, Fitzpatrick LA. Intimal estrogen receptor (ER)beta, but not ERalpha expression, is correlated with coronary calcification and atherosclerosis in pre- and postmenopausal women. J Clin Endocrinol Metab. 2006;91:2713–2720. doi: 10.1210/jc.2005-2672. [DOI] [PubMed] [Google Scholar]
- 35.Han G, Ma H, Chintala R, Fulton DJ, Barman SA, White RE. Essential role of the 90-kilodalton heat shock protein in mediating nongenomic estrogen signaling in coronary artery smooth muscle. J Pharmacol Exp Ther. 2009;329:850–855. doi: 10.1124/jpet.108.149112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer MR, Baretella O, Prossnitz ER, Barton M. Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology. 2010;86:58–64. doi: 10.1159/000315497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leung HS, Seto SW, Kwan YW, Leung FP, Au AL, Yung LM, Yao X, Huang Y. Endothelium-independent relaxation to raloxifene in porcine coronary artery. Eur J Pharmacol. 2007;555:178–184. doi: 10.1016/j.ejphar.2006.10.035. [DOI] [PubMed] [Google Scholar]
- 38.Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Mol Cell Endocrinol. 2007;265-266:138–142. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prossnitz ER, Barton M. Signaling, physiological functions and clinical relevance of the G protein-coupled estrogen receptor GPER. Prostaglandins Other Lipid Mediat. 2009;89:89–97. doi: 10.1016/j.prostaglandins.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer MR, Prossnitz ER, Barton M. The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function. Vascul Pharmacol. 2011;55:17–25. doi: 10.1016/j.vph.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- 42.Takada Y, Kato C, Kondo S, Korenaga R, Ando J. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem Biophys Res Commun. 1997;240:737–741. doi: 10.1006/bbrc.1997.7734. [DOI] [PubMed] [Google Scholar]
- 43.Kvingedal AM, Smeland EB. A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. 1997;407:59–62. doi: 10.1016/s0014-5793(97)00278-0. [DOI] [PubMed] [Google Scholar]
- 44.Haas E, Meyer MR, Schurr U, Bhattacharya I, Minotti R, Nguyen HH, Heigl A, Lachat M, Genoni M, Barton M. Differential effects of 17beta-estradiol on function and expression of estrogen receptor alpha, estrogen receptor beta, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension. 2007;49:1358–1363. doi: 10.1161/HYPERTENSIONAHA.107.089995. [DOI] [PubMed] [Google Scholar]
- 45.Gros R, Ding Q, Liu B, Chorazyczewski J, Feldman RD. Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am J Physiol Cell Physiol. 2013;304:C532–C540. doi: 10.1152/ajpcell.00203.2012. [DOI] [PubMed] [Google Scholar]
- 46.Yu X, Ma H, Barman SA, Liu AT, Sellers M, Stallone JN, Prossnitz ER, White RE, Han G. Activation of G protein-coupled estrogen receptor induces endothelium-independent relaxation of coronary artery smooth muscle. Am J Physiol Endocrinol Metab. 2011;301:E882–E888. doi: 10.1152/ajpendo.00037.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li F, Yu X, Szynkarski CK, Meng C, Zhou B, Barhoumi R, White RE, Heaps CL, Stallone JN, Han G. Activation of GPER Induces Differentiation and Inhibition of Coronary Artery Smooth Muscle Cell Proliferation. PLoS One. 2013;8:e64771. doi: 10.1371/journal.pone.0064771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batenburg WW, Jansen PM, van den Bogaerdt AJ, J Danser AH. Angiotensin II-aldosterone interaction in human coronary microarteries involves GPR30, EGFR, and endothelial NO synthase. Cardiovasc Res. 2012;94:136–143. doi: 10.1093/cvr/cvs016. [DOI] [PubMed] [Google Scholar]
- 49.Lindsey SH, Cohen JA, Brosnihan KB, Gallagher PE, Chappell MC. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology. 2009;150:3753–3758. doi: 10.1210/en.2008-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haas E, Bhattacharya I, Brailoiu E, Damjanović M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res. 2009;104:288–291. doi: 10.1161/CIRCRESAHA.108.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Broughton BR, Miller AA, Sobey CG. Endothelium-dependent relaxation by G protein-coupled receptor 30 agonists in rat carotid arteries. Am J Physiol Heart Circ Physiol. 2010;298:H1055–H1061. doi: 10.1152/ajpheart.00878.2009. [DOI] [PubMed] [Google Scholar]
- 52.Lindsey SH, Carver KA, Prossnitz ER, Chappell MC. Vasodilation in response to the GPR30 agonist G-1 is not different from estradiol in the mRen2.Lewis female rat. J Cardiovasc Pharmacol. 2011;57:598–603. doi: 10.1097/FJC.0b013e3182135f1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harada N, Sasano H, Murakami H, Ohkuma T, Nagura H, Takagi Y. Localized expression of aromatase in human vascular tissues. Circ Res. 1999;84:1285–1291. doi: 10.1161/01.res.84.11.1285. [DOI] [PubMed] [Google Scholar]
- 54.Figtree GA, Lu Y, Webb CM, Collins P. Raloxifene acutely relaxes rabbit coronary arteries in vitro by an estrogen receptor-dependent and nitric oxide-dependent mechanism. Circulation. 1999;100:1095–1101. doi: 10.1161/01.cir.100.10.1095. [DOI] [PubMed] [Google Scholar]
- 55.Simoncini T, Genazzani AR, Liao JK. Nongenomic mechanisms of endothelial nitric oxide synthase activation by the selective estrogen receptor modulator raloxifene. Circulation. 2002;105:1368–1373. doi: 10.1161/hc1102.105267. [DOI] [PubMed] [Google Scholar]
- 56.Carrier GO, Fuchs LC, Winecoff AP, Giulumian AD, White RE. Nitrovasodilators relax mesenteric microvessels by cGMP-induced stimulation of Ca-activated K channels. Am J Physiol. 1997;273:H76–H84. doi: 10.1152/ajpheart.1997.273.1.H76. [DOI] [PubMed] [Google Scholar]
- 57.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 58.Hosono M, Ueda M, Suehiro S, Sasaki Y, Shibata T, Hattori K, Kinoshita H. Neointimal formation at the sites of anastomosis of the internal thoracic artery grafts after coronary artery bypass grafting in human subjects: an immunohistochemical analysis. J Thorac Cardiovasc Surg. 2000;120:319–328. doi: 10.1067/mtc.2000.106328. [DOI] [PubMed] [Google Scholar]
- 59.Kawagoe J, Ohmichi M, Tsutsumi S, Ohta T, Takahashi K, Kurachi H. Mechanism of the divergent effects of estrogen on the cell proliferation of human umbilical endothelial versus aortic smooth muscle cells. Endocrinology. 2007;148:6092–6099. doi: 10.1210/en.2007-0188. [DOI] [PubMed] [Google Scholar]
- 60.Dubey RK, Jackson EK, Gillespie DG, Zacharia LC, Imthurn B, Keller PJ. Clinically used estrogens differentially inhibit human aortic smooth muscle cell growth and mitogen-activated protein kinase activity. Arterioscler Thromb Vasc Biol. 2000;20:964–972. doi: 10.1161/01.atv.20.4.964. [DOI] [PubMed] [Google Scholar]
- 61.Takahashi K, Ohmichi M, Yoshida M, Hisamoto K, Mabuchi S, Arimoto-Ishida E, Mori A, Tsutsumi S, Tasaka K, Murata Y, et al. Both estrogen and raloxifene cause G1 arrest of vascular smooth muscle cells. J Endocrinol. 2003;178:319–329. doi: 10.1677/joe.0.1780319. [DOI] [PubMed] [Google Scholar]
- 62.Sivritas D, Becher MU, Ebrahimian T, Arfa O, Rapp S, Bohner A, Mueller CF, Umemura T, Wassmann S, Nickenig G, et al. Antiproliferative effect of estrogen in vascular smooth muscle cells is mediated by Kruppel-like factor-4 and manganese superoxide dismutase. Basic Res Cardiol. 2011;106:563–575. doi: 10.1007/s00395-011-0174-z. [DOI] [PubMed] [Google Scholar]
- 63.Selzman CH, Gaynor JS, Turner AS, Whitehill TA, Horwitz LD, Harken AH. Estrogen replacement inhibits intimal hyperplasia and the accumulation and effects of transforming growth factor beta1. J Surg Res. 1998;80:380–385. doi: 10.1006/jsre.1998.5487. [DOI] [PubMed] [Google Scholar]
- 64.Foegh ML, Asotra S, Howell MH, Ramwell PW. Estradiol inhibition of arterial neointimal hyperplasia after balloon injury. J Vasc Surg. 1994;19:722–726. doi: 10.1016/s0741-5214(94)70047-8. [DOI] [PubMed] [Google Scholar]
- 65.Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, Hodgin J, Shaul PW, Thoren P, Smithies O, et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science. 2002;295:505–508. doi: 10.1126/science.1065250. [DOI] [PubMed] [Google Scholar]
- 66.Iafrati MD, Karas RH, Aronovitz M, Kim S, Sullivan TR, Lubahn DB, O’Donnell TF, Korach KS, Mendelsohn ME. Estrogen inhibits the vascular injury response in estrogen receptor alpha-deficient mice. Nat Med. 1997;3:545–548. doi: 10.1038/nm0597-545. [DOI] [PubMed] [Google Scholar]
- 67.Mårtensson UE, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grände PO, et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology. 2009;150:687–698. doi: 10.1210/en.2008-0623. [DOI] [PubMed] [Google Scholar]
- 68.Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2319–2330. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chandrasekar B, Nattel S, Tanguay JF. Coronary artery endothelial protection after local delivery of 17beta-estradiol during balloon angioplasty in a porcine model: a potential new pharmacologic approach to improve endothelial function. J Am Coll Cardiol. 2001;38:1570–1576. doi: 10.1016/s0735-1097(01)01552-2. [DOI] [PubMed] [Google Scholar]
- 70.Holm A, Baldetorp B, Olde B, Leeb-Lundberg LM, Nilsson BO. The GPER1 agonist G-1 attenuates endothelial cell proliferation by inhibiting DNA synthesis and accumulating cells in the S and G2 phases of the cell cycle. J Vasc Res. 2011;48:327–335. doi: 10.1159/000322578. [DOI] [PubMed] [Google Scholar]
- 71.Filardo EJ, Quinn JA, Bland KI, Frackelton AR. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 72.Filardo EJ, Quinn JA, Frackelton AR, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16:70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 73.Leung HS, Yung LM, Leung FP, Yao X, Chen ZY, Ko WH, Laher I, Huang Y. Tamoxifen dilates porcine coronary arteries: roles for nitric oxide and ouabain-sensitive mechanisms. Br J Pharmacol. 2006;149:703–711. doi: 10.1038/sj.bjp.0706921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Figtree GA, Webb CM, Collins P. Tamoxifen acutely relaxes coronary arteries by an endothelium-, nitric oxide-, and estrogen receptor-dependent mechanism. J Pharmacol Exp Ther. 2000;295:519–523. [PubMed] [Google Scholar]
- 75.Patel VH, Chen J, Ramanjaneya M, Karteris E, Zachariades E, Thomas P, Been M, Randeva HS. G-protein coupled estrogen receptor 1 expression in rat and human heart: Protective role during ischaemic stress. Int J Mol Med. 2010;26:193–199. doi: 10.3892/ijmm_00000452. [DOI] [PubMed] [Google Scholar]
- 76.Weil BR, Manukyan MC, Herrmann JL, Wang Y, Abarbanell AM, Poynter JA, Meldrum DR. Signaling via GPR30 protects the myocardium from ischemia/reperfusion injury. Surgery. 2010;148:436–443. doi: 10.1016/j.surg.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 77.Zhang B, Subramanian S, Dziennis S, Jia J, Uchida M, Akiyoshi K, Migliati E, Lewis AD, Vandenbark AA, Offner H, et al. Estradiol and G1 reduce infarct size and improve immunosuppression after experimental stroke. J Immunol. 2010;184:4087–4094. doi: 10.4049/jimmunol.0902339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patkar S, Farr TD, Cooper E, Dowell FJ, Carswell HV. Differential vasoactive effects of oestrogen, oestrogen receptor agonists and selective oestrogen receptor modulators in rat middle cerebral artery. Neurosci Res. 2011;71:78–84. doi: 10.1016/j.neures.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 79.Meyer MR, Field AS, Kanagy NL, Barton M, Prossnitz ER. GPER regulates endothelin-dependent vascular tone and intracellular calcium. Life Sci. 2012;91:623–627. doi: 10.1016/j.lfs.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li S, Li Y, Ning H, Na L, Niu Y, Wang M, Feng R, Liu L, Guo F, Hou S, et al. Calcium supplementation increases circulating cholesterol by reducing its catabolism via GPER and TRPC1-dependent pathway in estrogen deficient women. Int J Cardiol. 2013;168:2548–2560. doi: 10.1016/j.ijcard.2013.03.057. [DOI] [PubMed] [Google Scholar]
- 81.Teng J, Wang ZY, Prossnitz ER, Bjorling DE. The G protein-coupled receptor GPR30 inhibits human urothelial cell proliferation. Endocrinology. 2008;149:4024–4034. doi: 10.1210/en.2007-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Collins P, Mosca L, Geiger MJ, Grady D, Kornitzer M, Amewou-Atisso MG, Effron MB, Dowsett SA, Barrett-Connor E, Wenger NK. Effects of the selective estrogen receptor modulator raloxifene on coronary outcomes in the Raloxifene Use for The Heart trial: results of subgroup analyses by age and other factors. Circulation. 2009;119:922–930. doi: 10.1161/CIRCULATIONAHA.108.817577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.El-Ashmawy NE, Khalil RM. A review on the role of L-carnitine in the management of tamoxifen side effects in treated women with breast cancer. Tumour Biol. 2014;35:2845–2855. doi: 10.1007/s13277-013-1477-5. [DOI] [PubMed] [Google Scholar]