

Abstract
Vascular calcifications (VCs) are actively regulated biological processes associated with crystallization of hydroxyapatite in the extracellular matrix and in cells of the media (VCm) or intima (VCi) of the arterial wall. Both patterns of VC often coincide and occur in patients with type II diabetes, chronic kidney disease, and other less frequent disorders; VCs are also typical in senile degeneration. In this article, we review the current state of knowledge about the pathology, molecular biology, and nosology of VCm, expand on potential mechanisms responsible for poor prognosis, and expose some of the directions for future research in this area.
Keywords: Vascular calcifications, Mönckeberg's media sclerosis, Vascular molecular biology and genetics, Vascular function
Introduction
Vascular calcifications (VCs) are of similar composition to bone minerals. Vascular calcification of the media (VCm) are, in principal, deposits of hydroxyapatite with a high degree of crystallization.1 Initially, VCs were thought to be the result of passive degenerative processes; however, recent studies illustrate that VC is an active process initiated and regulated via a variety of molecular signalling pathways.2 While considerable progress elucidating the signalling pathways regulating VC formation has been achieved, the exact molecular basis of VC still remains elusive.3
With incoming new research data, the already large number of molecular mechanisms suggested to contribute to VC formation continues to grow. It appears that while deposition of hydroxyapatite represents the resulting commonality of VC, different initiating and propagating molecular mechanisms, as well as diverse crystalline compositions of calcium apatite crystals may be present in various forms of VC.4–6 For example, it seems likely that VC processes associated with atheroma formation may be triggered by specific biochemical cascades that are altogether different from the cascades initiated by primary damage to elastic fibres which occurs in medial calcification; however, both ultimately result in ectopic VC. While experimental conditions in a given model may replicate parts of the calcification process, they may not provide the whole picture in any of these specific conditions.
We believe that some of the emerging molecular complexity of VC may be accounted for by differences in experimental designs, tendencies to unite a host of different findings associated with different types of VC under a single umbrella hypothesis, undue focus on specific molecular cascades replicating parts of the calcification process in a given model, as well as disregard for specific aetiologies of VC types.
In this article, we review the current state of knowledge about the clinical pathology, molecular biology, and nosology of VCm and expose potential directions for future research in this exciting and clinically important area.
Nomenclature
Establishing common nomenclature is not only the first step to sharing wisdom but also critical to focus research directions and interpret the results. While molecular pathogenesis-based nomenclature of VC is not available, provisory descriptive terminology should supply the need at present (Figure 1). While VCi is commonly associated with atherosclerotic plaques, VCm represents a group of distinct pathological conditions of differing aetiologies but a common final consequence (media calcification). Mönckeberg medial sclerosis (MMS) is the most common VCm variant; it is localized in the arteries of the extremities, and it is frequently associated with type 2 diabetes (T2D) and end-stage renal disease (ESRD). Some of the distinctive features between the calcifications of the intima (VCi) and VCm are reviewed in Table 1. However, both forms of VC often coincide and overlaps are frequent.
Figure 1.

Simplified nomenclature of biological calcifications. Medial vascular calcifications comprise in humans a number of diseases; in the absence of knowledge of molecular pathogenesis of different types of medial vascular calcifications, the proposed nomenclature is based on the presence or absence of known risk factors. Mönckeberg's media sclerosis represents the most common variety of medial calcifications; it is frequently associated with type II diabetes and chronic kidney disease, yet, in some cases none of these diseases and no other known risk factors for vascular calcifications are present. In the latter case genetic or other as yet not known risk factors are likely.
Table 1.
Main characteristics of media- and intima-related vascular calcifications
| VCs types | Topography |
Lesion calcify |
Risk factors |
Main molecular cascade |
||||
|---|---|---|---|---|---|---|---|---|
| Systemic | Territorial | Early | Late | Traditional | Other | Inflammatory | Varied | |
| Media | (X) | XX | XXX | X | XX | (X) | XXX | |
| Intima | XXX | XXX | XXX | XXX | ||||
Inflammatory cascades are largely linked to lipoproteins and cytokines, varied cascades are primarily related to systemic and local disturbances of calcium and phosphate metabolism, metabolism of glucose and possibly elastin degeneration.
Prevalence and relevance
The prevalence of VCm of MMS type—based on ankle-brachial index (ABI) >1.3—has been estimated to occur in ∼0.5% of adults, is more prevalent in men than women (3 : 2) and the highest prevalence of VCm has been observed in patients with T2D.7 The prevalence of VCm in patients with newly diagnosed T2D was 17%,8 and among patients with established T2D receiving oral anti-diabetics, the prevalence of VCm was as high as 41.5%.9 In patients with ESRD, the prevalence of VCm was 27%. Compared with ESRD patients with VCi, the patients with VCm were younger, had less conventional risk factors for atherosclerosis, greater prevalence of calcium–phosphate disorder, and a length of dependence on haemodialysis.10 In patients awaiting renal transplantation, moderate-to-severe calcifications were seen in 39.5% of individuals;11 in all cited studies plain X-ray radiography of the pelvis and/or thigh was used for detection and differentiation between VCi and VCm. Vascular calcification of the media have been less frequently reported in association with other disorders (Table 2)8–36. While VCm are common to all of these conditions phenotypes may vary. For example, in calciphylaxis VCm is associated with extensive ischaemic necrotic skin and skeletal muscle lesions due to microthrombi, thrombotic occlusions, endovascular fibrosis, and massive ‘metastatic’ calcifications of small arteries (30–600 µm; average 100 µm);33–35 all features absent in the other varieties of VCm.
Table 2.
Diseases associated with media calcifications
| Condition | Markers and findings | Comments | Reference |
|---|---|---|---|
| Atherosclerosis | Lipoprotein and glucose metabolism analysis, genetic testing; duplex carotis ABI | Frequent coincidence between intima and media calcifications | 8–11 |
| Diabetes mellitus II | Fasting glucose profile; HbA1c quantification; CT | 7–9 | |
| Chronic renal disease | Creatinine, glomerular filtration rate, urea, phosphate, calcium | 10–12 | |
| Ageing | Pyrophosphate? | 13–15 | |
| Hyperparthyreoidism | Phosphate, Calcium analysis; quantification of parathormon (PTH) | 16,17 | |
| Vitamin D disorders | Phosphate, calcium, 25(OH)D2, 25(OH)D3, 1,25(OH)2D2, 1,25(OH)2D3 | 18 | |
| Vitamin K deficiency | Coagulation studies, F VII, IX, and X, osteocalcin | 19 | |
| Vitamin K–antagonist anticoagulants | Coagulation studies | 20 | |
| Osteoporosis | Bone mineral density analysis | Deficiency of OPG, dietary essential fatty acids, and hyperlipidaemia | 21 |
| Kawasaki disease | Persistant fever; elevated TNF-α, IL-1, IL-6, B/T lymphocytes | Infants and children <5 years of age | 22 |
| ACDC | Genetic testing for NT5E mutations; calcification in joint capsules; decreased ABI; decreased limb perfusion | Autosomal recessive pattern of inheritance | 23 |
| GACI | Genetic testing for ENPP1 mutations. Calcification can be detected in utero, in newborn or in early childhood | Clinical overlap with PXE, autosomal recessive pattern of inheritance | 24 |
| IBGC | Genetic testing for SLC20A2, PDGF-B mutations. Cerebral vascular calcifications and progressive neurological defects | Autosomal dominant pattern of inheritance | 25 |
| PXE | Skin biopsy; genetic testing for ABCC6 mutations | Adolescence; clinical overlap with GACI. Autosomal recessive pattern of inheritance | 26–28 |
| Rheumatoid arthritis | Anti-CCP antibodies, ERS, CRP | Treated with steroids, NSAIDs, DMARDs; coronary artery calcification | 29 |
| Singleton–Merten syndrome | Aortic calcification, dental dysplasia and skeletal changes resembling severe anaemia | 30,31 | |
| β-Thalassemia | Complete blood count, Hb tests, Hb-electrophoresis | Patients also exhibit angioid streaks and skin lesions similar to PXE patients | 32 |
| Calciphylaxis | Coagulation studies, blood count | Arteriolopathy, associated thrombosis, ischaemic dermal damage; uraemia and non-uraemic causes | 33–35 |
| ? | Laboratory screening for traditional and non-traditional risk factors | No known risk factors, no associated diseases | 36 |
ACDC, arterial calcification due to deficiency of CD73; GACI, generalized arterial calcification of infants; IBGC, idiopathic basal ganglia calcification; PXE, Pseudoxanthoma elasticum.
Vascular calcifications are considered an independent marker of cardiovascular risk.8–11,37 Nevertheless, as highlighted by issues concerning biomechanical stability of calcified coronary atherosclerotic plaques38,39 generalizations regarding detrimental effects of this form of VC may be fallacious and to decide on these matters requires careful re-evaluation of different pathologies.
Pathology
Traditionally, two major forms of ectopic (pathologic) calcifications were distinguished; dystrophic refers to VC occurring in damaged tissues while metastatic was associated with systemic disorders of calcium and phosphate metabolism; these descriptions reflect the differences between vascular ossifications (active process) and petrifications (passive process) described by Virchow.40 While Virchow also differentiated between VCi and VCm, the latter were systematically studied in the arteries of the extremities by Mönckeberg.41 Subsequently, VCm confirmed by histology were observed in large elastic type arteries (ascending aorta), medium-sized visceral and kidney arteries, and small transitional arteries (coronary, temporal, uterine, ovarian, parathyroid, mammary gland, and other) with diameter of at least 0.5 mm.42–45 In calciphylaxis predominantly arteriolar VCm are present.33–35 A systemic distribution of VCm appears uncommon,46 yet the true incidence has not been established. Interestingly, on autopsy, patients with ESRD had a high degree of VCi but no evidence of VCm in coronary arteries.47
Vascular calcification of the media lesions, whether in elastic, transitional or muscular-type arteries, appear indiscernible when examined microscopically. The four stages of lesion progression distinguish the extent and severity of MMS VC type. In stage 1, calcifications appear on haematoxylin–eosin (H&E) staining as irregular blue or violet deposits embedded within the media. On a high-resolution light microscopy using H&E, Elastica-van-Gieson, von Kossa, or Alizarin- staining deposits consisting of fine granulations, which increase in size and become confluent, are revealed. Both intra- and extracellular deposits are present. Intracellular deposits are located in vascular smooth muscle cells (VSMCs); extracellular deposits are largely associated with damaged and fractured elastic fibres embedded within the extracellular matrix. In muscular and transitional arteries, with H&E-staining granular calcifications develop alongside the internal elastic membrane (IEM) and nearby VSMC. In our experience, the involvement of the IEM is common. These bands of calcium-rich deposits may thicken, becoming solid plates extending deep into the inner layer of the media. With further progression of the disease calcifications may distort the junctions of the innermost and outermost layers of the media, spanning up to three quadrants of the cross section (stage 2) or it may involve the entire circumference (stage 3). In stages 2 and 3, large conglomerates of calcifications may form solid plates or sheaths, progressively distorting the architecture of the media; intrusions upon the intima are then common.48 In the absence of atherosclerotic lesions, the intima shows a subendothelial hyperplasia. Figure 2A–D demonstrates the characteristic features of stages 1–3 on histological specimen. In stage 4 of VCm foci of bone formation within the arterial media may be found, calcifications may undergo osseous metaplasia giving rise to true bony trabeculae. These structures delineate true medullary spaces harbouring haematopoietic cells interspersed with adipocytes49 (Table 3).
Figure 2.
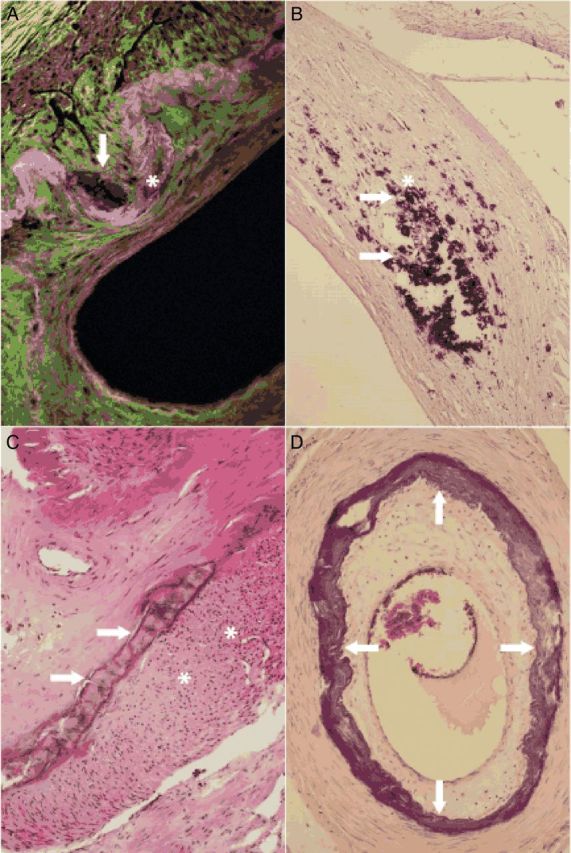
Microscopical images of medial calcifications. Small calcifications (arrow) alongside the internal elastic membrane (asterisk) characteristic of Mönckeberg medial sclerosis stage 1 are shown (femoral artery, fluorescence microscopy, 400×) (A). Larger amounts of medial calcifications (arrow) in vicinity of vascular smooth muscle cells (asterisk) in Mönckeberg medial sclerosis stage 1 lesion are demonstrated (femoral artery, haematoxylin–eosin stain, 200×) (B). Calcifications becoming confluent and forming solid plates (arrows) and subendothelial hyperplasia (asterisks) in the intima are seen; the findings correspond to Mönckeberg medial sclerosis stage 2 (femoral artery, haematoxylin–eosin stain, 40×) (C). Mönckeberg medial sclerosis stage 3 is characterized by calcifications distorting the media spanning the entire circumference (arrows) (tibial anterior artery, haematoxylin–eosin stain, 40×) (D).
Table 3.
Stages of medial calcifications and their histological aspects
| Stage | Histological aspects of vascular calcifications; MMS type |
|---|---|
| I | Granular calcifications alongside the internal elastic membrane Calcification nearby vascular smooth muscle cells |
| II | Calcifications increasing in size and becoming confluent Solid plates distorting the media spanning up to the incomplete circumference Association of subendothelial hyperplasia in the intima |
| III | Calcifications distorting the media spanning the entire circumference Association of subendothelial hyperplasia in the intima |
| IV | Calcifications and foci of bone formation (osseous metaplasia) |
MMS, Mönckeberg' media sclerosis.
In the arterial wall, calcification deposits associated with VCm may be perceived as foreign bodies and induce granuloma formation within the media; these structures often contain multinucleated giant cells. Other inflammatory components such as foam cells, lymphocytes, and mast cells may also be present. In advanced stages, large calcifications may induce secondary changes in the intima such as subendothelial hyperplasia characterized by an increase in cellularity (e.g. myofibroblasts, fibroblasts, fibrocytes) and ulcerations characterized by infiltrations of the intima or even protrusions into the lumen. It should be noted that VCm lesions do not spontaneously regress and the clinical complications may vary according to the site and the amount of calcification. Vascular calcification of the media lesions may become sites of thrombosis or they may detach and cause peripheral embolism.49 Although VCm are not necessarily indicative of peripheral artery disease (PAD), the co-incidence is common. Anatomically, VCm lesions may also involve the microcirculation, e.g. arterioles (10–500 µm in diameter). However, to our knowledge, systematic studies concerning the extent of VCm lesions towards the microcirculation have not been performed. In VCm, the affected arteries display linear calcifications of the media and IEM; VCi are patchy clusters located in the intima. While calcifications are present in all VCm lesions, they do not represent consistent finding in atheroma (Stary stages IV–VII).50–52
Biochemistry and molecular biology
The discovery of the expression of the osteoblast-related bone morphogenetic protein (BMP2) in calcified human atherosclerotic lesions2 has pioneered the hypothesis that VC is an active biological process reminiscent of osteogenesis. This notion has been reinforced with the observation of bone-forming events, including the production of matrix vesicles and apoptotic bodies in VC.53 In addition to this pathogenic view, results from several groups supported an additional hypothesis, that calcification is the consequence of the loss of local and/or circulating calcification inhibitors, such as vitamin-K dependent matrix-Gla protein (MGP),54 inorganic pyrophosphate (PPi),55 fetuin A,56 and osteoprotegerin (OPG).57 While both pathogenic models are not mutually exclusive58 determining whether the expression of bone-related genes is the cause or the consequence of the calcification process59 is critical, as therapies will differ depending on which mechanism is the initiating process.3,5
In recent years, these two concepts have been expanded and edited based on new evidence comprising a range of data concerning molecular genetic cascades and cellular transformations (for expert review, see3,5,12,60,61). Rather than recapitulating these complex biological scenarios, here we wish to focus on specific aspects particularly relevant to VCm.
Study of chronic kidney disease (CKD)-related hyperphosphataemia refocused attention to the importance of inorganic phosphate (Pi)/PPi homoeostasis. Patients with CKD-induced hyperphosphataemia develop rapid and extensive VCm, due in part to mineral dysregulation stemming from the primary renal disorder. Under these morbid conditions, vasculotoxicity of excess Pi, calcium, and other ions form nanocrystals that nucleate in soft tissues, including the vascular wall.62 In the presence of physiological cytoplasmic concentrations of Pi (∼1 mM Pi), calcification is only observed in specific genetic conditions (see below) or after long periods of exposure, either in vitro58,63 or in vivo as in ageing.64,65 However, in the presence of pathological hyperphosphataemic (>2 mM Pi) conditions calcium phosphate deposition is accelerated and easily observed. This strongly suggests that deposition is prevented by the presence and activity of calcification inhibitors, and that calcification is therefore accelerated when the limited capability of calcification inhibitors is overcome.58,66
In contrast to bone, calcium phosphate in the arteries is not predominantly deposited on type I collagen but rather on the amorphous elastin that comprises the elastic lamellae.67 This process is augmented when elastin is degraded by elastase and other proteases.68 While the elastin fibres can calcify both in vitro and in vivo as in elastocalcinosis,65,69 the role of the VSMCs and other cellular determinants (e.g. vascular progenitor cells, pericytes, calcifying vascular cells) is more complicated. There are at least three key aspects to be considered and elucidated regarding the role of these cells in the pathogenesis of VC: (i) the transformation into an osteochondrogenic phenotype, (ii) the generation of nucleating structures for calcium phosphate deposition, and (iii) the endocytosis and toxicity of calcium phosphate nanocrystals. The phenotypic plasticity of the mesenchymal-derived VSMC allows them to trans-differentiate in vitro and in vivo, gaining osteochondrogenic-like characteristics and expression of bone-related genes – e.g. Cbfa1/Runx2, Msx2, BMP2, alkaline phosphatase, osteopontin (OPN).69 The identity of these cells and impetus for their trans-differentiation has yet to be elucidated.70 Recent findings demonstrating that persistent DNA damage signalling associated with cellular senescence can activate osteogenic pathways in VSMCs are of significance, as ageing is the most dominant risk factor for the progression of VCm.71,72 In addition, recent evidence also supports the view that osteogene expression can be a consequence of the nucleation of calcium phosphate crystals, as bone-related gene expression can be completely prevented with calcification inhibitors such as PPi or phosphonoformic acid, even in the presence of high concentrations of calcium or phosphate.58,73 Furthermore, VC is associated with a decrease in the concentration of extracellular PPi and an increase of Pi.74 In order to activate osteogenic gene expression, nanoparticles of 30–500 nm need to be endocytosed and accumulated in the lysosomes, where the crystals are then dissolved. Increased calcium concentration in the cytosol can additionally be a cause of apoptosis and necrosis.73,75
Vascular smooth muscle cells also participate in the nucleation of calcium phosphate crystals through the formation of extracellular vesicles. Classification of these vesicles is a controversial area of research; recent attempts have been made to standardize nomenclature and classification.76 Matrix vesicles (30–300 nm in diameter) and fragmented apoptotic bodies (50–5000 nm in diameter) are the most studied vesicular structures in calcification.12 Matrix vesicles seem to be released by macrophages and VSMCs in calcified atheroma77 and by VSMCs in medial calcification.78
Apoptotic bodies released by VSMCs can act as nucleating agents for the deposition of crystals in the aortic wall.53 Normal vesicles contain inhibitors of calcification, especially in the presence of serum, and do not initiate calcification while in vitro79 and ex vivo77 data suggest that calcification due to the production of apoptotic bodies and matrix vesicles is dependent on the extracellular concentrations of calcium and phosphate.77,78 Apoptosis of VSMCs is also associated with calcification and medial degeneration;80 cell death can be caused by lysosome-dependent dissolution of internalized calcium phosphate crystals <1 µm.75 Cell death is not restricted to apoptosis, as necrotic cell debris also has been shown to induce calcification in a mesenchymal in vitro model.80
Recently an additional cellular phenomenon called autophagy81 has been described that counteracts the pro-calcific effect of apoptosis. In addition, recent findings suggest that the initiating events and the early pathogenic steps in the various ectopic calcification disorders may be different. Calcifying atherosclerosis, for example, occurs in the absence of hyperphosphataemia, and the strong inflammatory component is sufficient to create the conditions for nucleating calcium phosphates.82 This localized inflammatory response has not been observed in VCm, in particular macrophage accumulation does not occur at sites of medial calcification. During CKD (a major cause of VCm), a generalized, non-localized pro-inflammatory status is observed, with overexpression of TNFα, which could activate the osteochondrogenic programme in VSMCs.12
Many other general cellular mechanisms have been related to VC, such as microRNA control of gene expression,83 endoplasmic reticulum stress,84 and activation of the inflammasome.85 Clearly, VC represents a highly complex biological process. Elucidation of the main regulatory mechanisms and differentiating them from secondary phenomena is one of the urgent pending questions that must be addressed. Specifically related to the involvement of VSMCs in VC, we must identify which cell types are involved in the osteochondrogenic trans-differentiation and under what conditions; why do some cells respond apoptotically to endocytosis of nanocrystals, while other cells simply modify gene expression? And what are the specific conditions that determine apoptosis, necrosis, and other types of cell death as well as autophagy in the vessel wall?
The success of on-going and future studies will enhance our understanding of these specific questions, add to our knowledge of VC pathogenesis and identify therapeutic targets. Figure 3 shows potential molecular pathogenic pathways associated with VCm. The event responsible for triggering precipitation of hydroxyapatite from the metastable supersaturated solution of calcium and phosphate within the environment of the media—i.e. VSMCs and matrix—may result from either the presence of toxins, genetic predisposition, and/or acquired deficiency in calcification inhibitors.
Figure 3.

Suggested pathogenesis of medial calcifications. Shown are simplified molecular pathways potentially associated with initiation and propagation of medial calcifications. Nanocrystals of calcium phosphates could be nucleated and either deposited on elastine fibres and extracellular vesicles or endocysted and directed to lysosomes where they are then dissolved at low pH and Ca²+ are released to the cytosol. Cell death or osteochodrotic transdifferentiation could follow the transient increase in calcium concentration.
Molecular genetics
There are several genetically defined vascular diseases associated with primary vascular media calcification and understanding the role of these genes in this pathogenic process will help to uncover the shared mechanism regulating VCm. One monogenetic autosomal recessive disease that seems most closely to resemble the classical description of MMS is arterial calcification due to deficiency of CD73 (ACDC), a rare vascular disease hallmarked by tortuous arteries with accompanied medial calcifications and medial hyperplasia.23,86 In retrospect, it appears that ACDC patients were described as having atypical VCm, however, genetic confirmation on these patients cannot be pursued.87–89 Arterial calcification due to deficiency of CD73 patients develop massive VC in the medial layer of muscular-type arteries primarily located in the lower-extremities.86 These patients have no indication of classic atherosclerosis, impaired kidney function, or diabetes. Arterial calcification due to deficiency of CD73 is caused by loss-of-function mutations in the gene encoding for CD73 protein. This cell membrane-bound extracellular enzyme generates adenosine and Pi from AMP. Loss of CD73 activity results in an increase in activity of tissue non-specific alkaline phosphatase (TNAP), a key protein in bone formation that is rescued in vitro via exogenous adenosine administration. Increase in TNAP activity can lead to the hydrolyzation of extracellular PPi, a potent steric inhibitor of hydroxyapatite formation, thus increasing the concentration of Pi and tipping the scales towards a pro-calcific microenvironment. The discovery of ACDC opens up a new field of research as to the role of adenosine signalling on this disease phenotype.
Generalized arterial calcification in infants (GACI, also referred to as idiopathic infantile arterial calcification) is another rare vascular disease with systemic progressive VC that develop in utero and during the early postnatal period.24 Histologically, it is characterized by medial calcifications that lead to neointimal formation and vessel occlusion. This systemic vascular proliferative disease results in multi-organ failure due to non-atherosclerosis-related myocardial infarction as early as 3 months of age. Generalized arterial calcification in infant is an autosomal recessive disease caused by mutations in the gene ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1). This cell membrane-bound extracellular enzyme generates AMP and PPi from ATP and is therefore directly upstream of CD73, the causal gene for ACDC, in the extracellular purine metabolic pathway. Pyrophosphate is a potent physiological inhibitor of TNAP and the lack of PPi and subsequent increase in TNAP activity is believed to be the main driver for this pathological malignant form of primary medial calcification.
Familial idiopathic basal ganglia calcification (IBGC, also commonly known as Fahr's disease) is an additional rare disease with a wide spectrum of neuropathological symptoms that have recently been attributed to the medial calcification of small blood vessel that supply the area of the basal ganglia.25 Genetic evaluations of a subset of IBGC families identified variants in the gene sodium-dependent phosphate transporter 2 (SLC20A2 or PiT-2). PiT-2 is a Na+/Pi co-transporter that plays an important role in Pi homoeostasis and may be functionally linked to ENPP1 and CD73 in extracellular purine metabolism. A comprehensive review of genetic diseases leading to VC is available.61
Overall these three distinct genetic vascular diseases, all with medial calcification as their common primary pathology, highlight the importance of extracellular purine metabolism and downstream adenosine signalling, and phosphate homoeostasis in the development of VCm. The clinical presentation of the garden variety of VCm as described by Mönckeberg is clearly distinct from the devastating pathologies seen in patients suffering from ACDC, GACI, and IBGC; however, it is therefore important to genetically evaluate VCm variants to understand their molecular pathogenesis and to develop specific treatment strategies targeting VCm. For murine models of VC, see Supplementary material online, Appendix.
Functional implications
Arterial compliance (C) and distensibility (C/A) are given by the slope of the non-linear relation between the transmural pressure (p) and the luminal cross-sectional area (A), an expression of the elastodynamic coupling between the blood flow dynamics and vessel wall mechanics. The speed of the pressure wave, which is inversely proportional to the square root of the wall distensibility, can be also computed using the Moens–Korteweg equation;90 arterial calcification adversely affects blood flow dynamics and vessel wall mechanics.
Arterial medial calcifications have several major consequences according to the diseased arterial compartment. In the macrocirculation stiffening of the arterial wall is associated with increase in pulse wave velocity, increase in pulse pressure and pulse wave deformation (premature wave reflection, diastolic decay steepening).10,91,92 Attenuation of the Windkessel effect results in increased pulsatility potentially affecting organ perfusion, whereas augmented afterload results in left ventricular hyperthrophy,93 diastolic dysfunction,94 and heart failure with preserved ejection fraction.95 In advanced stages of VCm of MMS type, loss of elasticity of arterial walls is associated with increasingly deteriorating peripheral tissue perfusion finally resulting in arterial flow stasis and diffuse thrombus formation that is similar in appearance to deep vein thrombosis. Pathophysiological consequences of an increased pulsatility are less well understood. In the microcirculation, the loss in elasticity and increase of arterial blood pressure primarily interferes with autoregulation secondary to calcifications surrounding and within VSMC. Metabolic and endothelium-related responses all require a normal adjoining VSCM.
Arterial wall stiffening represents a strong independent predictor of future cardiovascular events and all-cause mortality.96 While the impact of the loss of Windkessel function on LV function is well understood, the impact of the loss of elasticity in patients with VCm of MMS type appears less well understood. In addition to the decreasing propulsion of the antegrade blood flow with ultimate stasis and thrombus formation in the end-stage disease, we suggest three additional pathogenic principles potentially responsible for the poor prognosis of MMS patients. First, the ring-like calcifications present in advanced VCm are likely to interfere with positive arterial remodelling in the presence of atherosclerosis as described by Glagov et al.97 and with arterial remodelling in a broader sense of arterial adaptation to haemodynamic alterations, noxes, and senescence.98 For example, in cases of coincidence with atherosclerosis VCm would prevent compensatory remodelling and markedly accelerate progression of the disease. Second, stage 4 VCm with secondary invasion of the intima shall increase the risk of thrombo-embolic events. Third, VCm arteriolar lesions may alter mechanotransduction, disturb autoregulation, and impair peripheral tissue perfusion. Thus, to determine the full functional impact of VCm on vascular function not only are measurements of pressure wave velocity99 required, but also concurrent and reliable measurements of vascular dimensions over time,100 as well as assessments of microcirculatory and endothelial functions.101,102
Diagnostic evaluations
Vascular calcification of the media of the MMS type is indicated by measuring the ABI; MMS is diagnosed with an ABI ≥1.1. It has been proposed that ankle-brachial index readings of 1.1–1.3, 1.3–1.5, and >1.5 denote an early, intermediate, and late MMS, respectively.103 In patients with suggested MMS (ABI ≥1.1), the toe-brachial index (TBI) has been proposed to improve the specificity of segmental blood pressure measurements.104 However, questions have been raised about the validity of this approach, and the role of TBI in diagnostics of MMS remains uncertain.105 Nevertheless, despite limitations ABI remains the most important screening tool to suggest the presence of both, PAD and MMS.106
In clinical settings, MMS is frequently identified accidentally from conventional X-ray radiographs of pelvis or lower extremities; MMS lesions are visualized as uniform linear radiopaque ‘railroad tracks’ as opposed to the discrete irregular radio-opaque lesions seen in atherosclerosis.107 With progressing disease granulations become coarser and less regular. Figure 4A and B shows a typical radiographic appearance of MMS lesions on native and angiographic X-ray films. Figure 5A and B shows the progression of MMS on 16-year follow-up in a male patient.
Figure 4.

X-ray images of left femoral bifurcation. Shown are native X-ray image (A) and selective needle angiogram (B) of the femoral bifurcation of a 56-year-old male patient. The typical ‘railroad trucks’ pattern (arrows) of medial calcification (A) and smooth endothelial interface (arrows) (B) of the femoral superficial and profunda arteries typical for Mönckeberg's type of medial calcification can be appreciated.
Figure 5.

Native X-ray images of a left thigh. Shown are native X-ray images of the left thigh of a male patient acquired at baseline at the age of 46 years (A) and on 16-year follow-up at the age of 62 years (B). The typical ‘railroad trucks’ pattern of medial calcification of the superficial femoral artery is seen on both images (arrows), however, on the image at baseline the pattern appears finely granulated while on the follow-up image the pattern appears coarse and more prominent signifying the progression of the disease.
Vascular ultrasound-based imaging techniques allow relatively inexpensive and non-evasive widely available means to detect VC and to differentiation between MMS- and the atherosclerosis-related lesions. In B-mode ultrasound images, MMS lesions are seen as distinct echogenic granules located in the abluminal layers of the arterial walls. Depending on the stage of the disease lesions can appear as a ‘string of beads’ (Figure 6A) or they may be larger and confluent. Employing the colour flow duplex ultrasound in addition flow patterns can be visualized (Figure 6B). In patients undergoing endovascular interventions, MMS lesions may be visualized employing intravascular ultrasound (IVUS) or optical coherence tomography (OCT). On IVUS MMS lesions are seen as highly echogenic zones located within the media. Owing to the presence of fibrotic tissue typically no acoustic ‘shadowing’ is seen (Figure 7A). In contrast, calcifications of the intima, as seen in atherosclerosis, acoustic ‘shadowing’ is frequent. Compared with IVUS OCT provides higher resolution and substantially a better visualization of the innermost layers of arterial walls. Mönckeberg medial sclerosis lesions are distinctly visualized within the tunica media (Figure 7B). Computed tomography represents another excellent tool to detect VC;108 however, distinction between intima- and media- type VC is unreliable and improved technology is required.109
Figure 6.
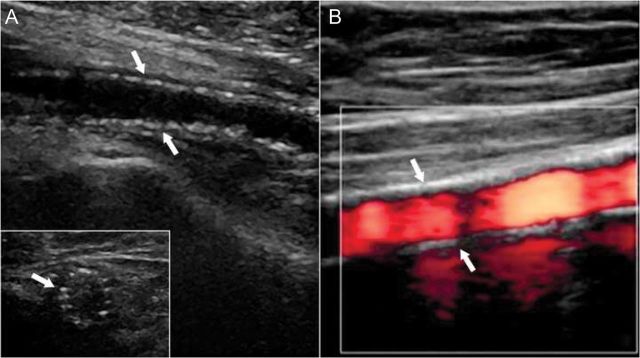
Ultrasound images of medial vascular calcifications. Shown are typical B-mode (A) and colour-mode (B) ultrasound images of a superficial femoral artery. B-mode images show the typical ‘string-of-beads’ pattern of media calcifications (arrows) with intact endothelial interface in both longitudinal sector and cross section (insert) images (courtesy C Garn). On colour-mode ultrasound image, the dense pattern of media calcifications (arrows), smooth endothelial interface, and homogeneous blood flow pattern (red colour) is shown.
Figure 7.
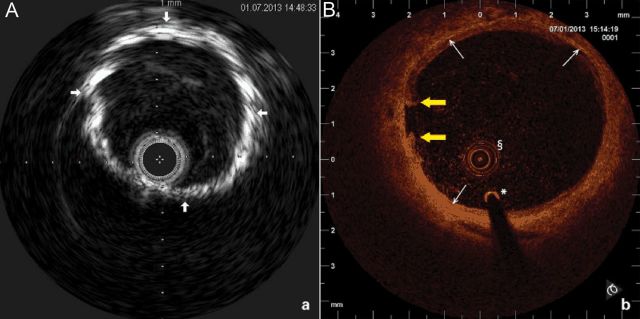
Typical images of medial vascular calcifications type Mönckeberg acquired by invasive imaging techniques. Intravascular ultrasound (A) and optical coherence tomography (B) cross-sectional images of a proximal superficial femoral artery are shown. On the intravascular ultrasound image dense tunica media calcification (arrows), the absence of acoustic shadowing and freedom from intimal atherosclerotic disease is demonstrated (Eagle Eye catheter, Volcano, 20 MHz, lateral resolution 200–250 µ, axial resolution 80–100 µ). On the optical coherence tomography image, the circumferential layer of medial calcifications (arrows) is documented. The intraluminal prolapsing fibrotic plaque is also seen (yellow arrows); §OCT catheter, *0.0.14 inch guidewire (Akquise, St Jude Medical, C7 Dragonfly catheter, lateral resolution 20 µ, axial resolution 10 µ).
While imaging studies employing conventional X-ray or ultrasound allow unequivocal definition of VCm, standardized laboratory tests to detect VC are not available at present. To assess high-risk patients or those with newly diagnosed with VCm, screening for cardiovascular risk factors for atherosclerosis including diabetes mellitus110,111 and screening for risk factors for CKD-mineral and bone disorder (CKD-MBD)112 should be performed. Laboratory evaluations of biomarkers specific for VC such as Pi, fibroblast growth factor 23 (FGF23), OPN, OPG, MGP, fetuin-A, alkaline phosphatase, and interleukin-6 (IL-6) have produced negative113 or equivocal results114–118 and have not yet been standardized for routine clinical evaluations.
Clinical management
Because of the typical absence of stenotic lesions until the late twentieth century, VCm has been considered rather a benign condition. Today, VCm is recognized as a key negative predictor of cardiovascular morbidity and mortality,8–11,96 requiring consequent prevention and, where possible, vigorous treatment. In all the patients with documented VCm regardless of potential cause and underlined pathogenesis secondary prevention and optimum treatment of associated diseases, if any, is required, in particular atherosclerosis, T2D, and CKD-MBD.
Based on current knowledge, disturbances in phosphate metabolism have been identified as a driving force for VCm and the number of potential triggers has been identified.5,12,23–25 Consequently, in patients with a documented disorder of phosphate homoeostasis such as those with CKD-MBD for prevention and treatment of VCm administration of phosphate binders, low-dose vitamin D, activators of calcium-sensing receptor (calcimimetics), magnesium, analogues of PPi (bisphosphonate), sodium thiosulfate, and more recently aldosterone antagonists has been proposed.119–122 Nevertheless, the utility of these treatment strategies has not yet been fully established and remains subject of clinical trials and investigations. In patients with T2D, the importance of inflammatory signalling123 and in particular receptors for advanced glycation end products124 direct the attention to the control of glucose metabolism; however, to our knowledge, no studies are available to document the impact of the quality of metabolic control on VCm.
Vascular calcification has been a well-documented co-morbidity in patients with osteoporosis, particularly in post-menopausal women, and those with CKD-MBD.125 Several common pathogenetic pathways including vitamin K deficiency126–128 have been proposed, yet the common links between these disease phenotypes remain tentative. In patients with vitamin K disorders depending on the cause discontinuation of vitamin K antagonists such as oral anticoagulants responsible for inhibition of carboxylation required for activation of a number of vitamin-K-dependent proteins including the potent inhibitor of VC MGP, coagulation and anticoagulation factors F II, VII, IX, and X, and protein C and protein S, respectively,19,129 or supplementation of the active form of vitamin K, mostly vitamins K2, such as menaquinone-4130 are indicated.
In patients with calciphylaxis, combined medical and surgical treatment including lowering calcium and phosphate concentration, increasing frequency of haemodialysis, i.v. administration of thiosulfate, hyperbaric oxygen, wound care, and debridement of necrotic tissue are indicated depending on clinical manifestation.131
The ageing process of arteries is characterized by a gradual increase in stiffness132 and calcification,13 potentially associated with numerous pathogenetic principles including generation of reactive oxygen species, systemic inflammation, endothelial dysfunction,133 disturbances in phosphate metabolism.74 Yet to date, no proven therapeutic principles exist to slow the ageing process of the arteries. In patients with VCm-type MMS without known risk factors, no treatment is available at present.
Perspectives
To improve our knowledge of pathogenesis and treatment of the various types of VCm, several steps are proposed.
Rules for the development of the standard nomenclature of VCs with and without the presence of extravascular tissue calcifications in human disease and in animal models should be established.
Morphological studies should be carried out to determine the systemic macro- and microcirculatory incidence of various types of VCm including MMS in human disease in different vascular beds.
Functional studies should be performed to determine the impact of various types of VCm including MMS on vascular health; specifically, the effect of various types of VCm on arterial remodelling, blood flow propulsion, and peripheral tissue perfusion should be determined.
Imaging and biomarker technology allowing sensitive detection of different stages of medial and intimal calcifications should be improved and implemented.
At the molecular and cellular level, several key issues should also be addressed. It is important, for example, to precisely define and refine the existing laboratory models of VCm, especially in vitro, and limit inappropriate generalizations from these studies. To coordinate the rapidly growing body of data, a dedicated database for mineral metabolism and VC research should be initiated, and this is the best accomplished by adopting an interdisciplinary approach.134 This will lead to the distinction between primary mechanisms of VCm and consequences, complications or accessory findings, in order to design appropriate therapies.
For example, it remains unclear whether the expression of bone-related genes is a cause or consequence of VCm in man; therefore, it is important that research focuses on the very early stages of VCm. In CKD where patients develop an extreme form of VCm, a mineral bone disorder seems to trigger the accumulation of calcium (but not Pi) in the aortic wall,135 and the same findings have been obtained with an electron microscopy and even in the presence of a Pi-rich diet.136 Calcium accumulation is accompanied by a concomitant increase in plasma concentrations of Pi, FGF23, and PTH among other agents.135 Therefore, in the very early stages increased calcium content must originate from calcium uptake or accumulation in extracellular structures of the media; these important issues also need to be studied.
To identify critical pathways and mechanisms regulating VCm, we believe that it is also important to utilize genetic approaches. In families exhibiting inborn errors leading to VCm next generation sequencing technologies should be applied to identify novel genes regulating this disease process. Further, cohorts of patients exhibiting VCm, such as primary MMS and MMS associated with chronic diseases mainly T2D and CKD-MBD, should be screened to identify SNPs that co-segregate with disease patients.
It is also important to understand the nature of the immediate consequences, once calcifications have been first established. Are extracellular and intracellular deposits of calcium phosphates in the vascular wall equally harmful, and will they differentially respond to the emerging therapies?
There is also the important issue of VCm reversal. Animal studies have shown that a very low Pi diet is able to modulate (reverse) the accumulation of both, calcium and Pi in the aorta of uraemic rats with established VCm.137 The Pi plasma concentration is therefore a key determinant of VCm in CKD and in the general population and further work on the cell biological effects of P on VSMCs is key to designing therapies.
Other areas that the scientific community should target are defining the factors that drive VCm in different disease states such as ageing,138 diabetes,60 and CKD5,12 as these may also be valid targets to slow the development of VCm, or even reverse it.
Supplementary material
Supplementary material is available at European Heart Journal online.
Conflict of interest: none declared.
Supplementary Material
References
- 1.Schlieper G, Aretz A, Verberckmoes SC, Krüger T, Behets GJ, Ghadimi R, Weinrich TE, Rohrmann D, Langer S, Tordoir JH, Amann K, Westenfeld R, Brandenburg VM, D'Haese PC, Mayer J, Ketteler M, McKee MD, Floege J. Ultrastructural analysis of vascular calcifications in uremia. J Am Soc Nephrol. 2010;21:689–696. doi: 10.1681/ASN.2009080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boström K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91:1800–1809. doi: 10.1172/JCI116391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sallam T, Cheng H, Demer LL, Tintut Y. Regulatory circuits controlling vascular calcification. Cell Mol Life Sci. 2013;70:3187–3197. doi: 10.1007/s00018-012-1231-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shanahan CM. Inflammation ushers in calcification: a cycle of damage and protection? Circulation. 2007;116:2782–2785. doi: 10.1161/CIRCULATIONAHA.107.749655. [DOI] [PubMed] [Google Scholar]
- 5.Wu M, Rementer C, Giachelli CM. Vascular calcification: an update on mechanisms and challenges in treatment. Calcif Tissue Int. 2013;93:365–373. doi: 10.1007/s00223-013-9712-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reid DG, Shanahan CM, Duer MJ, Arroyo LG, Schoppet M, Brooks RA, Murray RC. Lipids in biocalcification: contrasts and similarities between intimal and medial vascular calcification and bone by NMR. J Lipid Res. 2012;53:1569–1575. doi: 10.1194/jlr.M026088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krüger K. Epidemiologie der peripheren arteriellen Verschlusskrankheit in Deutschland; Was ist gesichert und was ist offen? Hämostaseol. 2006;26:193–196. [PubMed] [Google Scholar]
- 8.Niskanen L, Suhonen M, Sittonen O, Uusitupa MI. Medial artery calcification predicts cardiovascular mortality in patients with NIDDM. Diabet Care. 1994;17:1252–1256. doi: 10.2337/diacare.17.11.1252. [DOI] [PubMed] [Google Scholar]
- 9.Lehto S, Niskanen L, Suhonen M, Rönnemaa T, Laakso M. Medial artery calcification: a neglected harbinger of cardiovascular complications in non-insulin dependent diabetes mellitus. Arterioscler Thromb Vasc Biol. 1996;16:978–093. doi: 10.1161/01.atv.16.8.978. [DOI] [PubMed] [Google Scholar]
- 10.London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H. Arterial medial calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731–1740. doi: 10.1093/ndt/gfg414. [DOI] [PubMed] [Google Scholar]
- 11.Aitken E, Ramjug S, Buist L, Kingsmore D. The prognostic significance of iliac vessel calcification in renal transplantation. Transplant Proc. 2012;44:2925–2931. doi: 10.1016/j.transproceed.2012.06.058. [DOI] [PubMed] [Google Scholar]
- 12.Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphates. Circ Res. 2011;109:697–711. doi: 10.1161/CIRCRESAHA.110.234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliot RJ, McGrath LT. Calcification of the human thoracic aorta during aging. Calcif Tissue Int. 1994;54:268–273. doi: 10.1007/BF00295949. [DOI] [PubMed] [Google Scholar]
- 14.Atkinson J. Age-related medial elastocalcinosis in arteries: mechanisms, animal models and physiological consequences. J Appl Physiol. 2008;105:1643–1651. doi: 10.1152/japplphysiol.90476.2008. [DOI] [PubMed] [Google Scholar]
- 15.Leopold JA. Vascular calcification: an age-old problem of old age. Circulation. 2013;127:2380–2382. doi: 10.1161/CIRCULATIONAHA.113.003341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker MD, Silverberg SJ. Cardiovascular aspects of primary hyperparathyroidism. J Endocrinol Invest. 2008;10:925–931. doi: 10.1007/BF03346443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terai K, Nara H, Takahura K, Mizukami K, Sanagi M, Fukushima S, Fujimori A, Itoh H, Okada M. Vascular calcification and secondary hyperparathyroidism of severe chronic kidney disease and its relation to serum phosphate and calcium levels. Br J Pharmacol. 2009;156:1267–1278. doi: 10.1111/j.1476-5381.2008.00108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drücker TB, Massyi ZA. Role of vitamin D in vascular calcification: bad guy or good guy? Nephrol Dial Transplant. 2012;27:1704–1707. doi: 10.1093/ndt/gfs046. [DOI] [PubMed] [Google Scholar]
- 19.Danziger J. Vitamin K-dependent proteins, warfarin, and vascular calcification. Clin J Am Soc Nephrol. 2008;3:1504–1510. doi: 10.2215/CJN.00770208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schurgers LJ, Aebert H, Vermeer C, Bultmann B, Janzen J. Oral anticoagulant treatment: friend or foe in cardiovascular disease? Blood. 2004;104:3231–3232. doi: 10.1182/blood-2004-04-1277. [DOI] [PubMed] [Google Scholar]
- 21.Rodriquez-Garcia M, Gomez-Alonso C, Naves-Diaz M, Diaz-Lopez JB, Diaz-Corte C, Cannata-Andia JB the Asturias Study Group. Vascular calcifications, vertebral fractures and mortality in hemodialysis patients. Nephrol Dial Transplant. 2009;24:239–246. doi: 10.1093/ndt/gfn466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ino T, Shimazaki S, Akimoto K, Park I, Nishimoto K, Yabuta K, Tanaka A. Coronary artery calcifications in Kawasaki disease. Pediatr Radiol. 1990;20:520–523. doi: 10.1007/BF02011380. [DOI] [PubMed] [Google Scholar]
- 23.St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson-Donohoe H, Lederman RJ, Chen MY, Yang D, Siegenthaler MP, Arduino C, Mancini C, Freudenthal B, Stanescu HC, Zdebik AA, Chaganti RK, Nussbaum RL, Kleta R, Gahl WA, Boehm M. Nt5e mutations and arterial calcifications. N Engl J Med. 2011;364:432–442. doi: 10.1056/NEJMoa0912923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nürnberg P. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34:379–381. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- 25.Wang C, Li Y, Shi L, Ren J, Patti M, Wang T, de Oliveira JR, Sobrido MJ, Quintans B, Baquero M, Cui X, Zhang XY, Wang L, Xu H, Wang J, Yao J, Dai X, Liu J, Zhang L, Ma H, Gao Y, Ma X, Feng S, Liu M, Wang QK, Forster IC, Zhang X, Liu JY. Mutations in slc20a2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat Genet. 2012;44:254–256. doi: 10.1038/ng.1077. [DOI] [PubMed] [Google Scholar]
- 26.Lebwohl M, Halperin M, Phelps RG. Occult pseudoxanthoma elasticum in patients with premature cardiovascular disease. N Engl J Med. 1993;329:1237–1239. doi: 10.1056/NEJM199310213291705. [DOI] [PubMed] [Google Scholar]
- 27.LaRusso J, Li Q, Uitto J. Pseudoxanthoma elasticum, the paradigm of heritable ectopic mineralization disorder: can diet help? J Dtsch Dermatol Ges. 2011;9:586–593. doi: 10.1111/j.1610-0387.2011.07658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leftheriotis G, Omarjee L, LeSaux O, Henrion D, Abraham P, Prunier F, Willoteaux S, Martin L. The vascular phenotype in Pseudoxanthoma elasticum and related disorders: contribution of genetic disease to the understanding of vascular calcification. Front Gen. 2013;4:1–9. doi: 10.3389/fgene.2013.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paccou J, Brazier M, Mentaverri R, Kamel S, Fardellone P, Massy ZA. Vascular calcification in rheumatoid arthritis: prevalence, pathophysiological aspects and potential targets. Atherosclerosis. 2012;224:283–290. doi: 10.1016/j.atherosclerosis.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Singleton EB, Merten DF. An unusual syndrome of wiedened medullary cavities of the metacarpals and phalanges, aortic calcification and abnormal dentition. Pediatr Radiol. 1973;1:2–7. doi: 10.1007/BF00972817. [DOI] [PubMed] [Google Scholar]
- 31.Feigenbaum AM, Müller C, Yale C, Kleinheinz J, Jezewski P, Kehl HG, MacDougall M, Rutsch F, Hennekam RCM. Singleton-Merten syndrome: an autosomal dominant disorder with variable expression. Am J Med Genet. 2013;161A(Part A):360–370. doi: 10.1002/ajmg.a.35732. [DOI] [PubMed] [Google Scholar]
- 32.Aessopos A, Samarkos M, Voskaridou E, Papaioannou D, Kavoukis E, Vasopoulos G, Stamatelos G, Loukopoulos D. Arterial calcifications in beta-thalassemia. Angiology. 1998;49:137–143. doi: 10.1177/000331979804900206. [DOI] [PubMed] [Google Scholar]
- 33.Magro CM, Simman R, Jackson S. Calciphylaxis: a review. J Am Coll Certif Wound Spec. 2010;2:66–72. doi: 10.1016/j.jcws.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nigwekar SU, Wolf M, Sterns RH, Hix JK. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139–1143. doi: 10.2215/CJN.00530108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saifan C, Saad M, El-Charabaty E, El- Sayegh S. Warfarin-induced calciphylaxis: a case report and review of literature. Int J Gen Med. 2013;6:665–669. doi: 10.2147/IJGM.S47397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanzer P. Mediakalzinose Mönckeberg. Z Kardiol. 1998;87:586–593. doi: 10.1007/s003920050217. [DOI] [PubMed] [Google Scholar]
- 37.Renneberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcification as a marker for increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5:185–197. doi: 10.2147/vhrm.s4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burke AP, Weber DK, Kolodgie FD, Farb A, Taylor AJ, Virmani R. Pathophysiology of calcium deposition in coronary arteries. Herz. 2001;26:239–244. doi: 10.1007/pl00002026. [DOI] [PubMed] [Google Scholar]
- 39.Maldonado N, Kelly-Arnold A, Vengrenyuk Y, Laudier D, Fallon JT, Virmani R, Cardoso L, Weinbaum S. A mechanistic analysis of the role of microcalcification in atherosclerotic plaque stability: potential implications for plaque rupture. Am J Physiol Heart Circ Physiol. 2012;303:H619–H628. doi: 10.1152/ajpheart.00036.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Virchow R. Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebslehre. reprint: Hildesheim: Georg Olms Verlagsbuchhandlung; 1966. pp. 327–329. Verlag von August Hirschwald, Berlin: 1858. [Google Scholar]
- 41.Mönckeberg JG. Über die reine Mediaverkalkung der Extremitätenarterien und ihr Verhalten zur Arteriosklerose. Virchows Archiv für pathologische Anatomie und Physiologie, und für klinische Medicin, Berlin. 1903;171:141–167. [Google Scholar]
- 42.Nakamura S, Ishibashi-Ueda H, Niizuma S, Yoshihara F, Horio T, Kawano Y. Coronary calcification in patients with chronic kidney disease and coronary artery disease. Clin J Am Soc Nephrol. 2009;4:1892–1900. doi: 10.2215/CJN.04320709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aslanides IM, Pavlin CJ, Giavedoni LR. Moenckeberg’ sclerosis in temporal artery biopsy specimen. Br J Ophthalmol. 1999;83:1091–1092. doi: 10.1136/bjo.83.9.e1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saxena A, Waddell IC, Friesen RW, Michalski RT. Monckeberg medial calcific sclerosis mimicking malignant calcification pattern at mammography. J Clin Pathol. 2005;58:447–448. [PMC free article] [PubMed] [Google Scholar]
- 45.Castillo BV, Jr, Torczynski E, Edward DP. Monckeberg's sclerosis in temporal artery biopsy specimens. Br J Ophthalmol. 1999;83:1091–1092. doi: 10.1136/bjo.83.9.e1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lachman AS, Spray TL, Kerwin DM, Shugoll GI, Roberts WC. Medial calcinosis of Monckeberg. A review of the problem and a description of a patient with involvement of peripheral, visceral and coronary arteries. Am J Med. 1977;63:615–622. doi: 10.1016/0002-9343(77)90207-8. [DOI] [PubMed] [Google Scholar]
- 47.Schwarz U, Buzello M, Ritz E, Stein G, Raabe G, Wiest G, Mall G, Amann K. Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol Dial Transpl. 2000;15:218–223. doi: 10.1093/ndt/15.2.218. [DOI] [PubMed] [Google Scholar]
- 48.Janzen J, Vuong PN. Arterial calcifications: morphological aspects and their pathological implications. Z Kardiol. 2001;90(Suppl. 3):III/6–III/11. doi: 10.1007/s003920170044. [DOI] [PubMed] [Google Scholar]
- 49.Janzen J, Bültmann B, Leitritz M, Rothenberger-Janzen K, Vuong PN. Histopathological aspects of arterial calcifications. Perfusion. 2003;16:136–140. [Google Scholar]
- 50.Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Jr, Rosenfeld ME, Schaffer A, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. Arterioscler Thromb. 1994;14:840–856. doi: 10.1161/01.atv.14.5.840. [DOI] [PubMed] [Google Scholar]
- 51.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis: a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–1374. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 52.Stary HC. Natural history of calcium deposits in atherosclerosis progression and regression. Z Kardiol. 2000;89(Suppl. 2):II/28–II/35. doi: 10.1007/s003920070097. [DOI] [PubMed] [Google Scholar]
- 53.Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–1062. doi: 10.1161/01.res.87.11.1055. [DOI] [PubMed] [Google Scholar]
- 54.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- 55.Meyer JL. Can biological calcification occur in the presence of pyrophosphate? Arch Biochem Biophys. 1984;231:1–8. doi: 10.1016/0003-9861(84)90356-4. [DOI] [PubMed] [Google Scholar]
- 56.Schafer C, Heiss A, Schwarz A, Westenfeld R, Ketteler M, Floege J, Muller-Esterl W, Schinke T, Jahnen-Dechent W. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357–366. doi: 10.1172/JCI17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. 2010;9:528–536. doi: 10.1038/nrcardio.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villa-Bellosta R, Sorribas V. Calcium phosphate deposition with normal phosphate concentration. Role of pyrophosphate. Circ J. 2011;75:2705–2710. doi: 10.1253/circj.cj-11-0477. [DOI] [PubMed] [Google Scholar]
- 60.Hofmann-Bowman MA, McNally EM. Genetic pathways of vascular calcification. Trends Cardiovasc Med. 2012;22:93–98. doi: 10.1016/j.tcm.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rutsch F, Nitschke Y, Terkeltaub R. Genetics of arterial calcifications: pieces of a puzzle and cogs in a wheel. Circ Res. 2011;109:578–592. doi: 10.1161/CIRCRESAHA.111.247965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Razzaque MS. Phosphate toxicity and vascular mineralization. Contrib Nephrol. 2013;180:74–85. doi: 10.1159/000346784. [DOI] [PubMed] [Google Scholar]
- 63.Villa-Bellosta R, Millan A, Sorribas V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am J Physiol Cell Physiol. 2011;300:C210–C220. doi: 10.1152/ajpcell.00229.2010. [DOI] [PubMed] [Google Scholar]
- 64.Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME. Medial localization of mineralization-regulating proteins in association with Mönckeberg's sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation. 1999;100:2168–2176. doi: 10.1161/01.cir.100.21.2168. [DOI] [PubMed] [Google Scholar]
- 65.Mackenzie NC, MacRae VE. The role of cellular senescence during vascular calcification: a key paradigm in aging research. Curr Aging Sci. 2011;4:128–136. doi: 10.2174/1874609811104020128. [DOI] [PubMed] [Google Scholar]
- 66.Villa-Bellosta R, Sorribas V. Phosphonoformic acid prevents vascular smooth muscle cell calcification by inhibiting calcium-phosphate deposition. Arterioscler Thromb Vasc Biol. 2009;29:761–766. doi: 10.1161/ATVBAHA.108.183384. [DOI] [PubMed] [Google Scholar]
- 67.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi GP, Jaffer FA, Weissleder R. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hosaka N, Mizobuchi M, Ogata H, Kumata C, Kondo F, Koiwa F, Kinugasa E, Akizawa T. Elastin degradation accelerates phosphate-induced mineralization of vascular smooth muscle cells. Calcif Tissue Int. 2009;85:523–529. doi: 10.1007/s00223-009-9297-8. [DOI] [PubMed] [Google Scholar]
- 69.Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW, Gabbiani G, Giachelli CM, Parmacek MS, Raines EW, Rusch NJ, Speer MY, Sturek M, Thyberg J, Towler DA, Weiser-Evans MC, Yan C, Miano JM, Owens GK. Smooth muscle cell plasticity: fact or fiction? Circ Res. 2013;112:17–22. doi: 10.1161/CIRCRESAHA.112.281048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang Z, Wang A, Yuan F, Yan Z, Liu B, Chu JS, Helms JA, Li S. Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat Commun. 2013;3:875. doi: 10.1038/ncomms1867. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, Takahashi T, Matoba S, Yamada H, Okigaki M, Matsubara H. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol. 2009;297:H1673–H1684. doi: 10.1152/ajpheart.00455.2009. [DOI] [PubMed] [Google Scholar]
- 72.Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. 2013;112:e99–109. doi: 10.1161/CIRCRESAHA.111.300543. [DOI] [PubMed] [Google Scholar]
- 73.Sage AP, Lu J, Tintut Y, Demer LL. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011;79:414–422. doi: 10.1038/ki.2010.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Villa-Bellosta R, Rivera-Torres J, Osorio FG, Acin-Perez R, Enrique JA, Lopez-Otin C, Andres V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford Progeria Syndrome that is ameliorated on pyrophosphate treatment. Circulation. 2013;127:2442–2451. doi: 10.1161/CIRCULATIONAHA.112.000571. [DOI] [PubMed] [Google Scholar]
- 75.Ewence AE, Bootman M, Roderick HL, Skepper JN, McCarthy G, Epple M, Neumann M, Shanahan CM, Proudfoot D. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: a potential mechanism in atherosclerotic plaque destabilization. Circ Res. 2008;103:e28–e34. doi: 10.1161/CIRCRESAHA.108.181305. [DOI] [PubMed] [Google Scholar]
- 76.New SE, Goettsch C, Aikawa M, Marchini JF, Shibasaki M, Yabusaki K, Libby P, Shanahan CM, Croce K, Aikawa E. Macrophage-derived matrixvesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ Res. 2013;113:72–77. doi: 10.1161/CIRCRESAHA.113.301036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shroff RC, McNair R, Skepper JN, Figg N, Schurgers LJ, Deanfield J, Rees L, Shanahan CM. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol. 2010;21:103–112. doi: 10.1681/ASN.2009060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schoppet M, Kavurma MM, Hofbauer LC, Shanahan CM. Crystallizing nanoparticles derived from vascular smooth muscle cells contain the calcification inhibitor osteoprotegerin. Biochem Biophys Res Commun. 2011;407:103–107. doi: 10.1016/j.bbrc.2011.02.117. [DOI] [PubMed] [Google Scholar]
- 79.Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–2867. doi: 10.1097/01.ASN.0000141960.01035.28. [DOI] [PubMed] [Google Scholar]
- 80.Clarke MC, Littlewood TD, Figg N, Maguire JJ, Davenport AP, Goddard M, Bennett MR. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. 2008;102:1529–1538. doi: 10.1161/CIRCRESAHA.108.175976. [DOI] [PubMed] [Google Scholar]
- 81.Dai XY, Zhao MM, Cai Y, Guan QC, Zhao Y, Guan Y, Kong W, Zhu WG, Xu MJ, Wang X. Phosphate-induced autophagy counteracts vascular calcification by reducing matrix vesicle release. Kidney Int. 2013;83:1042–1051. doi: 10.1038/ki.2012.482. [DOI] [PubMed] [Google Scholar]
- 82.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850. doi: 10.1161/CIRCULATIONAHA.107.732867. [DOI] [PubMed] [Google Scholar]
- 83.Chen NX, Kiattisunthorn K, O'Neill KD, Chen X, Moorthi RN, Gattone VH, II, Allen MR, Moe SM. Decreased microRNA is involved in the vascular remodeling abnormalities in chronic kidney disease (CKD) PLoS One. 2013;8:e64558. doi: 10.1371/journal.pone.0064558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Duan X, Zhou Y, Teng X, Tang C, Qi Y. Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem Biophys Res Commun. 2009;387:694–699. doi: 10.1016/j.bbrc.2009.07.085. [DOI] [PubMed] [Google Scholar]
- 85.Wen C, Yang X, Yan Z, Zhao M, Yue X, Cheng X, Zheng Z, Guan K, Dou J, Xu T, Zhang Y, Song T, Wei C, Zhong H. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int J Cardiol. 2013;168:2242–2247. doi: 10.1016/j.ijcard.2013.01.211. [DOI] [PubMed] [Google Scholar]
- 86.Markello TC, Pak LK, St Hilaire C, Dorward H, Ziegler SG, Chen MY, Chaganti K, Nussbaum RL, Boehm M, Gahl WA. Vascular pathology of medial arterial calcifications in nt5e deficiency: implications for the role of adenosine in Pseudoxanthoma elasticum. Mol Genet Metab. 2011;103:44–50. doi: 10.1016/j.ymgme.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sharp J. Heredo-familial vascular and articular calcification. Ann Rheum Dis. 1954;13:15–27. doi: 10.1136/ard.13.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Top C, Cankir Z, Silit E, Yildirim S, Danaci M. Monckeberg's sclerosis: an unusual presentation—a case report. Angiology. 2002;53:483–486. doi: 10.1177/000331970205300418. [DOI] [PubMed] [Google Scholar]
- 89.Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nurnberg P. Mutations in enpp1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34:379–381. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- 90.Nichols W, O'Rourke MF, Vlachopoulos C. McDonald's Blood Flow in Arteries. 6th ed. London: Hodder Arnold; 2011. pp. 55–75. [Google Scholar]
- 91.Chirinos JA, Segers P. Measurements and basic principles of wave conduction and reflection noninvasive evaluation of left ventricular afterload: part 1: pressure and flow hypertension. Hypertension. 2010;56:555–562. doi: 10.1161/HYPERTENSIONAHA.110.157321. [DOI] [PubMed] [Google Scholar]
- 92.Chirinos JA, Segers P. Noninvasive evaluation of left ventricular afterload. Part 2: arterial pressure-flow and pressure-volume relationships in humans. Hypertension. 2010;56:563–570. doi: 10.1161/HYPERTENSIONAHA.110.157339. [DOI] [PubMed] [Google Scholar]
- 93.London GM. Cardiovascular disease in chronic renal failure: pathophysiologic aspects. Semin Dial. 2003;16:85–94. doi: 10.1046/j.1525-139x.2003.16023.x. [DOI] [PubMed] [Google Scholar]
- 94.Soldatos G, Jandeleit-Dahm K, Thomson J, Formosat M, D'orsa K, Calkin AC, Cooper ME, Ahimastos AA, Kingwell BA. Large artery biomechanics and diastolic dysfunction in patients with type 2 diabetes. Diab Med. 2011;28:54–60. doi: 10.1111/j.1464-5491.2010.03146.x. [DOI] [PubMed] [Google Scholar]
- 95.Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 96.Vlachopoulos C, Aznaouridis K, Stefanidis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness. J Amer Coll Cardiol. 2010;55:1318–1327. doi: 10.1016/j.jacc.2009.10.061. [DOI] [PubMed] [Google Scholar]
- 97.Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary artery disease. N Engl J Med. 1987;316:1371–1375. doi: 10.1056/NEJM198705283162204. [DOI] [PubMed] [Google Scholar]
- 98.Van Varik BJ, Rennenberg RJMW, Reutelingsperger CP, Kroon AA, de Leeuw PW, Schurgers LJ. Mechanisms of arterial remodeling: lessons from genetic diseases. Front Genet. 2012;3 doi: 10.3389/fgene.2012.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, Pannier B, Vlachopoulos C, Wilkinson I, Struijker-Boudier H. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27:2588–2605. doi: 10.1093/eurheartj/ehl254. [DOI] [PubMed] [Google Scholar]
- 100.Wang T, Wieser W, Springeling G, Beurskens R, Lancee CT, Pfeiffer T, van der Steen AF, Huber R, van Soest G. Intravascular optical coherence tomography imaging at 3200 frames per second. Opt Lett. 2013;38:1715–1717. doi: 10.1364/OL.38.001715. [DOI] [PubMed] [Google Scholar]
- 101.Nohria A, Gerhard-Herman M, Creager MA, Hurley S, Mitra D, Ganz P. Role of nitric oxide in the regulation of digital pulse volume amplitude in humans. J Appl Physiol. 2006;101:545–548. doi: 10.1152/japplphysiol.01285.2005. [DOI] [PubMed] [Google Scholar]
- 102.Morris AA, Patel RS, Binogno JNG, Poole J, al Mheid I, Ahmed Y, Stoyanova N, Vaccarino V, Din-Dzietham R, Gibbsons GH, Quyyumi A. Racial differences in arterial stiffness and microcirculatory function between black and white Americans. J Am Heart Assoc. 2013;2:002154. doi: 10.1161/JAHA.112.002154. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fritsch I. Untersuchungen zum Zusammenhang zwischen Mediasklerose und diabetischer Neuropathie bei Patienten mit Diabetes mellitus. http://edoc.ub.uni-muenchen.de/5698/1/Fritsch_Insa.pdf. 20 July 2013. [Google Scholar]
- 104.Niskanen L, Siitonen O, Suhonen M, Uusitupa MI. Medial artery calcification predicts cardiovascular mortality in patients with NIDDM. Diabetes Care. 1994;17:1252–1256. doi: 10.2337/diacare.17.11.1252. [DOI] [PubMed] [Google Scholar]
- 105.Romanos MT, Raspovic A, Perrin BM. The reliability of toe systolic pressure and toe brachial index in patients with diabetes. J Foot Ankle Res. 2010;3:31–38. doi: 10.1186/1757-1146-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Aboyans V, Criqui MH, Abraham P, Allision MA, Creager MA, Diehm C, Fowkes MA, Hiatt WR, Johansson B, Lacroix P, Marin B, McDermott MM, Norgren L, Pande RL, Preux P-M, Stoffers J. Measurement and Interpretation of the Ankle-Brachial Index: a Scientific Statement from the American Heart Association. Circulation. 2012;126:2890–2909. doi: 10.1161/CIR.0b013e318276fbcb. [DOI] [PubMed] [Google Scholar]
- 107.Lidbom A. Arteriosclerosis and arterial thrombosis in the lower limb: a roentgenological study. Acta Radiol Suppl. 1950;80:1–80. [PubMed] [Google Scholar]
- 108.Rumberger JA, Kaufman L. A rosetta stone for coronary calcium risk stratification: Agatson, volume, and mass scores in 11,490 individuals. Am J Roentgenol. 2003;181:743–748. doi: 10.2214/ajr.181.3.1810743. [DOI] [PubMed] [Google Scholar]
- 109.Hjortnaes J, New SE, Aikawa E. Visualizing novel concepts of cardiovascular calcification. Trends Cardiovasc Med. 2013;23:71–79. doi: 10.1016/j.tcm.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lim LS, Haq N, Mahmood S, Hoeksema L the ACPM Prevention Practice Committee:Atherosclerotic Cardiovascular Disease Screening in Adults American College of Preventive Medicine; Position Statement on Preventive Practice. J Prev Med. 2011;40:380–381. doi: 10.1016/j.amepre.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 111.Du F, Virtue A, Wand H, Yang X-F. Metabolomic analyses for atherosclerosis, diabetes, and obesity. Biomarker Res. 2013;1:17. doi: 10.1186/2050-7771-1-17. http://www.biomarkerres.org/content/1/1/17. 4 January 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.KDIGO Clinical Practice Guideline for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease–Mineral and Bone Disorder (CKD–MBD) http://www.kdigo.org/pdf/KDIGO%20CKD-MBD%20GL%20KI%20Suppl%20113.pdf. 4 January 2014. [DOI] [PubMed]
- 113.Liabeuf S, Okazaki H, Desjardins L, Fliser D, Goldsmith D, Covic A, Wiecek A, Ortiz A, Martinez-Castelao A, Lindholm B, Suleymanlar G, Mallacami F, Zoccali C, London G, Massy ZA. Vascular calcification in chronic kidney disease: are biomarkers useful for probing the pathobiology and the health risks of this process in clinical scenario? Nephrol Dial Transplant. 2013;28:2456–2463. doi: 10.1093/ndt/gft368. [DOI] [PubMed] [Google Scholar]
- 114.Lee C-T, Chua S, Hsu C-Y, Tsai Y-C, Ng H-Y, Kuo C-C, Wu C-H, Chen T-C, Chiu TT-Y, Lee Y-T. Biomarkers associated with vascular and valvular calcification in chronic hemodialysis patients. Dis Markers. 2013;34:229–235. doi: 10.3233/DMA-130965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eraso LH, Ginwala N, Qasim AN, Mehta NN, Dlugash R, Kapoor S, Schwartz S, Schutta M, Iqbal N, Mohler ER, 3rd, Reilly MP. Association of lower plasma fetuin-A levels with peripheral artery disease in type 2 diabetes. Diabet Care. 2010;33:408–410. doi: 10.2337/dc09-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lorant DP, Grujicic M, Hoebaus C, Brix JM, Hoellerl F, Schernthaner G, Koppensteiner R, Schernthaner GH. Fetuin-A levels are increased in patients with type 2 diabetes and peripheral artery disease. Diabet Care. 2011;34:156–161. doi: 10.2337/dc10-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jung C-H, Kim B-Y, Kim C-H, Kang S-K, Jung S-H, Mok J-O. Association of serum fetuin-A levels with insulin resistance and vascular complications in patients with type 2 diabetes. Diab Vasc Dis Res. 2013;10:459–466. doi: 10.1177/1479164113490766. [DOI] [PubMed] [Google Scholar]
- 118.Aoki A, Murata M, Asano T, Ikoma A, Sasaki M, Saito T, Otani T, Jinbo S, Ikeda N, Kawakami M, Ishikawa S-e. Association of serum osteoprogesterin with vascular calcification in patients with type 2 diabetes. Cardiovasc Diabetol. 2013;12:11. doi: 10.1186/1475-2840-12-11. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.O'Neill WC, Lomashvili KA. Recent progress in the treatment of vascular calcification. Kidney Int. 2010;78:1232–1239. doi: 10.1038/ki.2010.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Disthabanchong S. Lowering vascular calcification burden in chronic kidney disease: is it possible? World J Nephrol. 2013;2:49–55. doi: 10.5527/wjn.v2.i3.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Riccardi D, Kemp PJ. The calcium-sensing receptor beyond extracellular calcium homeostasis: concept, development, adult physiology, and disease. Annu Rev Physiol. 2012;74:271–297. doi: 10.1146/annurev-physiol-020911-153318. [DOI] [PubMed] [Google Scholar]
- 122.Lang F, Ritz E, Voelkl J, Alesutan I. Vascular calcification: is aldosterone a culprit? Nephrol Dial Transplant. 2013;28:1080–1084. doi: 10.1093/ndt/gft041. [DOI] [PubMed] [Google Scholar]
- 123.Shao J-S, Cheng S-L, Sadhu J, Towler DA. Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension. 2010;55:579–592. doi: 10.1161/HYPERTENSIONAHA.109.134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hoffmann-Bowman MA, Schmidt AM. The next generation of RAGE modulators: implications for soluble RAGE therapies in vascular inflammation. J Mol Med. 2013;91:1329–1331. doi: 10.1007/s00109-013-1097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hofbauer LC, Brueck CC, Shanahan CM, Schoppet M, Dobnig H. Vascular calcification and osteoporosis from clinical observation towards molecular understanding. Osteoporosis Int. 2007;18:251–259. doi: 10.1007/s00198-006-0282-z. [DOI] [PubMed] [Google Scholar]
- 126.Fang Y, Ginsberg C, Sugatani T, Monier-Faugere M-C, Malluche H, Hruska KA. Early chronic kidney disease-mineral disorder stimulates vascular calcification. Kidney Int. 2013;85:142–150. doi: 10.1038/ki.2013.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hampson G, Edwards S, Conroy S, Blake GM, Fogelman I, Frost ML. The relationship between inhibitors of the Wnt signaling pathway [Dickkopf-1(DKK1) and sclerotin], bone mineral density, vascular calcification and arterial stiffness in post-menopausal women. Bone. 2013;56:42–47. doi: 10.1016/j.bone.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 128.Shea MK, Holden RM. Vitamin K status and vascular calcification: evidence from observational and clinical studies. Adv Nutr. 2012;3:158–165. doi: 10.3945/an.111.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hristova M, van Beek C, Schurgers LJ, Lanske B, Danziger J. Rapidly progressive severe calcification sparing the kidney allograft following warfarin initation. Am J Kidney Dis. 2010;56:1158–1162. doi: 10.1053/j.ajkd.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Flore R, Ponziani FR, Di Rienzo TA, Zocco MA, Flex A, Gerardino L, Lupascu A, Santoro L, Santoliquido A, Di Stasio E, Chierici E, Lanti A, Tondi P, Gasbarrini A. Something more to say about calcium homeostasis: the role of vitamin K2 in vascular calcification and osteoporosis. Eur Rev Med Pharmacol Sci. 2013;17:2433–2440. [PubMed] [Google Scholar]
- 131.Don BR, Chin AI. A strategy for the treatment of calcific uremic arteriolopathy (calciphylaxis) employing a combination of therapies. Clin Nephrol. 2003;59:463–470. doi: 10.5414/cnp59463. [DOI] [PubMed] [Google Scholar]
- 132.Wang M, Jiang L, Monticone RE, Lakatta EG. Proinflammation; the key to arerial aging. Trends Endocrinol Metab. 2014;25:72–79. doi: 10.1016/j.tem.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cattan V, Kakou A, Louis H, Lacolley P. Pathophysiology, genetic, and therapy of arterial stiffness. Biomed Mater Eng. 2006;14(4 Suppl):S155–S161. [PubMed] [Google Scholar]
- 134.Epple M, Lanzer P. How much interdisciplinarity is required to understand vascular calcifications? Formulation of four basic principles of vascular calcification. Z Kardiol. 2001;90(Suppl. 3):2–5. doi: 10.1007/s003920170036. [DOI] [PubMed] [Google Scholar]
- 135.Fang Y, Ginsberg C, Sugatani T, Monier-Faugere MC, Malluche H, Hruska KA. Early chronic kidney disease-mineral bone disorder stimulates vascular calcification. Kidney Int. 2014;85:142–150. doi: 10.1038/ki.2013.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Martín-Pardillos A, Sosa C, Millán A, Sorribas V. Effect of water fluoridation on the development of medial vascular calcification in uremic rats. Toxicology. 2014;318:40–50. doi: 10.1016/j.tox.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 137.Finch JL, Lee DH, Liapis H, Ritter C, Zhang S, Suarez E, Ferder L, Slatopolsky E. Phosphate restriction significantly reduces mortality in uremic rats with established vascular calcification. Kidney Int. 2013;84:1145–1153. doi: 10.1038/ki.2013.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fritze O, Romero B, Schleicher M, Jaco MP, Oh J-Y, Starcher B, Schenke – Layland K, Bujan J, Stock UA. Age-related changes in the elastic tissue of the human aorta. J Vasc Res. 2012;49:77–86. doi: 10.1159/000331278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.