

Abstract
Background
Previous studies have investigated differential gene expression in gastrointestinal (GI) epithelial cancers by microarray. The aim of the present study was to use data from the Oncomine database to identify genes that share a similar differential expression in two or more primary GI cancer sites.
Methods
Five thousand of the most differentially expressed genes in epithelial cancers (compared to normal tissue) arising in the pancreas, liver, stomach, esophagus or colorectum were identified (1,000 per primary site) from Oncomine. Using Venn diagrams, genes common to two or more primary GI sites were identified. Functional and pathway analysis was performed on genes that were similarly expressed in ≥3 of the five areas of the GI tract.
Results
Forty six studies comprising 5,876 samples were included. Overall, 90.6% genes were unique to the respective primary sites, 7.4% shared between two GI primary sites, 1.8% between three and 0.2% between four GI primary sites. Pancreatic and hepatocellular cancers (HCC) shared most number of upregulated genes (N=66) while HCC and gastric cancer shared most downregulated genes (N=59). Genes encoding enzymes comprised the most commonly shared genes between GI primary sites (30.4% of upregulated and 63.2% of downregulated genes). Those genes that were shared between three or more GI primary sites also showed significant differential expression in the same direction in other non-GI cancers.
Conclusion
The present study has identified several genes that show similar differential expression in cancers arising from two or more sites in the GI tract. These genes can be potentially useful as novel therapeutic targets.
Keywords: Oncomine, pathway, network, database, cancer, gastrointestinal, therapy
Introduction
The Oncomine database is one of the largest collections of microarray data. The current research edition has 715 datasets with 86,733 samples. According to the American Cancer Society, cancers of the digestive tract comprise about 290,000 new cancers each year while accounting for nearly 145,000 cancer related deaths annually. Thus they are expected to account for nearly 17.5% of all new cancer cases and about 25% of cancer related deaths in 2013. While there are numerous studies that have investigated the global gene expression in various gastrointestinal (GI) cancers compared to normal GI tissue, there are differences in the results from study to study. Thus, analysis of results from multiple studies provides the opportunity to get a more accurate picture of differential gene expression in cancer tissues. The GI tract is embryologically derived from the endoderm with the esophagus and stomach derived from the foregut, duodenum from the foregut and midgut, liver from the ventral mesentery, pancreas from the septum transversum, transverse and ascending colon from the midgut and the transverse colon and rectum from the hindgut. If we could identify a set of genes that are common between cancers arising from multiple GI sites, this would help identify pathways and in turn targets for therapy of GI malignancies. The Oncomine database provides us the opportunity to test this hypothesis owing to the large number of samples available for analysis. In this study, we compared gene expression between epithelial cancers arising from five different GI primary sites (pancreas, liver, esophagus, stomach and colorectum). We discovered that there were multiple genes common to at least three and four GI primary sites but none to all five primary sites.
Methods
The Oncomine database (www.oncomine.org) was queried for studies comparing normal tissue from a part of the GI tract to tissue from an epithelial malignancy arising from the same tissue. All studies in the database which compared gene expression in GI cancer tissue to the corresponding normal tissue were included. For instance there were fifteen studies involving 2,208 samples that had compared gene expression in colorectal cancer to the corresponding normal tissue. The Oncomine database allows us to compare gene expression across multiple studies to identify the genes that are differentially over or under expressed in majority of the studies. Only those genes with P-value <0.05 were included. The first 500 most over and 500 most under expressed genes for each primary site were included in the analysis. Five primary sites in the GI tract, i.e. esophagus, stomach, pancreas, liver, and colorectum, were included in the study. To identify genes that were commonly expressed between different GI malignancies, we used VENNTURE, a freely available tool to compare genes among up to six groups (http://www.irp.nia.nih.gov/branches/lci/nia_bioinformatics_software.html). To investigate the function of a given gene we used the PANTHER database (http://www.pantherdb.org/). Interactions of one or more genes with other genes were investigated using the GeneMania online tool (http://genemania.org/). To investigate chemicals targeting a particular gene product, we used the STITCH database (http://stitch.embl.de/).
Results
A total of 5,000 genes (1,000 genes per primary GI site) were included in the analysis. For each primary site, we identified 500 genes that were upregulated and 500 that were most downregulated in majority of the included studies. There were fifteen studies comparing colorectal cancer to normal colon, seven comparing esophageal cancer (includes both squamous and adenocarcinoma) to normal esophagus, seven on gastric cancer, eight on hepatocellular (HCC) and nine comparing pancreatic cancer to respective normal tissue. These studies included 2208, 514, 1408, 1324 and 422 samples respectively.
To compare gene expression between the five primary GI sites (pancreas, liver, stomach, esophagus and colorectum) and in turn identify genes that were common between two or more primary sites, we used the VENNTURE Venn diagram tool. Separate comparisons were made for upregulated (Fig. 1) and downregulated genes (Fig. 2).
Figure 1.
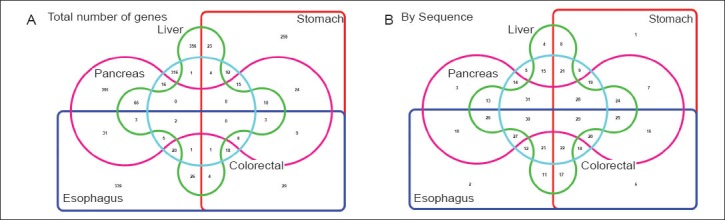
Venn diagram analysis of genes upregulated in five primary gastrointestinal cancer sites. A color represents a particular primary site of GI cancer- for instance, pink is Pancreas, green is for liver, light blue for colorectum, red for stomach and dark blue for esophagus. In Panel A, Venn diagram method has been used to identify the number of genes that are shared between two or more cancers. For instance, cancers arising in the pancreas and liver share 66 common upregulated genes while 306 genes are unique to the pancreas. In Panel B, we have sequence numbers corresponding to the column numbers in Suppl. File 1 (website). Refer to the corresponding column number for the names of specific genes. For instance, the 66 upregulated genes common between pancreatic and liver cancer are in column 13
Figure 2.
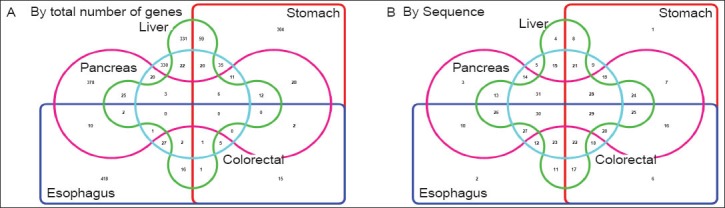
Venn diagram analysis of genes downregulated in five primary GI cancer sites. A color represents a particular primary site of GI cancer- for instance, pink is Pancreas, green is for liver, light blue for colorectum, red for stomach and dark blue for esophagus. In Panel A, Venn diagram method has been used to identify the number of genes that are shared between two or more cancers. For instance, cancers arising in the pancreas and liver share 25 common downregulated genes while 378 genes are unique to the pancreas. In Panel B, we have sequence numbers corresponding to the column numbers in Suppl. File 2 (website). Refer to the corresponding column number for the names of specific genes. For instance, the 25 downregulated genes common between pancreatic and liver cancer are in column 13
Upregulated genes
Table 1 shows the number of genes that were commonly upregulated in epithelial cancers arising from two or more primary GI sites (the genes that are differentially upregulated in GI cancers can be found in the website Suppl. File 1). Among the five GI sites, gastric cancer shared the most number of upregulated genes with the other four sites (only 51.8% of upregulated genes were unique to gastric cancer). In comparison, HCC shared the least number of upregulated genes with other sites (71.2% genes were unique to HCC).
Table 1.
Summary of common gene expression between two or more primary gastrointestinal primary cancer sites
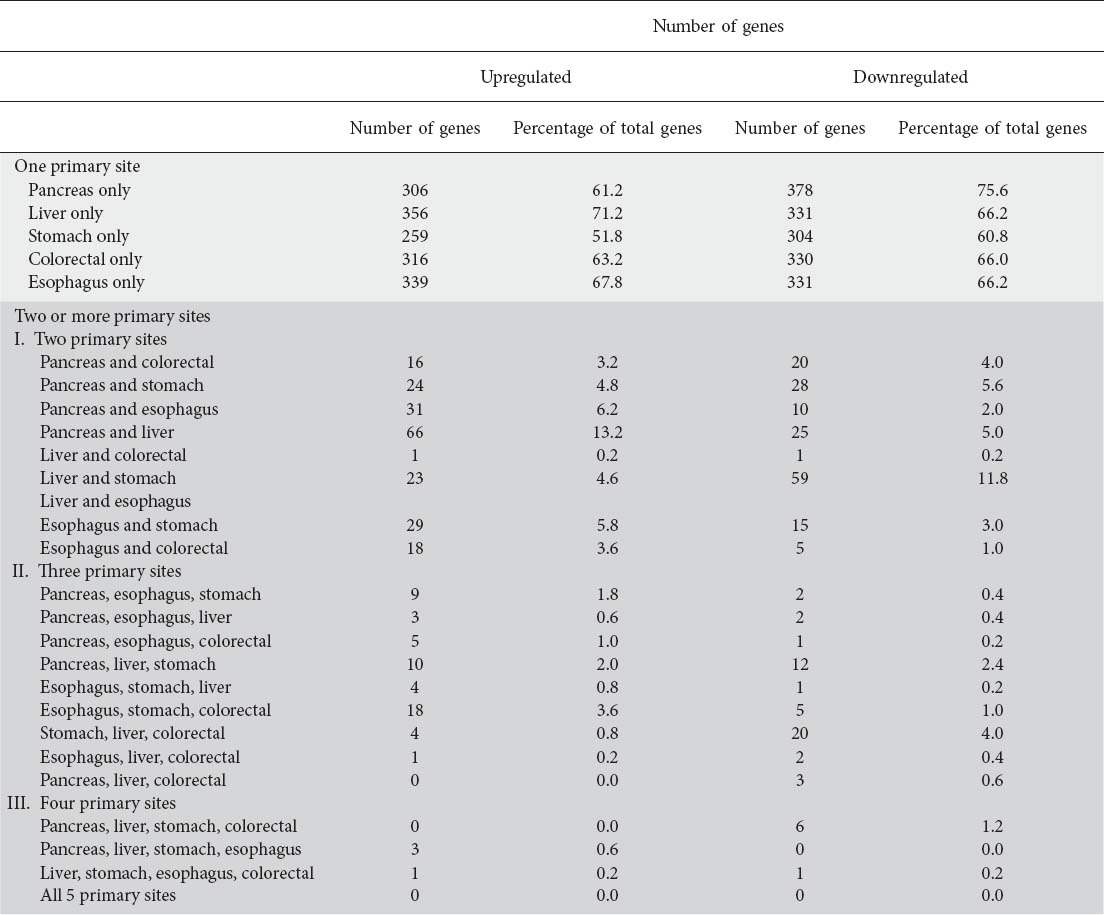
Pancreatic cancer and HCC had sixty six (13.2%) of upregulated genes in common, making them the two cancers most similar in this category. They were followed by pancreatic/esophageal cancer with 31 (6.2%) and esophageal/gastric cancer with 29 (5.8%) genes in common.
When we examined three primary GI sites that shared common upregulated genes, esophageal, gastric and colorectal cancer with 18 common genes (3.6% of total) were first. Pancreatic, HCC and gastric cancer with 10 common genes (2% of total) were second.
When we examined over expressed genes common to four primary GI sites, the pancreas, liver, stomach and esophageal cancers had three genes in common (0.6% of total). There was no gene upregulated in all five primary sites.
Downregulated genes
Gastric cancer shared the most number of downregulated genes with other cancers with only 60.8% unique genes, while pancreatic cancer shared the least (75.6% unique genes) (Table 1). The list of genes that are differentially downregulated in GI cancers can be found in the website Suppl. File 2.
With fifty nine genes (11.8% of total) commonly downregulated genes, HCC and gastric cancer were most similar, followed by pancreatic/gastric cancer with 28 (5.6% of total) and pancreatic cancer/HCC with 25 (5% of total) genes in common.
Gastric cancer, HCC and colorectal cancer had twenty genes in common making them the most similar trio. When comparing commonly downregulated genes in four primary GI sites, pancreatic, HCC, gastric and colorectal cancer had six genes in common making them the most similar quartet. There was no gene common to all five primary GI sites.
Functional classification of genes shared between different primary GI sites
Having identified the genes that were commonly up- or down-regulated in different primary GI cancers, we next sought to investigate the pathways that these common genes represented.
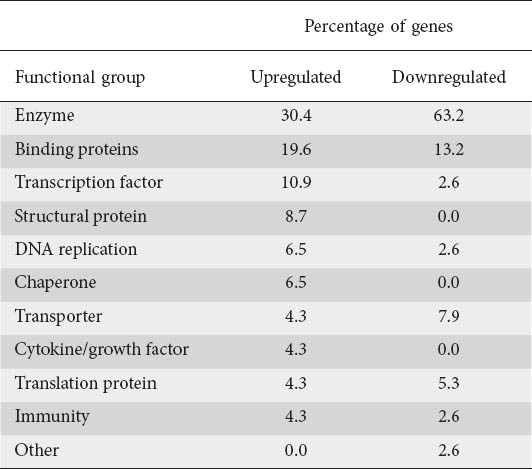
We chose genes that were shared between three or more GI primary sites for this analysis. We hypothesized that this would lead to an enrichment of genes (and therefore pathways) that were key in the development of cancer in GI and potentially in other non-GI cancers as well. To assign biological function to genes, we used the PANTHER program. Overall, we were able to identify the function of a gene in 90% of upregulated and 69% of downregulated genes that were common in three or more primary GI sites. We divided these genes into eleven functional groups, i.e. transporter proteins, enzymes, cytokines and growth factors, proteins (Excluding enzymes) involved in DNA replication, transcription factors, binding proteins, proteins involved in translation, chaperones, immunity associated proteins, structural proteins, and others (that did not fit into any of the previous ten groups). As a group, enzymes comprised the bulk of genes differentially expressed in cancer cells, making up 30.4% of upregulated and 63.2% of downregulated genes (website Suppl. Fig. 1 and 2). Binding proteins were the second most common group, accounting for 19.6% of upregulated and 13.2% of downregulated genes. The functional classification of the genes commonly up or downregulated in three or more primary GI cancer sites is summarized in Table 2.
Table 2.
Functional classification of genes common between three or more primary gastrointestinal malignancies
In order to investigate the cellular pathways regulated by genes common to three or more GI cancers, we used the GeneMania online program to investigate the interactions of these genes with other genes. For the upregulated genes, we noticed interactions with cell cycle regulators, Ras and Rho GTPases and components of the extracellular matrix. These genes were also involved in pathways that regulated cell division, adhesion, motility and assembly of the cell junctions (website Suppl. Fig. 4). The genes that were downregulated were involved in pathways that inhibited cell motility and cell growth, and regulated metabolism (including steroid, cholesterol, alcohol metabolism) and cellular biosynthesis (synthesis of glycolipids, lipoproteins and phosphatidylinositol). These genes (and their interacting partners) included several enzymes important in metabolic pathways including transferases, hydrolases and oxidoreductases (website Suppl. Fig. 4).
Next, we investigated whether the genes that were most commonly over or under expressed in GI cancers were also similarly expressed in non-GI cancers. For this, we chose genes that were common to four or more GI sites. Like before, we felt this would give us the best enrichment of genes commonly expressed in GI malignancies. Thus, we examined the expression of TMEM220, ACADVL, RNF222, ZZEF1, RILP, and G protein suppressor (GPS)2 all of which were downregulated in pancreatic, HCC, gastric and esophageal cancer compared to the corresponding normal tissue. Oncomine has twenty one categories of primary tumor sites. We analyzed the percentage of primary sites where each gene was underexpressed compared to the corresponding normal tissue. The resulting percentage was underexpression in 100%, 87.5%, 100%, 75%, 100%, 100% of primary non-GI cancer tissues compared to the corresponding normal tissue. For this analysis, we chose a P-value of 0.0001, a fold change of 2 or more and included top 10% of all genes. For GPS2 we chose a P-value of 0.05. From among the upregulated genes, we chose COL4A1 (upregulated in HCC, gastric, esophageal and colorectal cancer), UBE2C, OLFML2B and ANP32E (upregulated in pancreatic, HCC, gastric and esophageal cancer). They were significantly upregulated in cancer tissues compared to corresponding normal tissues in 90.5%, 95%, 100% and 76% of studies. Further, we searched the literature for role of the aforementioned genes in both GI and non-GI malignancies. UBE2C was the most upregulated gene in primary colon cancer and liver metastases in one study [1]. It was also upregulated nearly 150-fold in human thyroid cancer cell lines and in anaplastic thyroid cancer tissues compared to the non neoplastic thyroid cells [2]. Short interfering RNAs (siRNAs) against UBE2C when combined with inhibitors of the DR5/TRAIL pathway induced cell death in the cancer, but not in normal cells [3]. No data exists on functional role of OLFML2B, ANP32E in cancer. Among the downregulated genes, ACADVL (Acyl coenzyme-A dehydrogenase very long chain) expression was significantly downregulated in cervical squamous cell cancer [4] and adrenocortical carcinomas [5]. RILP (Rab7-interacting lysosomal protein) has been shown to be important for lysosome trafficking, which in turn was found to be a key determinant of tumor cell invasion [6]. GPS2 downregulation by siRNA stimulated proliferation in breast cancer cells, suggesting that it is a tumor suppressor [6]. There is currently no literature on the role of TMEM220, RNF222 and ZZEF1 in cancer.
Finally, we investigated the possibility of identifying novel inhibitors for the above identified genes. As an example, we chose UBE2C. The STITCH online database identifies chemicals that have or predicted to have an interaction with a given protein. These are linked to databases like PubChem where further information regarding the compound can be obtained. For UBE2C, there were two chemicals identified (Fig. 3).
Figure 3.
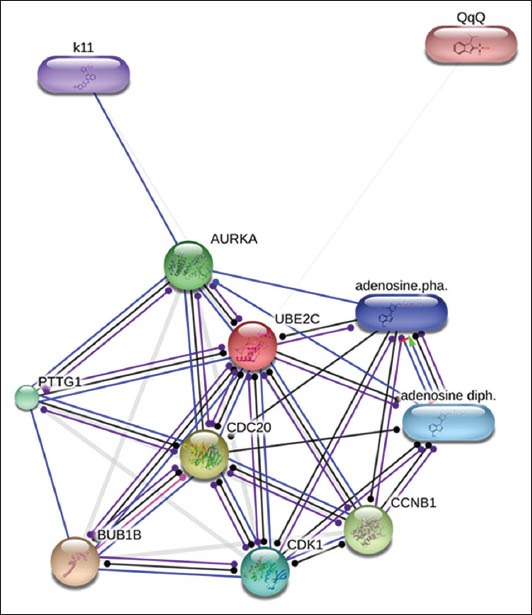
Identification of potential inhibitors of target genes using STITCH (http://stitch.embl.de/) online tool. Interaction of UBE2C with other proteins; thicker lines represent stronger interaction. Protein-protein interactions are in blue, chemical protein interactions in green and interactions between chemicals in red. Thus, k11 (CID 16731782) and QqQ (PubChem CID 2772579) are inhibitors of UBE2C
Discussion
In the present study, we compared five hundred of the most over- and under-expressed genes in five primary GI sites. The percentage of genes that were shared between two or more GI cancers ranged between 29-48% for the upregulated genes and 24-39% for the downregulated genes. About 7.5% of the five thousand genes were common to two primary GI sites, 1.8% to three primary GI sites and 0.2% to four primary GI sites. Genes encoding for enzymes, receptors and nucleic acid binding proteins were most commonly upregulated while those for enzymes, receptors, nucleic acid binding proteins and cytoskeletal proteins were most commonly downregulated in GI cancers.
Oncomine has been used previously to investigate the differential expression of specific genes in cancer including pore domain potassium channels, calcium channels, and RGS (regulators of G protein signaling) family of proteins [7-9]. Rhodes and colleagues in 2004 used Oncomine to identify a transcriptional profile that was common to most cancers and suggested that this likely reflected a common set of genes that were differentially overexpressed in most malignancies (compared to the normal tissue) [10]. Oncomine has also been used to identify gene signatures to classify human tumors [7,8,11]. In this study, we focused on GI malignancies. We used the advantages of large sample size and ability to compare gene expression across multiple studies to identify the top 1000 differentially expressed genes in different GI primary sites. We then investigated genes that were common between two or more of five GI primary sites. Once these were identified, we further investigated the pathways they are involved it. The genes identified in this study could potentially be targeted for development of novel therapeutics. We hypothesize that the resultant inhibitors would potentially be effective in malignancies arising from different GI primary sites. An interesting observation in the present study was that genes encoding enzymes were the most differentially altered in cancer cells compared to normal cells. This underlines the importance of alterations in cellular metabolism in cancer.
Previous studies have used microarray technology to identify therapeutic targets that were then verified by biologic studies. Turner and colleagues for instance, used high resolution microarray based comparative genomic hybridization to identify 40 genes that were significantly over expressed in triple negative breast cancer (TNBC). Of these, the authors demonstrated in vitro inhibition of breast cancer cell growth using an inhibitor to FGFR2, one of the oncogenes identified as being upregulated in TNBC [12]. Saito and co-workers used gene microarray to identify CD32 and CD25 as two genes that were highly expressed in leukemia-specific stem cells. The expression of these genes was however suppressed in normal hematopoietic stem cells [13]. These studies illustrate how microarray studies are very helpful to sift through large amounts of genetic information to identify targets that are commonly over or under expressed. The translation of genomic studies into therapeutic discoveries has been discussed in a recent article by Crews and colleagues [14]. The authors discuss how using data available in the public domain, researchers have identified genes and in turn potential drugs that targeted the differentially expressed genes. We used the STITCH database and identified two potential inhibitors of UBE2C, one of the genes shared between multiple GI cancers and upregulated in cancer cells. Further in vitro studies are needed to confirm the inhibitory activity of this drug on cancer cells.
In our study, we have not only identified genes that have previously been shown to play a role in GI malignancies (e.g. UBE2C) but also new ones about whom little is known regarding their role in cancer (e.g. TMEM220 and RNF222). Further studies are needed to investigate the role of these genes in the pathogenesis of GI cancers and as diagnostic, prognostic or therapeutic targets.
Thus, in summary we have used the Oncomine database to compare differential gene expression between different GI cancers to identify common genes that in turn could be useful as targets for diagnosis and therapy.
Summary Box.
What is already known:
Different cancers have similarities in gene expression patterns
Gastrointestinal (GI) tract arises from the endoderm
Multiple studies comparing gene expression in normal tissue vs. cancer arising from different parts of the GI tract have been performed, many of which are archived in the Oncomine database
What the new findings are:
Of the 5,000 most commonly differentially expressed genes in GI cancer arising from five different primary sites, nearly 7.4% were common to two primary sites, 1.8% to three and 0.2% to four primary GI sites
Subset analysis reveals that among the genes common to three or more GI cancers, those that are part of pathways regulating cell cycle, adhesion, motility and cell junction formation were upregulated while those that inhibited motility, growth and metabolism were downregulated
Genes that were expressed in cancers arising from three or more GI sites were also differentially expressed in a similar direction in most of the non-GI malignancies
Biography
University of Nebraska Medical Center, Omaha, NE, USA
Footnotes
Conflict of Interest: None
References
- 1.Takahashi Y, Ishii Y, Nishida Y, et al. Detection of aberrations of ubiquitin-conjugating enzyme E2C gene (UBE2C) in advanced colon cancer with liver metastases by DNA microarray and two-color FISH. Cancer Genet Cytogenet. 2006;168:30–35. doi: 10.1016/j.cancergencyto.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 2.Lee JJ, Au AY, Foukakis T, et al. Array-CGH identifies cyclin D1 and UBCH10 amplicons in anaplastic thyroid carcinoma. Endocr Relat Cancer. 2008;15:801–815. doi: 10.1677/ERC-08-0018. [DOI] [PubMed] [Google Scholar]
- 3.Wagner KW, Sapinoso LM, El-Rifai W, et al. Overexpression, genomic amplification and therapeutic potential of inhibiting the UbcH10 ubiquitin conjugase in human carcinomas of diverse anatomic origin. Oncogene. 2004;23:6621–6629. doi: 10.1038/sj.onc.1207861. [DOI] [PubMed] [Google Scholar]
- 4.Pan Z, Chen S, Pan X, et al. Differential gene expression identified in Uigur women cervical squamous cell carcinoma by suppression subtractive hybridization. Neoplasma. 2010;57:123–128. doi: 10.4149/neo_2010_02_123. [DOI] [PubMed] [Google Scholar]
- 5.Soon PS, Libe R, Benn DE, et al. Loss of heterozygosity of 17p13, with possible involvement of ACADVL and ALOX15B, in the pathogenesis of adrenocortical tumors. Ann Surg. 2008;247:157–164. doi: 10.1097/SLA.0b013e318153ff55. [DOI] [PubMed] [Google Scholar]
- 6.Steffan JJ, Cardelli JA. Thiazolidinediones induce Rab7-RILP-MAPK-dependent juxtanuclear lysosome aggregation and reduce tumor cell invasion. Traffic. 2010;11:274–286. doi: 10.1111/j.1600-0854.2009.01012.x. [DOI] [PubMed] [Google Scholar]
- 7.Williams S, Bateman A, O’Kelly I. Altered expression of two-Pore domain potassium (K2P) channels in cancer. PLoS One. 2013;8:e74589. doi: 10.1371/journal.pone.0074589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen R, Zeng X, Zhang R, et al. Cav1.3 channel α1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urol Oncol. 2013 doi: 10.1016/j.urolonc.2013.05.011. doi: 10.1016/j.urolonc. [DOI] [PubMed] [Google Scholar]
- 9.Sethakorn N, Dulin NO. RGS expression in cancer: oncomining the cancer microarray data. J Recept Signal Transduct Res. 2013;33:166–171. doi: 10.3109/10799893.2013.773450. [DOI] [PubMed] [Google Scholar]
- 10.Rhodes DR, Yu J, Shanker K, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A. 2004;101:9309–9314. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner N, Lambros MB, Horlings HM, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29:2013–2023. doi: 10.1038/onc.2009.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saito Y, Kitamura H, Hijikata A, et al. Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells. Sci Transl Med. 2010;2:17ra9. doi: 10.1126/scitranslmed.3000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crews KR, Hicks JK, Pui CH, Relling MV, Evans WE. Pharmacogenomics and individualized medicine: translating science into practice. Clin Pharmacol Ther. 2012;92:467–475. doi: 10.1038/clpt.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]