

Abstract
Purpose
Estrogen receptor-α (ERα) targeted therapies including tamoxifen (TAM) or Faslodex (ICI) are used to treat ER+ breast cancers. Up to 50% of tumors will acquire resistance to these interventions. Autophagy has been implicated as a major driver of antiestrogen resistance. We have explored the ability of hydroxychloroquine (HCQ), which inhibits autophagy, to affect antiestrogen responsiveness.
Experimental Design
TAM-resistant MCF7-RR and ICI-resistant/TAM cross-resistant LCC9 ER+ breast cancer cells were injected into mammary fat pads of female athymic mice and treated with TAM and/or ICI in combination with oral low-dose HCQ.
Results
We show HCQ can increase antiestrogen responsiveness in MCF7-RR and LCC9 cells and tumors, likely through the inhibition of autophagy. However, the combination of ICI+HCQ was less effective than HCQ alone in vivo, unlike the TAM+HCQ combination. Antiestrogen treatment stimulated angiogenesis in tumors but did not prevent HCQ effectiveness. The lower efficacy of ICI+HCQ was associated with ICI effects on cell-mediated immunity within the tumor microenvironment. The mouse chemokine KC (CXCL1) and IFNγ were differentially regulated by both TAM and ICI treatments, suggesting a possible effect on macrophage development/activity. Consistent with these observations, TAM+HCQ treatment increased tumor CD68+ cells infiltration, whereas ICI and ICI+HCQ reduced peripheral tumor macrophage content. Moreover, macrophage elimination of breast cancer target cells in vitro was reduced following exposure to ICI.
Conclusion
HCQ restores antiestrogen sensitivity to resistant tumors. Moreover, the beneficial combination of TAM+HCQ suggests a positive outcome for ongoing neoadjuvant clinical trials using this combination for the treatment of ER+ ductal carcinoma in situ lesions.
Keywords: estrogen receptor-α; chloroquine; tamoxifen; TAM; fulvestrant; ICI 182,780; antiestrogen resistant breast cancer; autophagy; angiogenesis; macrophages
Introduction
One out of every eight American women will develop invasive breast cancer over the course of her lifetime. An estimated 230,000 new cases of breast cancer are diagnosed annually in the USA (1). Of these cases, 70% will express estrogen receptor-α (ERα). ERα targeted therapies include the selective estrogen receptor modulator tamoxifen (TAM), the selective estrogen receptor downregulator Faslodex (fulvestrant, ICI), or aromatase inhibitors that block the conversion of androgens to estrogens. Even with the success of these drugs, some tumors fail to respond (de novo resistance) or acquire resistance over time (2-4).
Autophagy is a process by which a double membrane vesicle surrounds cellular contents, such as damaged organelles and misfolded or protein aggregates, and recycles the material through lysosomal degradation (5). Studies in breast cancer cells show that the induction of autophagy by various therapeutics is usually prosurvival (6-8). Furthermore, TAM and ICI both induce autophagy in ER+ breast cancer cells (6, 9-13). Antiestrogen resistant cell lines exhibit increased basal autophagy when compared with their antiestrogen sensitive parental cells (10). Inhibiting autophagy through autophagy-related gene 5 (ATG5) silencing potentiated antiestrogen-mediated cell death, indicating that antiestrogen stimulated autophagy is prosurvival and a critical mechanism of therapy resistance (10). Analysis of publically available human data sets indicates that autophagy related genes ATG5, ATG7, and p62 (SQSTM1) are elevated in early recurring breast cancer when compared with breast cancer that never recurs. Moreover, elevated p62 is significantly correlated with poor survival in breast cancer patients (Supplementary Figure 1), suggesting a role for autophagy in breast cancer reoccurrence (14-18).
HCQ is a lysotropic chloroquine derivative that accumulates within lysosomes, resulting in lysosome neutralization and the inhibition of autophagic flux. Originally applied as an antimalarial medication, the use of chloroquine (or chloroquine derivatives) to inhibitor autophagy is currently being explored as possible chemotherapeutic interventions for the treatment of cancer (19). Here, we have explored the possible beneficial effect of combining antiestrogen therapies with hydroxychloroquine (HCQ) for the treatment of antiestrogen resistant ER+ breast cancers. We used several cellular models of endocrine resistance: MCF7-RR [ER+, estrogen independent, TAM resistant, ICI sensitive cells; derived from MCF-7 cells selected against low serum and TAM (20, 21)], LCC9 [ER+, estrogen independent, ICI resistant, TAM cross-resistant cells; derived from the estrogen independent antiestrogen sensitive LCC1 cells by selection against ICI (22)], and ZR-75-1/ICI-R [ER+, ICI-resistant cells; derived from estrogen sensitive antiestrogen sensitive ZR-75-1 cells by selection against ICI]. We also used two different categories of antiestrogen therapy:tamoxifen, a selective estrogen receptor modulator (SERM) and ER partial agonist, and ICI, a selective estrogen receptor downregulator (SERD) and ER antagonist.
The combination of HCQ with TAM partially resensitized resistant MCF7-RR and LCC9 tumors, while a combination of ICI+HCQ was not as effective as HCQ given as a single agent therapy in treating ICI-resistant LCC9 orthotopic xenografts. Furthermore, we found that the tumor microenvironment plays an important role in the treatment of breast tumors with antiestrogens and HCQ. We show that monocytes that express ERα, and differentiated macrophages that express the estrogen and TAM responsive G-coupled protein receptor 30 (GPR30), may mediate some of the estrogen agonist and tumor-inhibitory activities of TAM. In contrast, the antineoplastic activity of ICI was reduced by its ability to inhibit monocyte differentiation and macrophage-mediated clearance of tumor cells. An ongoing clinical trial PINC (Preventing Invasive Neoplasia with Chloroquine) is investigating the ability of chloroquine alone or in combination with tamoxifen (in ER+ cases) to eliminate DCIS progenitor cells, highlighting the importance of our findings.
Material and Methods
Materials
The following materials were obtained as indicated: 4-hydroxytamoxifen and HCQ diphosphate salt (Sigma-Aldrich, St. Louis, MO); ICI 182,780 (Tocris Bioscience, Ellisville, MO); Improved Minimal Essential Medium (IMEM; Gibco Invitrogen BRL, Carlsbad, CA); fetal bovine serum (FBS) and bovine calf charcoal stripped serum (CCS) (Equitech-Bio Inc, Kerrville, TX); crystal violet (Fisher Scientific, Fairlawn, NJ). Growth factor reduced Matrigel (BD Biosciences, Franklin Lakes, NJ). Antibodies were obtained from the following sources: LC3A/B, VEGFR2, phospho-VEGFR2 (Cell Signaling Technology); p62 (Western: BD Biosciences; IHC: MBL International); CD68 (AbD Serotec); CD31 (Abcam); ERα, β-actin, and polyclonal and HRP-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA).
Cell Culture
MCF7-RR and LCC9 human breast carcinoma cells were grown in phenol-red free IMEM media containing 5% charcoal-treated calf serum (CCS). MCF7 and ZR-75-1/ICI-R cells were grown in phenol-red IMEM media containing 5% fetal bovine serum. MCF7 cells were obtained from Dr. Marvin Rich at the Michigan Cancer Foundation. MCF7-RR cells were obtained from Indiana University of Pennsylvania (Indiana, Pennsylvania, USA) (21). LCC9 and ZR-75-1/ICI-R cells were generated by our group at Georgetown University (Washington, DC, USA) (22). Cells were grown at 37°C in a humidified, 5% CO2:95% air atmosphere.
Crystal Violet Assay
Human breast cancer cells (3 × 103 cells/mL) in IMEM containing 5% CCS were plated in 24-well tissue culture plates. On day 1 after plating, and every 3 days, thereafter cells were treated with varying doses (10 nM-1000 nM) of either tamoxifen or ICI and 1 μM HCQ. On day 6, media was aspirated and cells were stained with crystal violet. Cells were permeabilized using citrate buffer and absorbance was read at 480 nm on a plate reader.
Western Blot Hybridization
MCF7-RR and LCC9 cells were solubilized by sonication in RIPA lysis buffer. Proteins were size fractionated by polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane. Non-specific binding was blocked by incubation for one hour at room temperature with Tris-buffered saline with 5% powdered milk and 1% Triton X-100. Membranes were incubated overnight at 4°C with primary antibodies, followed by incubation with polyclonal horseradish peroxidase (HRP)-conjugated secondary antibodies (1:2000) for 1 h at room temperature. Immunoreactive products were visualized by chemiluminescence (SuperSignal Femto West, Pierce Biotechnology, Rockford, IL) and quantified by densitometry using the ImageJ digital densitometry software (http://rsbweb.nih.gov/ij/).
Orthotopic human breast cancer xenografts
Five week old, intact, athymic nude mice (Harlan Laboratories, Fredrick, MD) were injected with 1 × 106 LCC9 or MCF7-RR cells in Matrigel:IMEM (50:50%). Cells were injected orthotopically through a 3-mm skin cut into 4 different mammary fat pads per mouse (one abdominal and one thoracic mammary gland on each side). For the MCF7-RR xenografts, mice also received a low dose 17β-estradiol pellet (0.36 mg, 60-day release; Innovative Research of America, Sarasota, FL) implanted subcutaneously under isoflurane anesthesia. Tumors grew to approximately 25-35 mm2 before therapy initiation; 1 to 2 tumors per mouse successfully reached the required size for treatment initiation. Mice were treated with hydroxychloroquine (HCQ), TAM, ICI, or a combination of an antiestrogen and HCQ. HCQ was administered in the drinking water; 0.29 mg/ml results in approximately 1-2 mg daily dose per mouse (23). Tamoxifen was administered in a standard rodent chow 5053 diet containing 400-ppm tamoxifen citrate (Harlan-Teklad, Madison, WI), which corresponds to a TAM dose of approximately 32 mg/kg/day. ICI was administered by subcutaneous injection 0.5 mg/mouse/week in 70% EtOH/30% NaCl. Mice were euthanized after 5 weeks of treatment, and tumors removed at necropsy, fixed in neutral buffered 10% formalin, and processed using routine histological methods. The efficacy of antiestrogen treatment was confirmed by staging of the estrus cycle; mice on TAM or ICI therapy did not cycle.
Tissue Staining and Immunohistochemistry (IHC)
Mammary tumors were fixed in 10% phosphate buffered formalin for 24 h prior to embedding in paraffin. Embedded tissues were cut into 5 μm thick sections and stained with hematoxylin and eosin (H&E) to determine morphology. Immunostaining was performed with an antibody to CD31 (1:100), pVEGFR2 (1:100), p62 (1:1000), LC3 (1:100), CD68 (1:100) using the streptavidin-biotin method. Stained sections were visualized and photographed. Since CD31 and pVEGFR2 staining was heterogeneous, data were quantified using a common method of angiogenic marker quantification defined as “hotspot” quantification. In summary, three “hotspots” in the tumor were selected, the number of vessels/micro-vessels in each “hotspot” was counted at 200x magnification, and a final score was expressed as the mean vessel density (24, 25). The longest diameter of each CD31+ blood vessel was measured using ImageJ and shown as average vessel diameter (Supplemental Figure 3B).
Cytokine Analysis
Plasma from treated mice was collected at necropsy and immediately frozen. Quansys Biosciences (Logan, UT) Q-Plex Array™ kits were used to measure the following mouse cytokines and chemokines: IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-12p70, IL-17, TNFα, IFNγ, MCP1, RANTES, Eotaxin, KC, MDC, TARC, TCA3.
Macrophage Differentiation and Cytotoxicity Assay
U937 human monocyte cells were treated in the presence of IFNγ (to differentiate monocytes into activated macrophages) and either 100 nM TAM, 1 μM HCQ, 100 nM ICI, or the combination of antiestrogen and HCQ for 72 hours. Macrophages were counted and plated with 70% confluent MDA-MB-231 (triple negative, antiestrogen non-responsive breast cancer cells) in a 1:5 ratio of macrophages to cancer cells for 72 hours. Macrophage killing was then assessed by RTS-ACEA® through electrical impedance (26).
Statistics
Data are presented as the mean ± standard error of the mean (SEM). For most studies, Student’s t-test (pairwise) or one way analysis of variance (ANOVA) followed by Bonferoni post hoc tests (groupwise) was used (Prism software). Statistical differences for in vivo tumor area were evaluated by Dr. Fang in the Department of Biostatistics, Bioinformatics and Biomathematics at Georgetown University Medical Center. Tumor volumes were obtained from measurements of the longest perpendicular axes. We use the weighted F-test for comparison between two groups with two tumor volumes, since the behavior of two tumors growing in a single mouse are not independent events (27). All computation was performed in the R-environment. There are 26 mice for LCC9 xenografts, and 19 mice for MCF7RR xenografts. Criterion for statistical significance was set at p ≤ 0.05.
Results
Antiestrogen sensitive MCF7 cells and antiestrogen resistant MCF7-RR (TAM resistant), ZR-75-1 ICI-R (ICI resistant), and LCC9 (ICI resistant/TAM cross-resistant) breast cancer cells were plated in 24-well dishes and treated with vehicle, 1 μM HCQ, and/or various concentrations of TAM or ICI (vehicle, 10 nM, 100 nM, 1000 nM) for 6 days. The effect on cell density was determined by crystal violet assays (28). The combination of antiestrogen and HCQ potentiated TAM and ICI-mediated cell death in endocrine sensitive MCF7 cells (Figure 1A). Furthermore, HCQ and antiestrogen therapy inhibited LCC9 cells (Figure 1B), MCF7-RR (Figure 1C), and ZR-75-1 ICI-R (Figure 1D). Moreover, the addition of HCQ resulted in increased LC3-II formation (lapidated form of LC3 that is a marker of autophagosome formation) and accumulation of p62 (marker of autophagosomal flux) in both MCF7-RR (Figure 1E) and LCC9 (Figure 1F) as determined by Western blot hybridization. Increased LC3-II and p62 expression is indicative of inhibited autophagic flux resulting in the cellular accumulation of autophagosomes. Treatment of MCF7-RR cells with TAM or LCC9 cells with TAM or ICI resulted in increased LC3-II with a corresponding decrease in p62 levels confirming previous studies that antiestrogens stimulate autophagic flux (10).
Figure 1.
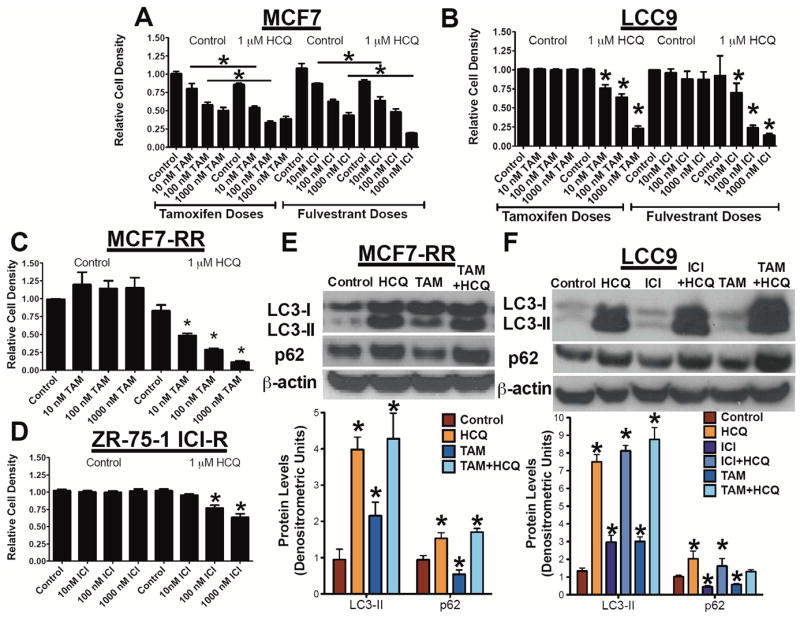
Hydroxychloroquine restores antiestrogen responsiveness in vitro. A. MCF7 cells were treated with 1 μM HCQ, and/or various doses (vehicle, 10 nM, 100 nM, 1000 nM) of either TAM or ICI for 6 days. Relative cell density was determined by crystal violet assay. B. LCC9 cells were treated with 1 μM HCQ, and/or various doses (vehicle, 10 nM, 100 nM, 1000 nM) of either TAM or ICI for 6 days and cell density determined by crystal violet assay. C. MCF7-RR cells were treated with 1 μM HCQ, and/or various doses (vehicle, 10 nM, 100 nM, 1000 nM) TAM for 6 days. Relative cell density was determined by crystal violet assay. D. ZR-75-1 ICI-R cells were treated with 1 μM HCQ, and/or various doses (vehicle, 10 nM, 100 nM, 1000 nM) ICI for 6 days. Relative cell density was determined by crystal violet assay. MCF7-RR (E) or LCC9 (F) cells were treated with 1 μM HCQ, 100 nM TAM or ICI, or a combination of antiestrogen and HCQ for 72 hours. Cells were harvested and Western blot hybridization was used to confirm levels of autophagy related proteins LC3A/B and p62. Equivalence of protein loading onto gels was confirmed by measuring β-actin expression. n=3, *p<0.05.
LCC9 and MCF7-RR cells were orthotopically injected into the mammary fat pads of female athymic mice. Tumors were grown to 20-35 mm2 before treatment with HCQ and/or TAM (MCF7-RR), or with HCQ and/or TAM or ICI (LCC9). In LCC9 xenografts TAM+HCQ was most effective at reducing initial tumor growth (Figure 2A and 2B) but after 3 weeks of treatment HCQ alone was just as effective at inhibiting LCC9 as the TAM+HCQ combination. Unexpectedly, the combination of ICI+HCQ was less effective than either TAM+HCQ or HCQ treatment alone. TAM or ICI treatment alone had no significant difference in tumor area when compared with control tumors. When tumor growth was measured as wet weight at necropsy, LCC9 tumors treated with HCQ alone, TAM+HCQ, or ICI+HCQ had significantly reduced tumor wet-weight when compared with control tumors (Figure 2C). However, HCQ alone was significantly more effective than ICI+HCQ, suggesting a potentially antagonist interaction between ICI and HCQ. In MCF7-RR orthotopic xenografts, control tumors continued to grow whereas TAM+HCQ significantly reduced tumor size (Figure 2D and 2E). HCQ or TAM alone had no significant effect on MCF7-RR tumor growth when compared with controls. The combination of TAM+HCQ significantly reduced MCF7-RR tumor wet weight when compared with control treated tumor weight measured at necropsy (Figure 2F).
Figure 2.

Low-dose oral HCQ resensitizes resistant breast tumors to antiestrogen therapy. A. LCC9 orthotopic tumors were grown to 25-35 mm2 before treated with TAM, ICI, HCQ, HCQ+ICI, or HCQ+TAM for 5 weeks. Tumors were measured weekly with calipers; % change in tumor area (A) and tumor area (B) was calculated. C. LCC9 tumor weight upon completion of study. MCF7-RR orthotopic tumors were untreated (control) or treated with TAM, HCQ, or HCQ+TAM for 5 weeks. Tumors were measured weekly with calipers and % change in tumor area (D) or tumor area growth curves (E) were calculated. F. Average wet weight of MCF7-RR upon sacrifice.
Formalin-fixed LCC9 tumors were embedded in paraffin and cut into 5 μm sections. Tumor sections were stained with specific antibodies against either LC3 (Figure 3A) or p62 (Figure 3B) using an avidin-biotin technique that reacts with peroxidase-conjugated streptavidin substrate to determine the effects of treatments on autophagy markers. LCC9 tumors from mice treated with ICI, TAM, or HCQ show elevated LC3A/B staining (Figure 3C). Measuring autophagic activity using immunohistochemistry can be challenging often because it is difficult to differentiate between LC3 and LC3-II by IHC in tissue sections. However, the high level of magnification (1000×) shows positive LC3 staining forming puncta-like structures broadly consistent with autophagy induction. Tumors from animals treated with ICI or TAM have reduced p62 expression, consistent with increased autophagic flux (Figure 3C). In contrast, tumors from mice treated with HCQ have elevated p62 expression, suggesting a block in the later stages of autophagy (29, 30). To corroborate the systemic effect of HCQ on autophagy, uterine tissue from control mice or mice treated with HCQ were isolated and Western blot hybridization was performed on protein lysates for LC3-II and p62 levels. As shown in Figure 3D, HCQ dosing increases LC3-II formation and results in the accumulation of p62, suggesting HCQ systemically inhibits autophagy.
Figure 3.
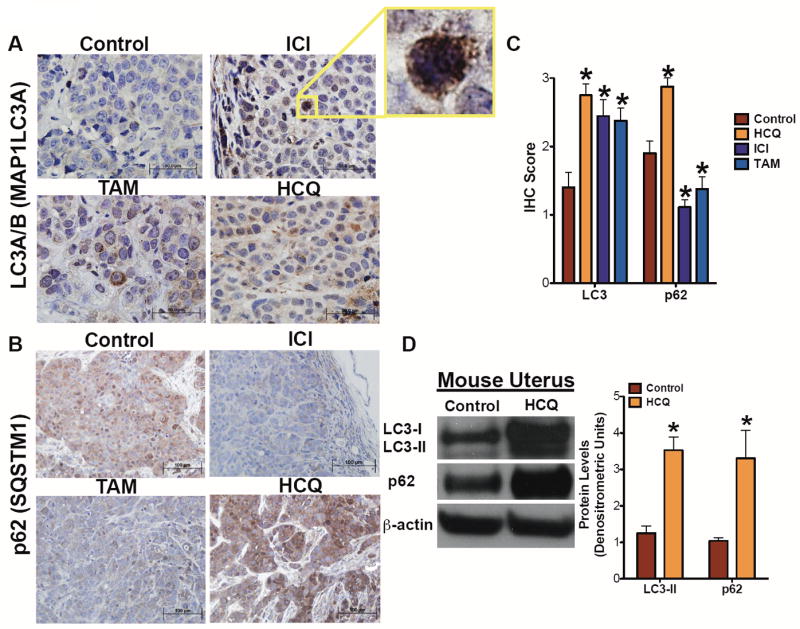
Immunohistochemical evaluation of autophagy in treated tumors. LCC9 tumor sections were stained using LC3A/B (A) or p62 (B) antibodies. LC3A/B is visualized at 1000× while p62 is visualized at 400×. C. Quantification of LC3 and p62 IHC. Stained positive cells were scored and averaged 3 frames per tumor. n=5-8 tumors, *p<0.05. D. Uterine tissue from control and HCQ treated mice were harvested and Western blot hybridization was used to confirm levels of autophagy related proteins LC3A/B and p62. Equivalence of protein loading onto gels was confirmed by measuring β-actin expression. n=4, *p<0.05.
To confirm antiestrogen drug effectiveness, LCC9 paraffin embedded tumor sections were stained for estrogen receptor-α (ERα) or progesterone receptor (PR) (Supplementary Figure 2). ICI reduces ERα staining and PR staining in LCC9 tumors, consistent with the known effects of this drug. TAM treatment resulted in a cytoplasmic diffuse distribution of ERα and reduced PR staining, consistent with observed drug activity.
Treatment of LCC9 cells with HCQ, ICI, or TAM in vitro increased pVEGFR2 expression determined by Western blot hybridization (Supplementary Figure 3A), suggesting that antiestrogen therapy stimulates pro-angiogenic signaling in drug-resistant breast cancer cells. Thus, LCC9 tumor sections were stained with antibodies against either CD31 (an endothelial cell marker) or phosphorylated vascular endothelial growth factor receptor 2 (pVEGFR2). TAM and ICI treatment increased CD31-positive vessel staining (Figure 4A and 4B) and elevated levels of pVEGFR2 (Figure 4C and 4D), implying increased angiogenesis. To confirm the effect of antiestrogen therapy on angiogenesis, mice were injected with 500 μL of matrigel between the abdominal wall and the skin to form a matrigel plug. Mice were either untreated (as control) or treated with TAM, ICI, HCQ, ICI+HCQ, or TAM+HCQ. After five days, the plug was removed, fixed in formalin, and embedded in paraffin. Sections of matrigel plug were stained with hematoxylin and eosin (H&E), as shown in Figure 5A. Mice treated with ICI showed increased number of infiltrating cells into the matrigel plug (Figure 5B), which was not observed in matrigel plugs from animals treated with TAM. However, treatment with HCQ in combination with either antiestrogen had no effect on CD31 staining or pVEGFR2 staining, suggesting that the stimulation of angiogenesis likely does not explain the differential effects observed between ICI+HCQ and TAM+HCQ treatment.
Figure 4.
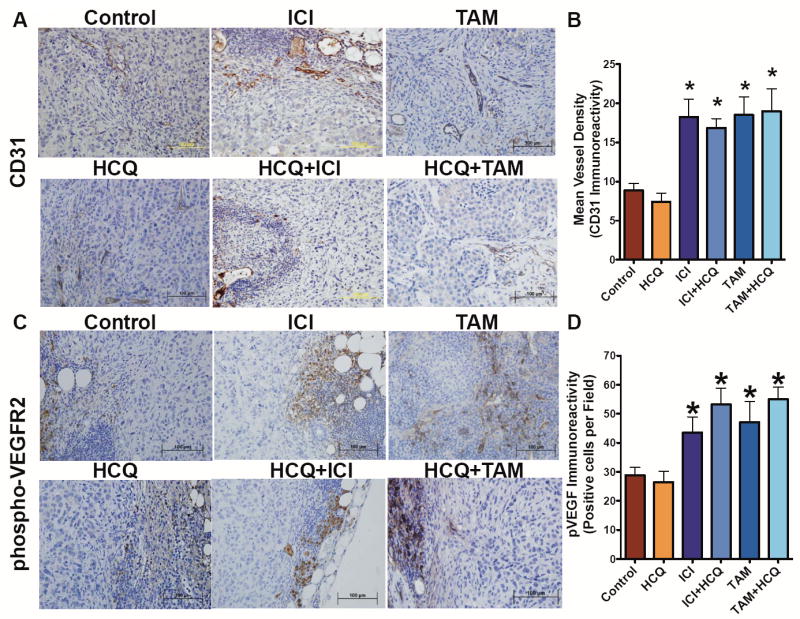
Antiestrogen therapy stimulates angiogenesis. A. LCC9 tumor sections from tumor treated with Control, HCQ, ICI, ICI+HCQ, TAM, or TAM+HCQ were stained for CD31 a marker of endothelial cells to visual blood vessels and quantified as average vessels per section (B). C. LCC9 tumors were also stained for pVEGFR2, visualized at 400x, and quantified as average positive cells per field (D). n= 6-10, *p<0.05
Figure 5.
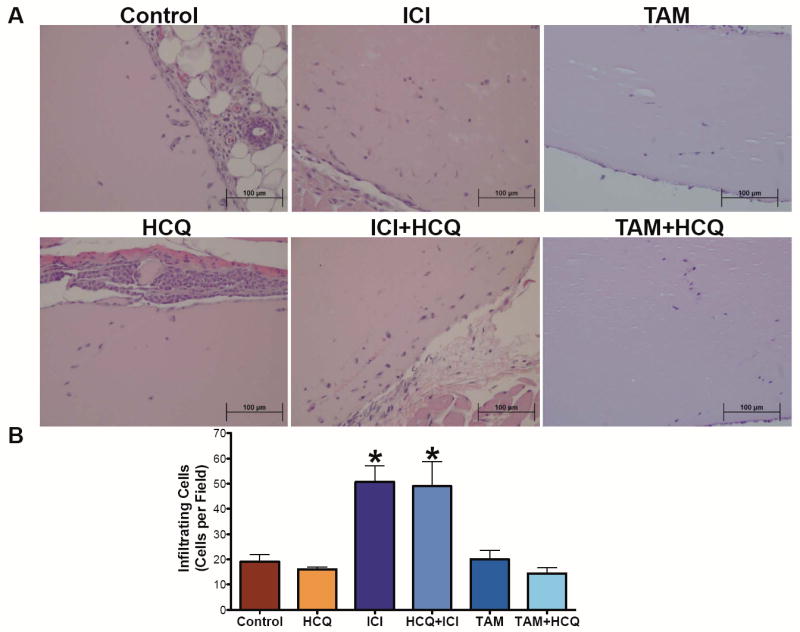
ICI treatment affects the extent of matrigel plug cell invasion. A. Matrigel plug was injected subcutaneous in mice treated with Control, HCQ, ICI, ICI+HCQ, TAM, TAM+HCQ. After 5 days, plugs were fixed with formalin and embedded in paraffin. Matrigel plug sections were stained with H&E to show infiltrating cells. B. Infiltrating cells into the matrigel plugs were quantified as cells per section. n=3-6 matrigel plugs, *p<0.05
Cytokine production and reticuloendothelial cell infiltrates are common in some breast cancers (31). Thus, blood was collected at the time of euthanasia to measure various cytokine concentrations. We measured the mouse cytokines and chemokines IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-12p70, IL-17, TNFα, IFNγ, MCP1, RANTES, Eotaxin, KC, MDC, TARC, and TCA3 using Q-Plex® Array kits from Quansys Biosciences (Supplementary Figure 4-6). While variability in plasma cytokines is high, several cytokine trends are evident between treatment groups. Of particular interest, the mouse chemokine KC (human analog: chemokine C-X-C motif ligand 1 (CXCL1)) is elevated in the plasma of TAM and TAM+HCQ mice and decreased in ICI and ICI+HCQ treated mice. Also, IFNγ is reduced in ICI and ICI+HCQ treated animals.
Since CXCL1 is expressed by macrophages and IFNγ stimulates macrophages, we investigated whether there were differential effects of antiestrogens and HCQ on macrophages. LCC9 tumor sections were stained with an antibody against the macrophage marker CD68 (Figure 6A). As quantified in Figure 6B, ICI and ICI+HCQ decreased peripheral tumor macrophage content when compared with control tumors. Moreover, TAM+HCQ treated mice significantly increased infiltrating CD68-positive cells when compared with control mice, indicating increased tumor macrophage infiltration. Next, U937 human monocyte cells were differentiated into macrophages in vitro in the presence of IFNγ and protein lysates from the precursor monocytes and differentiated macrophages were collected. As shown in Figure 6C, MCF7-RR, LCC9, U937 undifferentiated monocytes, and U937 differentiated macrophages were studied to confirm the presence of ERα and/or G-coupled protein receptor 30 (GPR30). The precursor U937 monocyte cells do not express GPR30 but express ERα, albeit at a lower level than the breast cancer cells. Conversely, differentiated U937 macrophage cells do not express detectable ERα but express higher levels of GPR30 than MCF7-RR, LCC9, or their parental U937 monocyte cells.
Figure 6.
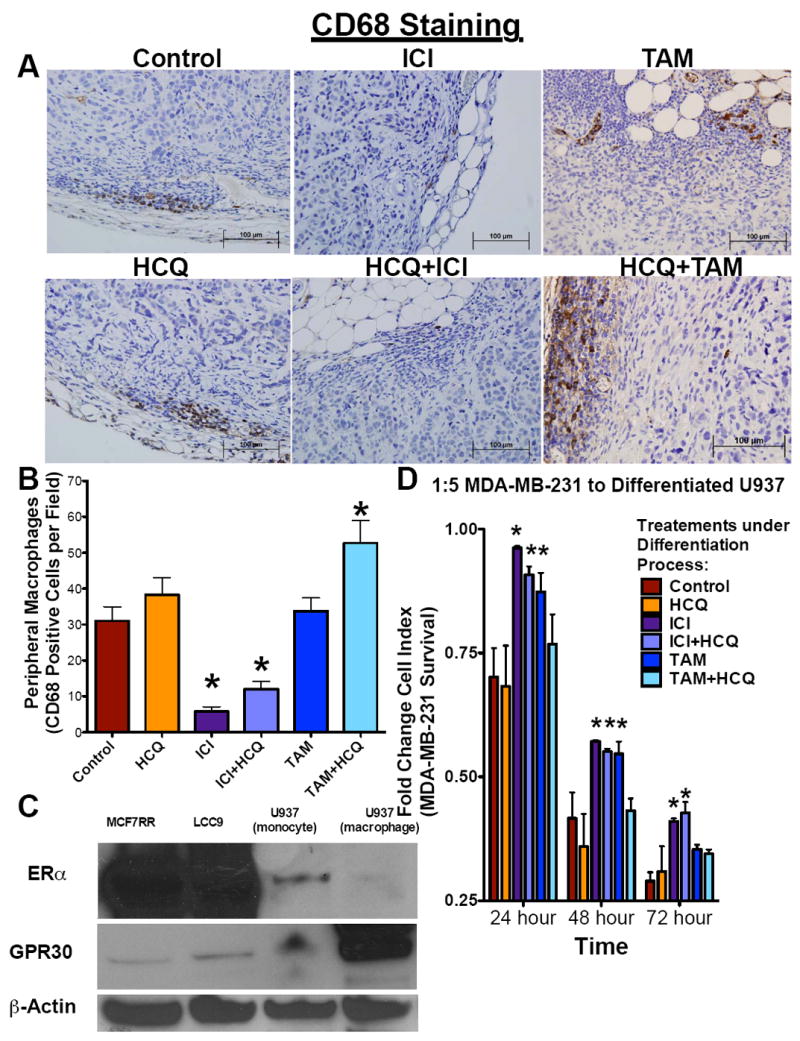
Systemic ICI treatment inhibits macrophage activation. A. LCC9 tumor sections from tumor treated with Control, HCQ, ICI, ICI+HCQ, TAM, or TAM+HCQ were stained for CD68 a marker of macrophages. B. Peripheral macrophages were quantified as cells per section. n=4-8; *p<0.05 C. ERα and GPR30 expression was determined in LCC9, MCF7-RR, U937 (undifferentiated monocytes), and U937 (differentiated macrophages) protein lysates by Western blot hybridization. D. U937 cells differentiated into macrophages in the presence of vehicle control, HCQ, ICI, ICI+HCQ, TAM, or TAM+HCQ were added to pre-plated MDA-MB-231 ER- breast cancer cells (1:5 ratio macrophages: cancer cells) and cell index was measured by electrical impedance over 72 hours. n=3; *p<0.05
To determine if antiestrogens or HCQ affect macrophages’ tumor cell killing capacity, U937 cells were differentiated in the presence of IFNγ and vehicle control, 1μM HCQ, 100 nM ICI, 1μM HCQ+100 nM ICI, 100 nM TAM, or 1μM HCQ+100 nM TAM for 72 hours. Macrophages were then collected, resuspended in fresh media to remove drug, and added to pre-plated MDA-MB-231 breast cancer cells. Use of these ER-negative cells prevented any remaining antiestrogen from inhibiting the target cells. Monocytes, differentiated into macrophages in the presence of ICI, ICI+HCQ, and TAM, had a decreased breast cancer cell killing capacity at 24 hours and 48 hours after plating when compared with their vehicle treated controls (Figure 6D). However, in the presence of HCQ or TAM+HCQ, macrophages had no significant difference in their cell killing capacity when compared with the vehicle-treated control macrophages. Seventy-two hours after the addition of macrophages, only macrophages in the presence of ICI and ICI+HCQ had significantly reduced breast cancer cell killing capacity as measured by cell index.
Discussion
Breast cancer remains the most prevalent cancer in women, with the majority of these tumors expressing ERα. Resistance to endocrine therapies remains a critical limitation in their ability of these agent to cure some patients. Autophagy is a key pathway in the development of endocrine resistance in breast cancer, and targeting autophagy can reverse antiestrogen resistance (6). Chloroquine, an anti-malarial drug, inhibits autophagy by preventing degradation of autolysosomes. Moreover, chloroquine derivatives, such as HCQ, in combination with anti-neoplastic chemotherapeutic drugs or radiotherapy treatments inhibit multiple cancer cell types (32, 33). We now show that using chloroquine-derived therapies in combination with antiestrogens increased the sensitivity of resistant breast cancer cells to endocrine therapies (Figure 1A-C).
Resensitization to antiestrogens is characterized by an increase in LC3-II levels, the lipidated form of LC3 that is found in the autophagosomal membrane, implying an increase in the rate of autophagy initiation. In contrast, increased levels of p62 indicate autophagosome accumulation (Figure 1E-F) leading to undegraded autophagosome accumulation in the cytoplasm (34), suggesting that the later steps in autophagy are not completed. When measured with changes in LC3, decreased p62 generally indicates that the autolysosome and its cargo have been degraded and that autophagic flux is intact. LCC9 or MCF7-RR cells treated with either TAM or ICI show increased LC3-II and reduced p62 expression, consistent with prior reports that endocrine-targeting therapy induces autophagic flux (6, 10, 35). A synthetic quinine analog often used for chloroquine-resistant malarial cases, mefloquine (Lariam®), was also shown to inhibit autophagy and induce cell death in MCF7 antiestrogen sensitive breast cancer cells when given as a single agent (36). Chloroquine prevented ductal carcinoma in situ xenografts’ outgrowth in athymic mice (37, 38) and inhibited N-methyl-N-nitrosurea-induced mammary carcinogenesis, suggesting chloroquine-based therapy as a possible agent in the prevention of initial premalignant lesions from progressing to breast cancer (39).
We show that oral low-dose HCQ given to tumor-bearing mice in combination with an antiestrogen resulted in the resensitization of resistant MCF7-RR (Figure 2A and 2C) and the potentiation of antiestrogen-mediated cell death in LCC9 breast tumors (Figure 2B and 2D). Overall, HCQ is likely to be an effective chemotherapeutic agent in combination with specific antiestrogen therapy for the treatment of some ER+ breast cancers. Nonetheless, since established tumors were not eliminated completely, i.e. “cured”, during the 5-week treatment, improvements in overall response rates to HCQ in adjuvant settings may be meaningful but modest. The efficacy of HCQ in prevention remains to be evaluated with the results of the ongoing Preventing Invasive Breast Neoplasia with Chloroquine (PINC) clinical trial. Importantly, since increased LC3A/B and p62 were observed in LCC9 tumors from mice treated with HCQ (Figure 3), consistent with an inhibition of autophagy, changes in expression of these markers in tumors may be worth evaluating as possible biomarkers for predicting treatment responses in patients.
While a combination of both HCQ+TAM and HCQ+ICI resensitized resistant breast cancers to therapy, the combination of HCQ and ICI was less effective at reducing tumor burden than the combination of TAM+HCQ at the doses tested. We hypothesized that systemic ICI therapy negatively affects the tumor microenvironment to reduce effectiveness of the HCQ+ICI combination. LCC9 tumors from mice treated with ICI, ICI+HCQ, TAM, or TAM+HCQ each displayed increased CD31 (Figure 4A-B) and phospho-VEGFR2 (Figure 4C-D) immunoreactivity. Thus, antiestrogens can stimulate tumor angiogenesis in endocrine-resistant tumors and HCQ has no effect on this response. Moreover, LCC9 cells treated with antiestrogens in vitro displayed increased expression of phospho-VEGFR2 (Supplementary Figure 3), suggesting that the effect of TAM and ICI on angiogenesis is mediated directly by the tumor cells. Since both TAM and ICI stimulated angiogenesis, increased blood vessel formation is unlikely to explain the differential antitumor effects observed between TAM+HCQ and ICI+HCQ treatments. Previous reports showed that TAM but not ICI increased VEGFR2 expression in antiestrogen sensitive MCF7 cells, further suggesting that the increase in angiogenesis observed in both TAM and ICI treated tumors is due to a direct effect mediated by the tumor epithelial cells (40). To determine the effect of antiestrogen therapy on cell invasion, standard matrigel plug assays were performed (Figure 5). While both TAM and ICI stimulated tumor angiogenesis (Figure 4) only ICI and ICI+HCQ treated mice exhibited increased cell invasion into their matrigel plugs. These data suggest that ICI and TAM have different effects on the tumor microenvironment.
Interestingly, analysis of circulating cytokines and chemokines in mice treated with HCQ and antiestrogens indicate that ICI and TAM may have a differential effect on the macrophage population (Supplementary Figure 4-6). Macrophages can be classified as M1-like (cytotoxic) or M2-like (immunosuppressive) and often serve opposing roles, highlighting the plasticity of these cell types (41). Estrogen was previously shown to affect the monocyte-macrophage system (42, 43), suggesting that antiestrogen therapy may also perturb these cells. When LCC9 tumors were stained for the macrophage marker CD68 (Figure 6A), mice treated with ICI and ICI+HCQ showed decreased staining when compared with other treatments (Figure 6B). Thus, ICI appears to effect negatively macrophage development and/or chemotaxis. Moreover, circulating levels of IFNγ in mice treated with ICI and ICI+HCQ were reduced when compared with TAM and TAM+HCQ treated mice. IFNγ can increase development of the M1-like cytotoxic macrophages (41), suggesting that systemic ICI treatment may reduce the cell-killing capacity of macrophages. As shown in Figure 6C, human monocytes express ERα albeit at a reduced level than the breast cancer cells, while ERα expression is lost in the macrophages. Moreover, macrophages have elevated GPR30 when compared with their parental monocyte cells and breast cancer cells. GPR30 is a G-coupled protein receptor that may act as an estrogen receptor and promotes cellular growth and activation. Tamoxifen’s partial estrogen agonist activity was shown to also activate GPR30 (44), which may explain in part the elevated macrophage number in the tumors from TAM+HCQ treated animals.
To confirm the effect of antiestrogen treatment on macrophage development and cytotoxicity, U937 monocytes were differentiated into macrophages in the presence of IFN and either ICI, TAM, HCQ, HCQ+TAM, or HCQ+ICI. Macrophages that were differentiated in the presence of ICI, ICI+HCQ, or TAM had decreased tumor cell cytotoxicity, indicating that systemic antiestrogen treatment reduces macrophage activity (Figure 6D). Furthermore, only ICI and ICI+HCQ treated macrophages had a significantly higher cell index (reduced cytotoxicity) at 72 hours post macrophage addition to breast cancer cells, while TAM and TAM+HCQ produced no significant difference in cell index when compared with vehicle treated controls. These data suggest that ICI but not TAM had deleterious effects on macrophage distribution, activation, and/or cytotoxicity that may result in the potentially antagonist interaction seen with the ICI + HCQ combination therapy.
An ongoing clinical trial is examining the effect of combining TAM and chloroquine for the treatment of ER+ ductal carcinoma in situ (DCIS) patients: Preventing Invasive Breast Neoplasia with Chloroquine (PINC) [See clinicaltrials.gov/show/NCT01023477]. Pre-clinical data showed that chloroquine treatment inhibited DCIS cell tumorgenicity in NOD/SCID mice, indicating the possibility of using chloroquine as a chemopreventive drug for breast cancer (38). In this study, we show that HCQ in combination with antiestrogens potentiates endocrine therapy sensitivity in acquired resistant ER+ breast tumors. More importantly, this study highlights a potentially non-beneficial effect of Faslodex (ICI) on the tumor microenvironment that reduces the value of combining chloroquine-based therapies with Faslodex. Future clinical trial study designs may consider using Tamoxifen as the preferred antiestrogen to optimize the effects of an antiestrogen in the tumor microenvironment. In our studies, a combination of HCQ + TAM was more effective than either drug alone.
Supplementary Material
Translational Impact.
Breast cancer affects over 230,000 American women each year. An estimated 70% of these cases have tumors that express the estrogen receptor and are eligible to be treated with antiestrogens such as tamoxifen or, for the treatment of advanced postmenopausal disease, Faslodex. However, 50% of these breast cancers will either fail to respond (de novo resistance) or lose therapy responsiveness over time (acquired resistance). We found that inhibiting autophagy through low-dose oral hydroxychloroquine (HCQ) administration increases the responsiveness of resistant mammary tumors to antiestrogens. Moreover, we show that the combination of tamoxifen and HCQ is more effective than that of Faslodex and HCQ due to activities within the tumor microenvironment. These preclinical data highlight the relevance of a clinical trial combining chloroquine and tamoxifen for the treatment of ER+ in situ breast lesions (PINC trial) and suggests a clinical benefit of the addition of HCQ to antiestrogen therapy for the treatment of ER+ breast cancer. Furthermore, this study predicts a more modest result for combining Faslodex and chloroquine-based therapies for prevention and/or treatment of breast cancer.
Acknowledgments
Katherine Cook is supported by a DOD Breast Cancer Research Program Postdoctoral Fellowship (BC112023). This research was also supported in part by awards from the US Department of Health and Human Services (R01-CA131465 and U54-CA149147) to Robert Clarke.
Footnotes
Disclosure of Potential Conflict of Interest: Authors have no conflict of interests.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Riggins RB, Bouton AH, Liu MC, Clarke R. Antiestrogens, aromatase inhibitors, and apoptosis in breast cancer. Vitamins and hormones. 2005;71:201–37. doi: 10.1016/S0083-6729(05)71007-4. [DOI] [PubMed] [Google Scholar]
- 3.Clarke R, Skaar TC, Bouker KB, Davis N, Lee YR, Welch JN, et al. Molecular and pharmacological aspects of antiestrogen resistance. The Journal of steroid biochemistry and molecular biology. 2001;76:71–84. doi: 10.1016/s0960-0760(00)00193-x. [DOI] [PubMed] [Google Scholar]
- 4.Clarke R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacological reviews. 2001;53:25–71. [PubMed] [Google Scholar]
- 5.Clarke R, Cook KL, Hu R, Facey CO, Tavassoly I, Schwartz JL, et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer research. 2012;72:1321–31. doi: 10.1158/0008-5472.CAN-11-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cook KL, Shajahan AN, Clarke R. Autophagy and endocrine resistance in breast cancer. Expert review of anticancer therapy. 2011;11:1283–94. doi: 10.1586/era.11.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas S, Thurn KT, Bicaku E, Marchion DC, Munster PN. Addition of a histone deacetylase inhibitor redirects tamoxifen-treated breast cancer cells into apoptosis, which is opposed by the induction of autophagy. Breast cancer research and treatment. 2011;130:437–47. doi: 10.1007/s10549-011-1364-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PloS one. 2009;4:e6251. doi: 10.1371/journal.pone.0006251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarke R, Shajahan AN, Riggins RB, Cho Y, Crawford A, Xuan J, et al. Gene network signaling in hormone responsiveness modifies apoptosis and autophagy in breast cancer cells. The Journal of steroid biochemistry and molecular biology. 2009;114:8–20. doi: 10.1016/j.jsbmb.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook KL, Shajahan AN, Warri A, Jin L, Hilakivi-Clarke LA, Clarke R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer research. 2012;72:3337–49. doi: 10.1158/0008-5472.CAN-12-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Molecular cancer therapeutics. 2008;7:2977–87. doi: 10.1158/1535-7163.MCT-08-0447. [DOI] [PubMed] [Google Scholar]
- 12.Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy. 2009;5:400–3. doi: 10.4161/auto.5.3.7784. [DOI] [PubMed] [Google Scholar]
- 13.Cook KL, Soto-Pantoja DR, Abu-Asab M, Clarke PA, Roberts DD, Clarke R. Mitochondria directly donate their membrane to form autophagosomes during a novel mechanism of parkin-associated mitophagy. Cell Biosci. 2014;4:16. doi: 10.1186/2045-3701-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:1239–46. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- 15.Loi S, Haibe-Kains B, Desmedt C, Wirapati P, Lallemand F, Tutt AM, et al. Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC genomics. 2008;9:239. doi: 10.1186/1471-2164-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loi S, Haibe-Kains B, Majjaj S, Lallemand F, Durbecq V, Larsimont D, et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10208–13. doi: 10.1073/pnas.0907011107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–9. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Sieuwerts AM, McGreevy M, Casey G, Cufer T, Paradiso A, et al. The 76-gene signature defines high-risk patients that benefit from adjuvant tamoxifen therapy. Breast cancer research and treatment. 2009;116:303–9. doi: 10.1007/s10549-008-0183-2. [DOI] [PubMed] [Google Scholar]
- 19.Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. European journal of pharmacology. 2009;625:220–33. doi: 10.1016/j.ejphar.2009.06.063. [DOI] [PubMed] [Google Scholar]
- 20.Butler WB, Berlinski PJ, Hillman RM, Kelsey WH, Toenniges MM. Relation of in vitro properties to tumorigenicity for a series of sublines of the human breast cancer cell line MCF-7. Cancer research. 1986;46:6339–48. [PubMed] [Google Scholar]
- 21.Butler WB, Fontana JA. Responses to retinoic acid of tamoxifen-sensitive and -resistant sublines of human breast cancer cell line MCF-7. Cancer research. 1992;52:6164–7. [PubMed] [Google Scholar]
- 22.Brunner N, Boysen B, Jirus S, Skaar TC, Holst-Hansen C, Lippman J, et al. MCF7/LCC9: an antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer research. 1997;57:3486–93. [PubMed] [Google Scholar]
- 23.Lewis MD, Pfeil J, Mueller AK. Continuous oral chloroquine as a novel route for Plasmodium prophylaxis and cure in experimental murine models. BMC research notes. 2011;4:262. doi: 10.1186/1756-0500-4-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soto-Pantoja DR, Menon J, Gallagher PE, Tallant EA. Angiotensin-(1-7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Molecular cancer therapeutics. 2009;8:1676–83. doi: 10.1158/1535-7163.MCT-09-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weidner N. Current pathologic methods for measuring intratumoral microvessel density within breast carcinoma and other solid tumors. Breast cancer research and treatment. 1995;36:169–80. doi: 10.1007/BF00666038. [DOI] [PubMed] [Google Scholar]
- 26.Martin-Manso G, Calzada MJ, Chuman Y, Sipes JM, Xavier CP, Wolf V, et al. sFRP-1 binds via its netrin-related motif to the N-module of thrombospondin-1 and blocks thrombospondin-1 stimulation of MDA-MB-231 breast carcinoma cell adhesion and migration. Archives of biochemistry and biophysics. 2011;509:147–56. doi: 10.1016/j.abb.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan M, Fang HB, Tian GL, Houghton PJ. Small-sample inference for incomplete longitudinal data with truncation and censoring in tumor xenograft models. Biometrics. 2002;58:612–20. doi: 10.1111/j.0006-341x.2002.00612.x. [DOI] [PubMed] [Google Scholar]
- 28.Frandsen TL, Boysen BE, Jirus S, Spang-Thomsen M, Dano K, Thompson EW, et al. Experimental models for the study of human cancer cell invasion and metastasis. Fibrinolysis. 1992;6:71–6. [Google Scholar]
- 29.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwartz-Roberts JL, Shajahan AN, Cook KL, Warri A, Abu-Asab M, Clarke R. GX15-070 (obatoclax) induces apoptosis and inhibits cathepsin D- and L-mediated autophagosomal lysis in antiestrogen-resistant breast cancer cells. Molecular cancer therapeutics. 2013;12:448–59. doi: 10.1158/1535-7163.MCT-12-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke R, Dickson RB, Lippman ME. Hormonal aspects of breast cancer. Growth factors, drugs and stromal interactions. Critical reviews in oncology/hematology. 1992;12:1–23. doi: 10.1016/1040-8428(92)90062-u. [DOI] [PubMed] [Google Scholar]
- 32.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8:200–12. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukhopadhyay A, Helgason GV, Karvela M, Holyoake TL. Hydroxychloroquine for chronic myeloid leukemia: complete cure on the horizon? Expert review of hematology. 2011;4:369–71. doi: 10.1586/ehm.11.34. [DOI] [PubMed] [Google Scholar]
- 34.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cook KL, Clarke R. Heat shock 70 kDa protein 5/glucose-regulated protein 78 “AMP”ing up autophagy. Autophagy. 2012;8:1827–9. doi: 10.4161/auto.21765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma N, Thomas S, Golden EB, Hofman FM, Chen TC, Petasis NA, et al. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer letters. 2012;326:143–54. doi: 10.1016/j.canlet.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 37.Espina V, Liotta LA. What is the malignant nature of human ductal carcinoma in situ? Nature reviews Cancer. 2011;11:68–75. doi: 10.1038/nrc2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Espina V, Mariani BD, Gallagher RI, Tran K, Banks S, Wiedemann J, et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PloS one. 2010;5:e10240. doi: 10.1371/journal.pone.0010240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loehberg CR, Thompson T, Kastan MB, Maclean KH, Edwards DG, Kittrell FS, et al. Ataxia telangiectasia-mutated and p53 are potential mediators of chloroquine-induced resistance to mammary carcinogenesis. Cancer research. 2007;67:12026–33. doi: 10.1158/0008-5472.CAN-07-3058. [DOI] [PubMed] [Google Scholar]
- 40.Patel RR, Sengupta S, Kim HR, Klein-Szanto AJ, Pyle JR, Zhu F, et al. Experimental treatment of oestrogen receptor (ER) positive breast cancer with tamoxifen and brivanib alaninate, a VEGFR-2/FGFR-1 kinase inhibitor: a potential clinical application of angiogenesis inhibitors. European journal of cancer. 2010;46:1537–53. doi: 10.1016/j.ejca.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, et al. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. The International journal of developmental biology. 2011;55:861–7. doi: 10.1387/ijdb.113371dl. [DOI] [PubMed] [Google Scholar]
- 42.Lang TJ. Estrogen as an immunomodulator. Clinical immunology. 2004;113:224–30. doi: 10.1016/j.clim.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 43.Harkonen PL, Vaananen HK. Monocyte-macrophage system as a target for estrogen and selective estrogen receptor modulators. Annals of the New York Academy of Sciences. 2006;1089:218–27. doi: 10.1196/annals.1386.045. [DOI] [PubMed] [Google Scholar]
- 44.Du GQ, Zhou L, Chen XY, Wan XP, He YY. The G protein-coupled receptor GPR30 mediates the proliferative and invasive effects induced by hydroxytamoxifen in endometrial cancer cells. Biochemical and biophysical research communications. 2012;420:343–9. doi: 10.1016/j.bbrc.2012.02.161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.