

Introductory Paragraph
Gliomas arising in the brainstem and thalamus are devastating tumors that are difficult to surgically resect. To determine the genetic and epigenetic landscape of these tumors, we performed exomic sequencing of 14 brainstem gliomas (BSG) and 12 thalamic gliomas. We also performed targeted mutational analysis of an additional 24 such tumors, and genome-wide methylation profiling of 45 gliomas. This study led to the discovery of tumor-specific mutations in PPM1D, encoding Wild type p53-Induced Protein Phosphatase 1D (Wip1), in 37.5% of the BSGs that harbored hallmark H3F3A K27M mutations. PPM1D mutations were mutually exclusive with TP53 mutations in BSG and attenuated p53 activation in vitro. The PPM1D mutations were truncating alterations in exon 6 that enhanced the ability for PPM1D to suppress the activation of DNA damage response checkpoint protein Chk2. These results define PPM1D as a frequent target of somatic mutation and as a potential therapeutic target in brainstem gliomas.
We explored the genetic landscape of gliomas arising in well-defined locations throughout the brainstem and thalamus, from both pediatric and adult patients (Supplementary Table 1). Whole exome sequencing was performed on 14 BSGs and 12 thalamic gliomas and matched normal blood (Online Methods). The coverage of bases in target regions with at least 10 reads is from 87% to 96% of all the samples (Supplementary Table 2). A total of 708 tumor-specific (somatic) mutations were discovered in these 26 samples (average 27 mutations per sample, Supplementary Table 3 and 4). 81 of 91 randomly selected mutated genes were validated by Sanger sequencing, reflecting an 89% sensitivity of our whole exome sequencing (Supplementary Table 5).
The mutational landscape of the 14 BSG and 12 thalamic gliomas samples is displayed in Fig. 1. This analysis confirmed that TP53 and H3F3A were frequently mutated in both BSGs and thalamic gliomas (>64% in both BSGs and thalamic gliomas)1–3. In addition, we confirmed the occurrence of PDGFRA alterations in these tumor types, with mutations identified in 1 BSG and 2 thalamic gliomas4. Also, an FGFR1 N546K mutation that frequently occurs in pilocytic astrocytomas was identified in one of each of the BSG and thalamic gliomas, each of which also contained an inactivating NF1 mutation5. Sanger sequencing for additional 8 samples shows two FGFR1 V664L mutations in BSG. In total 4 out of 34 (12%) samples indicate FGFR1 mutations are frequent in BSG and thalamic gliomas. Most importantly, this exome sequencing analysis led to the discovery of genes that had not previously been linked to the disease. Most notably, we found PPM1D mutations in 4 BSGs (29%) and IDH1 mutations in 5 BSGs (36%). All these important recurrent mutations in these significantly-mutated genes were validated by Sanger sequencing.
Figure 1.

Exome sequencing results for BSG and thalamic gliomas. (a) The frequency of mutations per case. A: Astrocytoma; O: Oligodendroglioma; OA: Oligoastrocytoma; GBM: Glioblastoma (b) The overview for mutation status of genes mutated in at least 2 cases. No deletions were detected in these genes. (c) The frequency of mutation per gene.
To establish the prevalence of PPM1D recurrent mutations in BSG and to explore the relationship between PPM1D mutations and other alterations, we performed targeted sequencing on an additional 24 BSGs and thalamic gliomas. Sanger sequencing was performed on PPM1D, IDH1, TP53, and H3F3A for a total of 33 BSGs and 17 thalamic gliomas when including samples used for exomic sequencing (Fig. 2). Among the extended series of 33 BSGs, 19 samples contained TP53 mutations, 16 contained H3F3A mutations, 8 contained IDH1 mutations, and 6 contained PPM1D mutations. In the extended series of 17 thalamic gliomas, we found 13 samples with TP53 mutations and 11 samples with H3F3A mutations. All 27 H3F3A mutations in both BSG and thalamic gliomas were the hallmark K27M hotspot mutations associated with these midline tumor types2,3. No HIST1H3B mutations, or H3F3A Arg34 mutations associated with supratentorial gliomas, were identified1–3. PPM1D mutations, as well as IDH1 mutations, were completely absent in thalamic gliomas. Patients with IDH1-mutated tumors were significantly older than those with IDH1-wild type tumors (median age 43 vs. 24, P = 0.0056), but neither H3F3A, TP53, nor PPM1D mutational status delineated significant differences in patient age (Supplementary Table 6).
Figure 2.
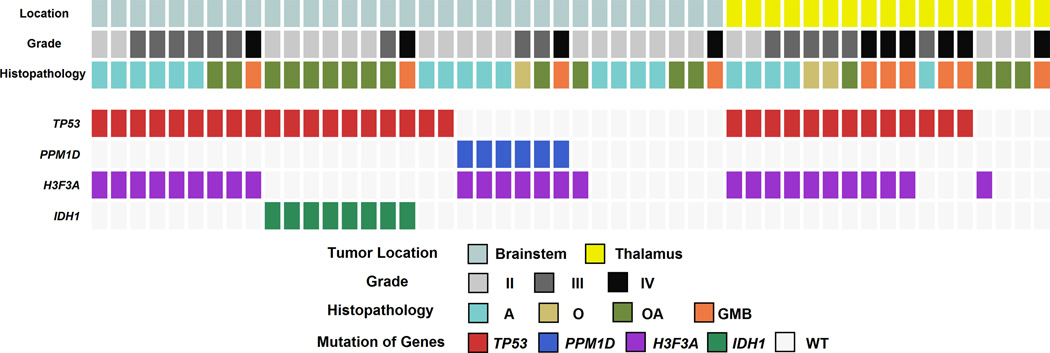
Mutation status of TP53, PPM1D, H3F3A, IDH1 in the extended series. Mutational status as determined by Sanger sequencing is shown for BSGs (n=33) and thalamic gliomas (n=17).
Analysis of the extended series of tumors revealed that PPM1D mutations were only present in the subgroup of H3F3A-mutated BSGs (16/33 BSGs). Almost all H3F3A-mutated BSGs (15/16) contained either a PPM1D mutation or a TP53 mutation in a completely mutually exclusive fashion (Fisher’s exact test: P < 10−5). Overall survival was not significantly different between patients with TP53-mutated and PPM1D-mutated brainstem gliomas (Supplementary Fig. 1, Supplementary Table 7). This mutual exclusivity and the similar clinical features of the patients suggest that TP53 and PPM1D mutations may have equivalent oncogenic functions in BSG. An additional PPM1D mutation was identified in one of 57 cerebral gliomas, indicating that PPM1D mutations are uncommon events in gliomas arising outside of the brainstem.
All PPM1D mutations identified here were truncating mutations in exon 6. Six of the PPM1D mutations were nonsense mutations targeting Glu472, Leu484, Ser516, Glu525 (2 cases), and Glu540, while one was a frameshift mutation that disrupted Asn448 and resulted in a de novo stop codon (Fig. 3a). Thus, all PPM1D mutations identified here truncate the C-terminal regulatory domain but leave the catalytic N-terminal PP2C phosphatase domain (residues 1-370) intact. PPM1D is a known oncogene that is amplified in breast and other cancers6. Also, remarkably similar truncating mutations in exon 6 of PPM1D have recently emerged as an unusual genetic risk factor for cancer after their discovery in germline tissues of rare (<1%) breast, ovarian, and colorectal cancer patients7,8. Additionally, both gain-of-function PPM1D genetic alternations and inactivating missense PPM1D mutations are found mutated at a low frequency in several types of cancer in available cancer data sets (http://www.cbioportal.org)9. The PPM1D mutations uncovered here establish truncating mutation in exon 6 of PPM1D as a frequent, tumor-specific (somatic) event in a type of cancer. To determine whether PPM1D mutation associates with different PPM1D protein expression patterns in tumors, we performed immunohistochemical analysis of PPM1D in a series of BSG tissues. No degradation or increased expression of PPM1D was identified in the PPM1D-mutated tumors compared to PPM1D-wild type tumors (Fig. 3b).
Figure 3.

Landscape of PPM1D mutations and IHC staining of PPM1D in tumor samples. (a) All mutations occurred in the C-terminal portion of PPM1D, leaving the phosphatase domain untouched. All mutations were identified in BSGs, except E472X which was identified in a cerebral glioma. (b) Tumors with or without PPM1D mutations were selected for IHC staining with an N-terminal anti-PPM1D antibody. PPM1D-WT (case ID); PPM1D-mutant (case ID, mutation type). Scale bar, 100 µm.
Our targeted sequencing analysis also revealed a subgroup of BSGs with IDH1 mutations (8/33, 24%). All IDH1 mutations in BSGs were R132 hotspot mutations that are a hallmark of the supratentorial intermediate grade gliomas that occur in adults10. Among BSGs, IDH1 mutations were mutually exclusive with H3F3A mutations (Fisher’s exact test: P < 10−6). Also, IDH1 mutations occurred solely in adult cases (median age 43), which may explain why previous studies that focused solely on pediatric BSG patients did not identify IDH1 alterations2,3. Our series of BSGs may have had different characteristics from previous studies because we examined the exophytic portions of diffuse intrinsic gliomas that were accessible for surgical resection, while previous studies examined intrinsic tissue from diffuse intrinsic gliomas that may not have had associated exophytic portions2,3.
We further determined whether BSG or thalamic gliomas exhibited epigenetic differences from supratentorial gliomas. To do so, methylation status of >485k genomic loci was determined for BSG (n=17), thalamic gliomas (n=10), and supratentorial glioma (n=18) samples. These samples were then subjected to unsupervised hierarchical clustering, consensus clustering, and principal component analysis based on methylation β-values for the 2% most variant loci (Fig. 4, Supplementary Fig. 2 and 3). Tumor location did not affect clustering of the samples based on these approaches. Rather, these clustering approaches identified three distinct groups corresponding almost perfectly to IDH1 and H3F3A mutation status. IDH1-mutated tumors tend to be hypermethylated compared with non-mutated tumors regardless of whether they are located in the brainstem or supratentorium, whereas H3F3A-mutated tumors showed a hypomethylation phenotype (Supplementary Fig. 4 and 5), consistent with previous reports11–14. Notably, PPM1D-mutated and non-PPM1D mutated samples were intermingled within the H3F3A cluster. Gene Expression data of selected samples were analyzed by Gene Set Enrichment Analysis15,16 (Supplementary Table 8). This analysis confirms the association between IDH1 mutation, H3F3A mutation, and methylation status11–14, and indicates that location and PPM1D mutation status are not major contributors to tumor methylation status.
Figure 4.

Heatmap of variant methylation probes in 45 glioma samples, with genetic information. Forty-five glioma samples (columns) and top 2% variant probes (rows) are arranged by unsupervised average-linkage hierarchical clustering. IDH1 mutated, H3F3A (K27)-mutated, and WT samples clustered separately by unsupervised average-linkage hierarchical clustering. Within the IDH1 mutated subcluster, two hemizygous IDH1 mutation samples described previously23 were included (marked as light green in the status of IDH1), which showed distinct methylation patterns compared with other IDH1-mutated samples. Gene expression-based classification into proneural, neural, classical, and mesenchymal subtypes by TCGA classification24 is shown for samples for which material was available for gene expression analysis.
We next sought to examine the function of the PPM1D mutations revealed in our genome-wide mutational analysis of BSGs, especially in the context of radiation treatment, a key therapeutic modality for gliomas. PPM1D functions as a phosphatase that dephosphorylates and inactivates many DNA damage response mediators such as Chk2, p53, and H2AX17–19. Cancer-associated PPM1D truncation mutants attenuate p53 and H2AX activation following irradiation, possibly because PPM1D mutants are stabilized at the protein level due to loss of a C-terminal degradation signal8. To extend these findings, we assessed the impact of truncated PPM1D on p53, H2Ax, and Chk2 activation and examined the effect of repairing PPM1D mutation in a cell line with a native PPM1D mutation.
Glioma-derived PPM1D-E472X and E540X truncated mutants were introduced to HEK293T cells, which are TP53 wild type. As controls, we also introduced wild type PPM1D, a phosphatase-dead PPM1D-D314A mutant20, or vector alone. Expression of the glioma-derived PPM1D mutants attenuated the increases in phospho-Thr68 Chk2, phospho-Ser15 p53, and γH2Ax following 10 Gy irradiation (Fig. 5a). This effect was also achieved by expression of PPM1D-WT, but not by the phosphatase-dead PPM1D-D314A mutant. These results demonstrate that cancer-associated PPM1D truncation mutants retain phosphatase activity toward Chk2 and confirm that they retain phosphatase activity against p53 and H2Ax7,8.
Figure 5.

The effect of cancer-derived truncated PPM1D on Chk2, p53, and H2AX activation and cell growth. (a) HEK293T cells transfected with full length wild type, E472X truncated PPM1D, E540X truncated PPM1D, and D314A PPM1D were exposed to ionizing radiation (IR, 10Gy). Cells were collected before IR and 1 h and 5 h after IR. Whole cell lysates were analyzed for Chk2 Thr68 phosphorylation, p53 Ser15 phosphorylation, γ-H2AX, and GAPDH. Western blot against PPM1D showed successful transfection. (b) Gene targeting to repair a native PPM1D mutation. HCT116 cells contain a heterozygous frameshift deletion that results in a truncated PPM1D mutant (L450X). Homologous recombination was used to replace the mutated PPM1D allele with wild type PPM1D in two HCT116 sublines. Western blot demonstrates expression of full length and truncated PPM1D in the parental cell line, but only full length PPM1D expression in the two repaired cell lines. PPM1D-F: Full length PPM1D. PPM1D-T: Truncated PPM1D. (c) HCT116 parental (PPM1D mutant) and two isogenic repaired lines (PPM1D wild type) were exposed to ionizing radiation (IR, 10 Gy). Cells were collected before IR and 1 h and 5 h after IR. Whole cell lysates were analyzed for Chk2 Thr68 phosphorylation, p53 Ser15 phosphorylation, γ-H2AX, and GAPDH. (d) Proliferation assay for HCT116 parental cell line (PPM1D mutant) and isogenic repaired lines (PPM1D wild type). 1000 cells were seeded in each well (triplicates) of 96-well plates on day 0. Relative cell number was determined each day after seeding. Error bars represent mean ± SEM. (e) Colony formation by PPM1D-mutant HCT116 parental cell line and isogenic wild type PPM1D lines. Colony formation was assessed in the absence of IR, and under 2 Gy, or 4 Gy IR. P < 0.05 (*) between parental cells and isogenic repaired line 1 or 2 under 0 Gy, 2 Gy, and 4 Gy IR. n=4, error bars represent mean ± SEM.
We next sought to dissect the contribution of PPM1D mutation to the biochemical and biological properties of PPM1D-mutated cancer cells. Since BSG-derived cell lines with PPM1D mutation have not been established, we focused on the HCT116 colorectal carcinoma cell line which contains a native PPM1D-L450X mutation8 at the same region of the mutations identified in BSGs. Using homologous recombination (Online Methods), we repaired the mutated PPM1D allele to create two isogenic PPM1D wild type cell lines (Fig. 5b and Supplementary Fig. 6). After irradiation, the repaired PPM1D wild type cell lines had higher levels of Chk2 and of p53 phosphorylation than the PPM1D-mutant parental cell line (Fig. 5c). γH2Ax was not increased by replacement of the mutant PPM1D allele, indicating that dynamic H2Ax phosphorylation may be regulated by other phosphatases in this cell line. Notably, cell growth and colony formation were decreased after repair of the mutant PPM1D allele (Fig. 5d, 5e, and Supplementary Fig. 7), indicating that mutant PPM1D drives growth of PPM1D-mutated cancer cells. These results demonstrate that mutant PPM1D contributes to cell growth, as well as to p53 and Chk2 dephosphorylation after radiation treatment.
In summary, our genomic and epigenomic analysis reveals H3F3A-PPM1D co-mutation and IDH1-TP53 co-mutation as common genetic subtypes of BSGs, and confirms that H3F3A-TP53 co-mutation commonly occurs in both BSGs and thalamic gliomas. We identified truncating mutations in exon 6 of PPM1D as frequent somatic events in a human tumor type, in the typical tumor-specific distribution expected for oncogenic mutations selected during tumor clonal evolution. The mutual exclusivity of PPM1D and TP53 mutations together with the functional study results suggest that somatic mutations in either PPM1D or TP53 are the genetic basis for dysfunction of the radiation-induced DNA damage response network in brainstem gliomas. Our data further show that these gain-of-function PPM1D mutants can suppress phosphorylation of Chk2 upon radiation-induced DNA damage stress and potentially abrogate cell cycle checkpoints.
This study adds another layer to the complex genetic role for PPM1D in cancer: in addition to the gain-of-function/truncating PPM1D mutations reported here in BSGs, germline mosaic gain-of-function/truncating PPM1D mutations have been linked to several other types of cancer7,8, and still other rare tumors have been reported with loss-of-function/missense PPM1D mutations9. Assessment of PPM1D mutational status in future prospective trials will be necessary to determine whether PPM1D status is predictive of response to radiation or other therapies. Future studies that employ whole genome sequencing may identify additional alterations in these tumor types, and further studies on genetically engineered models of PPM1D-mutated gliomas, which have yet to be developed, will determine the precise mechanism by which PPM1D mutations confer a selective advantage in the glioma setting. Also, since small chemical inhibitors of PPM1D phosphatase activity are being explored21,22, the finding of gain-of-function PPM1D mutations offers a potential therapeutic opportunity for a subgroup of patients with this challenging brain tumor type.
Online Methods
Patients
Brain tumor samples and peripheral blood of patients were collected from Beijing Tiantan Hospital, China from 2010 to 2012 with informed consent reviewed by Institutional Review Board. Total 107 samples were used in this analysis, 50 BSGs and thalamic gliomas and 57 supratentorial gliomas. Among 50 samples (33 BSGs and 17 thalamic gliomas), 26 samples were used for whole exome sequencing for preliminary screening, and total 50 samples were validated by Sanger sequencing. Survival data were displayed using Kaplan-Meier analysis and the log-rank test was used to test for survival differences between groups.
Sequencing and Copy Number Alterations
Whole exome sequencing and copy number analyses were performed by Personal Genome Diagnostics, Inc., Baltimore, MD, Beijing Pangenomics Technology, Co. Ltd, and Softgenetics. A targeted approach, using Sanger sequencing, was performed to screen for additional mutations in a larger series of samples for a subset of the genes identified. Copy number amplifications were confirmed in targeted genes of PPM1D and PDGFRA by qPCR analysis. Randomly selected mutations were also validated by Sanger Sequencing to evaluate the sensitivity of whole exome sequencing. Sequencing primers were selected as previously reported25. PolyPhen-2 was applied to predict the functional effects of missense mutations26.
DNA Methylation
Illumina HumanMethylation450 BeadChip was used for genome-wide methylation profiling of 18 BSG, 10 thalamic gliomas samples, and 20 supratentorial tumors including one duplicate sample for internal control and one cell line. GenomeStudio Methylation Module was used for data processing and quality check. The internal control showed almost perfect correlation with its duplicate (r=0.9978, P < 10−16). One BSG sample was excluded for obviously considered as an outlier from other samples. Consensus clustering by GenePattern and hierarchical clustering by R package pheatmap were performed for evaluation of subgroups27–30.
Gene Expression Profiling
Affymetrix Human Genome U133 Plus 2.0 Array was used for evaluation of gene profiling of samples (n=21) at Shanghai Biotechnology Corporation, China. Robust Multichip Average (RMA) method by R was used for data processing and normalization31. For gene expression subgroups classification, additional GBM samples (n=116) were obtained from the TCGA websites as training dataset. Classification to Nearest Centroids (ClaNC) was used for predicting the subtypes of our tumor samples32. Gene Set Enrichment Analysis was used to analyze the possible affected pathways15,16.
Plasmid construction
The plasmids contain full-length wildtype PPM1D cDNA (pCMV-Neo-Bam-PPM1D) and D314A mutated PPM1D (pCMV-Neo-Bam-PPM1D D314A) with C-terminal Flag tags were obtained from Addgene (plasmids 28105 and 28106)20. To generate the truncating mutations (PPM1D472 and PPM1D540), the primers were used along with the QuikChange II Site Directed Mutagenesis Kit (Agilent, 200521)(Supplementary Table 9).
Gene targeting of PPM1D mutant allele in HCT116 cells
The strategy for gene targeting in human cells has been described previously12. Briefly, targeting recombinant adenovirus associated virus (rAAV) construct was designed to replace the PPM1D L450X mutant allele with WT PPM1D allele. Homologous arms surrounding the insertion site were PCR-amplified from human blood DNA using platinum taq HIFI polymerase (Invitrogen, 11304). The infectious rAAV stock was prepared and applied to the HCT116 cell line as previously described33. DNA of G418-resistant clones were extracted and screened by PCR and PPM1D status of the resulting positive clones was validated by direct Sanger sequencing. Two validated clones of which the PPM1D L450X mutation was successfully repaired were further treated with Cre recombinase adenovirus (Vector Biolabs, 1045) to remove the drug-resistance cassette.
Cell culture, transfection and irradiation
HEK293T cells (American Type Culture Collection, Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, 11965) with 10% fetal bovine serum (FBS) (Gibco); HCT116 parental and isogenic cells were grown in McCoy5A medium (Invitrogen, 16600) with 10% FBS; for transfection experiments, 293T cells were plated in 6-well plates (50%–70% confluence) and were transfected with 2 µg of plasmid using Lipofectamine 2000 (Invitrogen, 11668027). After 24 h transfection, the cells were exposed to 10 Gy gamma irradiation (X-ray) and were harvested at 1 and 5 hours post-irradiation. The HCT116 parental and isogenic cell lines were exposed to gamma irradiation with the same settings. The Cell Counting Kit-8 (Dojindo, CK04-13) was used to determine relative cell counts according to the manufacturer’s protocol.
Colony formation assay
HCT116 parental and its derived isogeneic cells were seeded in quadruplicate. After 12 hours incubation, cells were exposed to gamma irradiation 0 Gy (500 cells per well on a 6-well plate), 2 Gy (500 cells), and 4 Gy (2000 cells) from an X-ray source. Cells were fixed with ice-cold methp.15anol and stained with 0.05% crystal violet solution after 8–10 days incubation. Colonies were counted and the p value was calculated by Wilcoxon rank sum test.
Western Blot and Immunohistochemistry
Protein expression and modification were determined by Western blotting using standard means. The cells were washed with ice cold 1 × PBS twice followed by treatment with RIPA lysis buffer (Santacruz, sc-364162) supplemented with protease inhibitor cocktail (Santacruz, sc-29130) and sodium orthovanadate. After sonication and centrifugation, the protein concentrations of supernatants were quantified by bicinchoninic acid (BCA) assay. A total of 20 µg of proteins were denatured at 70°C for 10 min with NuPAGE® LDS Sample buffer (Life Technology, NP0007) and resolved by electrophoresis. The proteins were transferred to a nitrocellulose membrane. Membranes were blocked by 5% BSA or 5% fat free milk according to the antibodies datasheet. After blocking, membranes were incubated with a primary antibody: mouse anti-PPM1D (Santacruz, sc-376257); rabbit anti-GAPDH (Santa Cruz, SC-25778); rabbit anti-P53 (Cell Signaling, 9282); rabbit anti-phospho-P15 Ser15 (Cell Signaling, 9284); rabbit anti-CHK2 (Santa Cruz, SC-9064); rabbit anti-CHK2 T68 (Cell Signaling, 2661); rabbit anti γ-H2AX (Cell Signaling, 9718) and rabbit anti-H2AX (Cell Signaling, 7631) at 4°C overnight at dilutions specified by the manufacturers. The membranes were washed three times with 0.5% Tween-20 (TBST) before incubated with secondary antibodies (ECL anti-rabbit IgG or ECL anti-mouse IgG, GE Life Science) and detected by ChemiDoc™ MP System (BIO-RAD). Immunohistochemisty was performed on 5-µm formalin-fixed, paraffin-embedded tissue sections using a rabbit polyclonal antibody raised against the N-terminus of PPM1D (Abcam, ab60243) at a 1:100 dilution according to the manufacturer’s instructions.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This study was partly supported by a National Cancer Institute grant R01CA140316 (HY), American Cancer Society (RSG-10-126-01-CCE)(HY), Pediatric Brain Tumor Foundation Institute grant (DB), a Voices Against Brain Cancer Foundation grant (DB), a James S. McDonnell Foundation grant (HY), The V Foundation (HY), and an Accelerate Brain Cancer Cure Foundation grant (HY), The Natural Science Foundation of China Grants 30772237 and 81241078 (LZ).
Footnotes
Accession Code
Methylation and Gene Expression Microarray data of this study are MIAME compliant and have been submitted to the MIAME compliant NCBI Gene Expression Omnibus (GEO) database, accession number GSE50774. Raw sequencing data will be available to qualified investigators upon request.
Author Contributions
L. Z., H. Y., Z. J. R., and L. H. C. designed research. L. H. C., Z. J. R., J. B. W., and H.Y. wrote the manuscript. L. Z., L. H. C., H. W., W. Z., J. F., S. J., R. Y., P. J. K., J. Z., Z. Wu., S. H., Y. W., and J. B. W. performed the experiments. Z. W. and H. Z. contributed to biological validation of PPM1D mutant functions. H. F., and A. F., contributed to the inspection of clinical data. L. H. C., S. Y., S. J., S. W., G. L., R. E. M., Y. H., Z. J. R., D. D. B., and H. Y. analyzed the data.
Competing financial interests
Under agreements between Duke University, Agios Pharmaceuticals, Blueprint Medicines and Personal Genome Diagnostics, H.Y. and D. D. B. are entitled to a share of the royalties received by the University on sales of products related to genes described in this manuscript. S.W. and H.Y. are co-founders and own stocks of Beijing Pangenomics Technology, Co. Ltd.
References
- 1.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 2.Khuong-Quang DA, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puget S, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One. 2012;7:e30313. doi: 10.1371/journal.pone.0030313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones DT, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927–932. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bulavin DV, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–215. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 7.Ruark E, et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature. 2013;493:406–410. doi: 10.1038/nature11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleiblova P, et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J Cell Biol. 2013;201:511–521. doi: 10.1083/jcb.201210031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudgeon C, et al. Genetic variants and mutations of PPM1D control the response to DNA damage. Cell Cycle. 2013;12:2656–2664. doi: 10.4161/cc.25694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turcan S, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duncan CG, et al. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012;22:2339–2355. doi: 10.1101/gr.132738.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sturm D, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 14.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 16.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliva-Trastoy M, et al. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene. 2007;26:1449–1458. doi: 10.1038/sj.onc.1209927. [DOI] [PubMed] [Google Scholar]
- 18.Lu X, Nannenga B, Donehower LA. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005;19:1162–1174. doi: 10.1101/gad.1291305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoda A, et al. Intrinsic kinase activity and SQ/TQ domain of Chk2 kinase as well as N-terminal domain of Wip1 phosphatase are required for regulation of Chk2 by Wip1. J Biol Chem. 2006;281:24847–24862. doi: 10.1074/jbc.M600403200. [DOI] [PubMed] [Google Scholar]
- 20.Moon SH, Nguyen TA, Darlington Y, Lu X, Donehower LA. Dephosphorylation of gamma-H2AX by WIP1: an important homeostatic regulatory event in DNA repair and cell cycle control. Cell Cycle. 2010;9:2092–2096. doi: 10.4161/cc.9.11.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yagi H, et al. A small molecule inhibitor of p53-inducible protein phosphatase PPM1D. Bioorg Med Chem Lett. 2012;22:729–732. doi: 10.1016/j.bmcl.2011.10.084. [DOI] [PubMed] [Google Scholar]
- 22.Gilmartin AG, et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat Chem Biol. 2014 doi: 10.1038/nchembio.1427. [DOI] [PubMed] [Google Scholar]
- 23.Jin G, et al. Disruption of wild type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-12-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reich M, et al. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 28.Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning. 2003;52:91–118. [Google Scholar]
- 29.Team, R.C. R. A language and environment for statistical computing. 2012 [Google Scholar]
- 30.Kolde R. pheatmap: Pretty Heatmaps. R package version 0.7.3. 2012 [Google Scholar]
- 31.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 32.Dabney AR. Classification of microarrays to nearest centroids. Bioinformatics. 2005;21:4148–4154. doi: 10.1093/bioinformatics/bti681. [DOI] [PubMed] [Google Scholar]
- 33.Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.