

Abstract
Background
Plasma levels of endothelial microparticles (EMPs), small membrane vesicles, shed from activated or apoptotic endothelial cells are elevated in patients with COPD and in smokers with normal lung function. Whether plasma EMPs levels are elevated in a rat exposed in cigarette smoke, whether the elevated EMPs derived from pulmonary endothelial cell apoptosis, and the relationship between EMP and lung function are obscure.
Methods
All 60 wister rats were divided into six groups, three groups of ten rats were exposed to cigarette smoke of ten non-filter cigarettes per day, 5 days a week, using a standard smoking machine (Beijing BeiLanBo Company, China) for a period of 2, 4 and 6 months (n=10, respectively). Age-matched three control groups were sham-smoked. Pulmonary function parameters, including the ratio of forced expiratory volume in 0.3 second over forced vital capacity (FEV0.3/FVC) and dynamic compliance (Cdyn), were tested at the end of each period (2, 4, 6 months). Blood samples were collected and platelet-free plasma was isolated. Then CD42b–/CD31+ EMPs were analysed by flow cytometry. In parallel, lungs were removed and Colocalization with terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL), Hoeschts and CD31 was performed to evaluate pulmonary capillaries-specific apoptosis and identify the origins of the EMPs.
Results
At 2, 4 and 6 months, in comparison with control groups, rats in cigarette smoke exposed groups had a significant increase in CD42b–/CD31+ EMPs (P<0.001, P<0.001, P<0.001, respectively), and Pulmonary function indicated that FEV0.3/FVC (P<0.05, P<0.01, P<0.01, respectively) and Cdyn (P<0.01, P<0.001, P<0.001 respectively) decreased. At the same time, CD42b–/CD31+ EMP counts were negatively correlated with Cdyn (P<0.05). Moreover, in vivo, TUNEL-positive cells co-localized with CD31 in whole lung tissue demonstrated a sequence of apoptosis signal in the cigarette smoke exposed groups.
Conclusions
CD42b–/CD31+ EMPs may be a potential biomarker for indicating the severity of impairment of pulmonary function in the rats exposed cigarette smoke. The increased EMPs may derive from pulmonary capillaries-specific apoptosis.
Keywords: Cigarette smoke, circulating endothelial microparticle, pulmonary capillary, pulmonary function, apoptosis
Background
Long-term cigarette smoke exposure induces an inflammatory process in the lung that may underlie the development of chronic obstructive pulmonary disease (COPD), which involves a mixture of pathomorphological changes (airways narrowing, emphysema and vascular abnormalities) (1,2). The pathogenesis of COPD is complex and includes the balance of proteases and antiproteases in the lung, tilted toward an excess of unopposed proteases that destroy the connective tissue backbone of the lung parenchyma (2-6). Despite major abnormalities taking place in the airway side, changes in pulmonary vessels represent an important component of the disease. In recent years, increased evidence has demonstrated that endothelial damage is closely connected to the pathophysiology of COPD (7-9). It was noted that there was a paucity of the pulmonary microvessel in lung parenchyma with emphysema, and alterations in pulmonary endothelial injury and apoptosis are highly prevalent.
Circulating endothelial microparticles (EMPs), small membrane vesicles (100 nm-1 μm in diameter), are shed into circulating blood that originate from activated and apoptotic endothelial cells (10,11). The number of EMPs increases in patients with vascular disorders, such as acute coronary disease (12), renal failure (13) and metabolic diseases (14), and it reflects endothelial damage occurring in these patients. Gordon and coworkers presented their finding that smokers, even in the absence of obvious radiographic, spirometric, or clinical evidence of emphysema or airflow obstruction, had increased levels of circulating endothelial micro-particles (CD42b–/CD31+), which demonstrate an apoptotic phenotype (reduced CD42b–/CD62+ or CD42b–/CD31+ ratio and increased annexin V1 expression), and appeared to originate from the pulmonary vasculature (evident by positive immunostaining for angiotensin-converting enzyme) (15). In addition, Takahashi et al. found that VE-cadherin (CD144), PECAM (CD31) and E-selectin (CD62E) EMPs were significantly more numerous in the stable COPD patients than in the healthy non-COPD volunteers, and their numbers further increased in the exacerbated phase. And higher E-selectin EMP levels may predict the COPD patients who are susceptible to exacerbation (16).
Based on the above knowledge, we hypothesize that the number of EMPs is elevated in rats exposed in cigarette smoke, and the elevated EMPs are derived from pulmonary capillaries. By observing changes in EMP levels and pulmonary function of two cohorts of laboratory rats (cigarette smoke exposure groups and sham exposure groups), and calculated the correlations between levels of EMP and pulmonary function parameters (FEV0.3/FVC and Cdyn). EMP, quantified in plasma as particles that are CD31 (PECAM) possitive, but CD42b (the constitutive platelet-specific glycoprotein Ib) negative. We also explored the role of pulmonary endothelial injury and apoptosis on the pathogenesis of COPD.
Some of the results from this study have been previously presented in abstracts.
Methods
Animals
Sixty male wistar rats, 8 weeks old, and initially weighing (180±5) g, were purchased from Nantong University Laboratory Animal Center. Upon arrival, all animals were given a diet of standard chow and water, and were acclimated to the new housing environment for 1 week before cigarette smoke exposure began. Rats were removed from the study due to lethargy and significant weight loss at the end of each time period. All procedures involving animals and their care were approved by the Ethics Committee of Nantong University and were conducted following institutional guidelines that comply with national laws and policies.
Cigarette smoke exposure
Rats were divided into six groups, three groups of ten rats was exposed to the smoke of ten nonfilter cigarettes per day, 5 days a week, using a standard smoking machine (Beijing BeiLanBo Company, China) for a period of 2, 4 and 6 months (n=10, respectively). Age-matched control groups were sham-smoked.
Pulmonary function tests
At the end of each time period, the rats were anesthetized intraperitoneally with 12% urethane at a dose of 1 mL per 100 mg. After their tracheas were cannulated, the rats were placed supine in a small animal plethysmograph (Beijing BeiLanBo Company, China). Lung function tests were performed using the modification method of Wright et al. (17). The lungs were inflated with air at a volume of 5-fold tidal volume and then rapidly deflated to a pressure of –35 cm H2O. The ratio of forced expiratory volume in 0.3 second to forced vital capacity (FEV0.3/FVC) and dynamic compliance (Cdyn) were calculated by computer.
Assay of EMP
After all pulmonary function tests were completed, blood samples were collected by the heart punctures in 5 mL sodium citrate tubes and, within 1 hour, centrifuged 10 minutes (160 g, 23 °C) to prepare platelet rich plasma. Within 5 minutes, the supernatant was further centrifuged 8 minutes (1,000 g, 23 °C) to obtain platelet-poor plasma. Within 5 minutes, 50 μL aliquots of platelet-poor plasma were incubated (45 min, 4 °C) with 4 μL of fluorescein-conjugated anti-rat PECAM (FITC) and 4 μL of phycoallocyanine-conjugated anti-rat CD42b (PE) was added (45 min, 4 °C) to each sample to exclude platelet-derived microparticles. CD42b–/CD31+ microparticle levels were corrected for correlating isotype control antibodies. EMP phenotype analysis was performed within 15 minutes based on size and fluorescence. The method of flow cytometry were performed using the method of Gordon et al. (15).
Co-localization assay for apoptosis
Co-localization assays were performed referencing the method of Hassoun et al. (18). Briefly, after the rats were exsanguinated, the right pulmonary were inflated with 0.5% agarose, fixed in formalin, and embedded in paraffin blocks. Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assays for apoptosis were performed referencing per manufacturer’s instructions (Roche, Indianapolis IN, USA), and subsequent staining with rat anti-CD31 monoclonal antibody (1:1,000, BD, USA) and Hoeschts dye (BisBenzimide, 1 mg/100 mL) were co-localized endothelial cells undergoing apoptosis by confocal microscopy.
Statistical analysis
All data were reported as means ± standard deviation (SD), and statistical analyzed by SPSS vs.17.0 (SPSS, Inc, Chicago, USA). For comparison between smoke exposed groups and sham-smoked groups, a paired student’s t-test was performed. The Pearson test was used to calculate correlations between levels of EMP and pulmonary function parameters (FEV0.3/FVC and Cdyn). P values of less than 0.05 were considered statistically significant.
Results
Weight and lung function
Two rats from the 2 months smoke exposure group, two from the 4 months smoke exposure group, four from the 6 months smoke exposure group, and each two rats from the 4 months, 6 months control groups were removed from the study due to lethargy and significant weight loss at the end of each time period. At the end of the 6 months of smoke exposure, the remaining six rats were included in flowing analysis, and the six sets of data from each other groups was drawn randomly.
Rats of the smoke-exposed groups weighed less than rats of the sham-smoked groups did. At the end of 2, 4, and 6 months, they weighed 225±10, 250±10, and 285±10 g, respectively, whereas the sham-smoked rats weighed 240±5, 280±8, and 340±8 g, respectively (P<0.05, 0.05, and 0.01, respectively).
The pulmonary function test data are shown in Figures 1,2. After 2 month of smoke exposure, the smoke-exposed rats began to show low-grade obstructive ventilatory disorder with decrease in the ratio of FEV0.3 to FVC (P<0.05). This was accompanied by significantly decreased Cdyn (P<0.01). At the end of 4 and at 6 months, the obstructive ventilatory disorder was marked (4 months, P<0.01 FEV0.3/FVC and P<0.001 Cdyn; 6 months, P<0.01 FEV0.3/FVC, and P<0.001 Cdyn) with the smoke exposure time.
Figure 1.
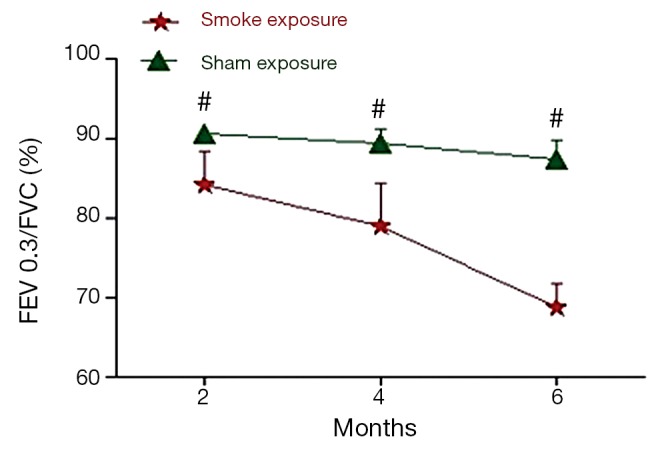
FEV0.3/FVC% are shown expressed in percentage for each of the 2-, 4-, and 6-month time periods. Values = mean ± SD. #, indicate P<0.05 or greater significant difference between sham exposed (n=6) and smoke-exposed (n=6) rats; SD, standard deviation.
Figure 2.
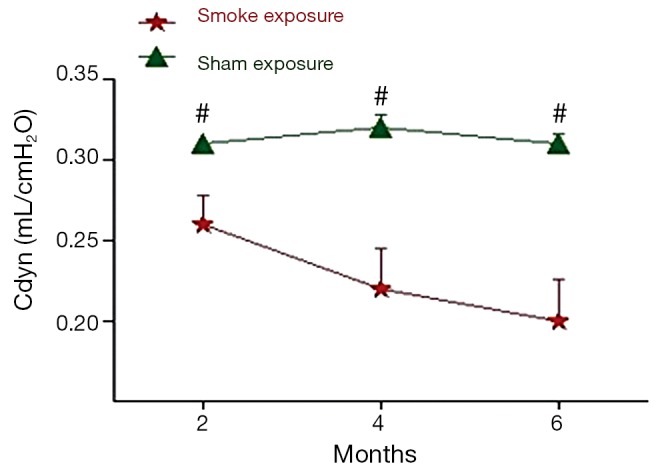
Cdyn are shown expressed in mL/cm H2O for each of the 2-, 4-, and 6-month time periods. Values = mean ± SD. #, indicate P<0.05 or greater significant difference between sham exposed (n=6) and smoke-exposed (n=6) rats; SD, standard deviation.
Levels of EMP
CD42b–/CD31+ EMP were measured at the end of each period. Results are shown in Figure 3. This demonstrates that EMPs measured with the special marker discriminate rats of the smoke-exposed from rats of the sham exposure groups.
Figure 3.
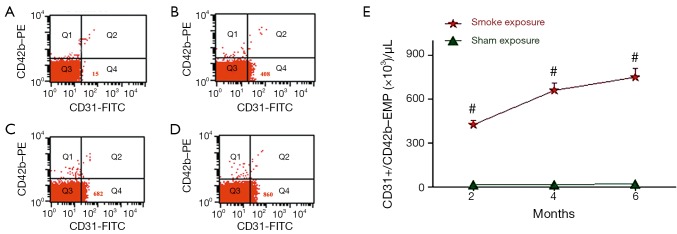
Representative examples of flow cytometry of CD42b–/CD31+ EMP in a rat exposed to smoke 2 months (B) 4 months (C) and 6 months (D) and a rat of sham exposure groups (A). The points represented in region Q4 of both examples are CD31+ (positive) and CD42- (negative); therefore they are considered to be EMPs. In region Q4 (lower right of histogram), there is a visibly more dense cluster of EMPs in the rat exposed to smoke (B, C, D) compared with in the rat of sham exposure groups (A). In these examples, the rat exposed to smoke 2 months had 408 EMPs (B), the rat exposed to smoke 4 months had 682 EMPs (C), the rat exposed to smoke 6 months had 860 EMPs (D), and the rat of sham exposure groups had 15 EMPs (A). (E) CD42b–/CD31+ EMP counts are shown for each of the 2-, 4-, and 6-month time periods. Values = mean ± SD. #, indicate P<0.05 or greater significant difference between sham exposed (n=6) and smoke-exposed (n=6) rats; EMP, endothelial microparticle; SD, standard deviation.
Figure 3E summarizes data on EMPs detected with CD31and CD42. It shows that, at the end of 2, 4 and 6 months, CD42b–/CD31+ EMP levels are markedly higher in rats of the smoke-exposed groups than in rats of the control groups (P<0.001, respectively). It also illustrates the significant elevation in rats of the 6 months exposed group with the 2, 4 months exposed groups (P<0.001, respectively), and 4 months exposed group with the 2 months exposed group (P<0.001). The CD42b–/CD31+ EMP level was highest in 6 months exposed group, followed by 4 months exposed group and 2 months exposed group.
Co-localization assay for pulmonary endothelium cells apoptosis
TUNEL-positive cells co-localized with CD31 in rat lung tissue after cigarette smoke exposed 2 months, demonstrating an increase signal of cell death of pulmonary endothelium cells, but negative in lung tissue of sham exposure rats (Figure 4).
Figure 4.

Lung endothelial cell apoptosis after smoke exposure and sham exposure. Lung immunofluorescence micrographs (×200) stained with CD31 (green), TUNEL (red), Hoechst (blue), and merged CD31-TUNEL- Hoechst. The pink signals indicated by the arrows suggesting lung endothelial cell apoptosis in representative examples of smoke exposed rats (A1, A2 and A3). No signal indicated in the lung stain in sham exposed rats (B1, B2 and B3). TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling.
Correlations between levels of EMP and pulmonary function parameters (FEV0.3/FVC and Cdyn)
There were no correlations between CD42b–/CD31+ EMP counts and FEV0.3/FVC (r=–0.907, P=0.276). CD42b–/CD31+ EMP counts were negatively correlated with Cydn (r=–0.998, P=0.038) (Figure 5).
Figure 5.
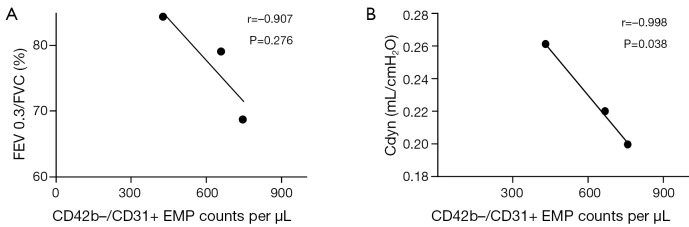
Correlations between levels of EMP and pulmonary function parameters [FEV0.3/FVC (A) and Cdyn (B)] are shown and P values of less than 0.05 were considered statistically significant. EMP, endothelial microparticle.
Discussion
Cigarette smoke exposure will induce emphysema and vascular alterations (17,19). Despite pulmonary hypertension is a major component of vascular abnormalities of COPD patients, pulmonary endothelial injury and apoptosis are becoming more and more important in the aetiopathogenesis of COPD/emphysema (2,4,8,20-24). On the basis of previous research (15,16), we have rats exposed to a standard smoking machine for a period of 2, 4 and 6 months. To evaluate the alterations of pulmonary function (FEV0.3/FVC and Cdyn) and the endothelial damage induced by cigarette smoke exposure in rats, we compared circulating EMP numbers in rats of the smoke exposure groups and the sham exposure groups. In this study, we found that exposure of rats to cigarette smoke induced decreases in FEV0.3/FVC and Cdyn compared with rats of sham exposure groups, similar to what has been described of Wright et al. (17). By measuring levels of EMP with the special marker, we found that exposure of rats had high levels of circulating EMPs compared with rats of sham exposure groups, and the levels of circulating EMPs higher with the time of smoke exposure. The EMPs likely derived from endothelial cells undergoing apoptosis, and likely mostly from pulmonary endothelium.
Microparticles are submicron membrane vesicles shed from the plasma membranes of different cell types in response to cell activation, injury, and/or apoptosis (25-28). As what mentioned before, there are various subtypes of EMPs. Reviewing previous researches, apoptosis-induced EMPs are more likely to express only CD31 and show the presence of phosphatidylserine (annexin V) as an apoptotic parameter. Healthy smokers and symptomatic smokers with normal spirometry and DLco had mildly elevated levels of circulating EMPs compared with healthy nonsmokers. Strikingly, however, healthy smokers with normal spirometry but an isolated reduction in DLco had high levels of circulating EMPs compared with all other groups (15). In our study, the levels of circulating EMPs in rats are higher with the time of smoke exposure.
The following correlations between the Levels of EMP and lung function parameters were observed: (I) there were no correlations between CD42b–/CD31+ EMP counts and FEV0.3/FVC; (II) CD42b–/CD31+ EMP counts were negatively correlated with Cydn.
FEV0.3/FVC expressed the pulmonary ventilation function well, especially in COPD rat model (15,16), whereas CD42b–/CD31+ EMP counts showed no correlations with FEV0.3/FVC. Because we only made rat models of the early period of COPD, it may require a longer time of study.
Cdyn is the lung compliance measured in respiratory cycle when the flow is not blocked. In small airway disease, such as the early period of COPD, Cdyn declined, while the rising of intrapleural pressure with the Specific changes in lung capacity. CD42b–/CD31+ EMP counts showed negative correlations with Cdyn. These indicate that the high levels of circulating EMPs may reflect the decline of small airway function indirectly in the early of COPD.
Through co-localization stain of pulmonary endothelium cells, we also demonstrated that the evaluated EMPs mostly derived from pulmonary endothelial cells undergoing apoptosis. CD31 EMPs may measure as a preferable marker of pulmonary endothelium injury and destruction of pulmonary function.
Pulmonary endothelial apoptosis as a primary mechanism in the development of COPD is supported by the observation of endothelial apoptosis in the lungs of humans with emphysema (8,9,20-23). Therefore, high EMP levels may indicate a role for endothelial damage in COPD progression. Because quicker responses can be seen in circulating EMP levels compared with an annual lung function decline, monitoring EMP levels is valuable for estimating COPD progression, and can be a useful index for drug discovery.
There are limitations in this study, we used experimental groups of only six animals because of the disqualification of weight and the spirit. While this somewhat limits statistical power, for almost all the measurements we made there were clearly significant differences between smoke and sham smoke. Overall, however, we do not believe that using a group size of six has posed a serious problem to our data interpretation.
In conclusion, pulmonary endothelial damage occurs during the smoke exposure, and has been a primary mechanism in the development of COPD. Plasm levels of CD42b–/CD31+ EMPs is useful for evaluating the degree of COPD progression.
Acknowledgements
This study was supported by Nantong social development project [NO: S2009023] and Nantong fourth period “226 high-level personnel training project” project.
Disclosure: The authors declare no conflict of interest.
References
- 1.Rivera RM, Cosio MG, Ghezzo H, et al. Comparison of lung morphology in COPD secondary to cigarette and biomass smoke. Int J Tuberc Lung Dis 2008;12:972-7 [PubMed] [Google Scholar]
- 2.Roth M.Pathogenesis of COPD. Part III. Inflammation in COPD. Int J Tuberc Lung Dis 2008;12:375-80 [PubMed] [Google Scholar]
- 3.Abboud RT, Vimalanathan S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int J Tuberc Lung Dis 2008;12:361-7 [PubMed] [Google Scholar]
- 4.Hogg JC, Senior RM. Chronic obstructive pulmonary disease - part 2: pathology and biochemistry of emphysema. Thorax 2002;57:830-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moraes TJ, Chow CW, Downey GP. Proteases and lung injury. Crit Care Med 2003;31:S189-94 [DOI] [PubMed] [Google Scholar]
- 6.Spurzem JR, Rennard SI. Pathogenesis of COPD. Semin Respir Crit Care Med 2005;26:142-53 [DOI] [PubMed] [Google Scholar]
- 7.Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev 2004;56:515-48 [DOI] [PubMed] [Google Scholar]
- 8.Kasahara Y, Tuder RM, Cool CD, et al. Expression of 15-lipoxygenase and evidence for apoptosis in the lungs from patients with COPD. Chest 2000;117:260S. [DOI] [PubMed] [Google Scholar]
- 9.Morissette MC, Parent J, Milot J. Alveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to know. Int J Chron Obstruct Pulmon Dis 2009;4:19-31 [PMC free article] [PubMed] [Google Scholar]
- 10.Diamant M, Tushuizen ME, Sturk A, et al. Cellular microparticles: new players in the field of vascular disease? Eur J Clin Invest 2004;34:392-401 [DOI] [PubMed] [Google Scholar]
- 11.Freyssinet JM. Cellular microparticles: what are they bad or good for? J Thromb Haemost 2003;1:1655-62 [DOI] [PubMed] [Google Scholar]
- 12.Nozaki T, Sugiyama S, Koga H, et al. Significance of a multiple biomarkers strategy including endothelial dysfunction to improve risk stratification for cardiovascular events in patients at high risk for coronary heart disease. J Am Coll Cardiol 2009;54:601-8 [DOI] [PubMed] [Google Scholar]
- 13.Amabile N, Guérin AP, Leroyer A, et al. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol 2005;16:3381-8 [DOI] [PubMed] [Google Scholar]
- 14.Pirro M, Schillaci G, Paltriccia R, et al. Increased ratio of CD31+/CD42- microparticles to endothelial progenitors as a novel marker of atherosclerosis in hypercholesterolemia. Arterioscler Thromb Vasc Biol 2006;26:2530-5 [DOI] [PubMed] [Google Scholar]
- 15.Gordon C, Gudi K, Krause A, et al. Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. Am J Respir Crit Care Med 2011;184:224-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi T, Kobayashi S, Fujino N, et al. Increased circulating endothelial microparticles in COPD patients: a potential biomarker for COPD exacerbation susceptibility. Thorax 2012;67:1067-74 [DOI] [PubMed] [Google Scholar]
- 17.Wright JL, Churg A. Cigarette smoke causes physiologic and morphologic changes of emphysema in the guinea pig. Am Rev Respir Dis 1990;142:1422-8 [DOI] [PubMed] [Google Scholar]
- 18.Hassoun HT, Lie ML, Grigoryev DN, et al. Kidney ischemia-reperfusion injury induces caspase-dependent pulmonary apoptosis. Am J Physiol Renal Physiol 2009;297:F125-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright JL, Sun JP. Effect of smoking cessation on pulmonary and cardiovascular function and structure: analysis of guinea pig model. J Appl Physiol (1985) 1994;76:2163-8 [DOI] [PubMed] [Google Scholar]
- 20.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 2000;106:1311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kasahara Y, Tuder RM, Cool CD, et al. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 2001;163:737-44 [DOI] [PubMed] [Google Scholar]
- 22.Aoshiba K, Yokohori N, Nagai A.Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am J Respir Cell Mol Biol 2003;28:555-62 [DOI] [PubMed] [Google Scholar]
- 23.Plataki M, Tzortzaki E, Rytila P, et al. Apoptotic mechanisms in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis 2006;1:161-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013;187:347-65 [DOI] [PubMed] [Google Scholar]
- 25.Jimenez JJ, Jy W, Mauro LM, et al. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res 2003;109:175-80 [DOI] [PubMed] [Google Scholar]
- 26.Chironi GN, Boulanger CM, Simon A, et al. Endothelial microparticles in diseases. Cell Tissue Res 2009;335:143-51 [DOI] [PubMed] [Google Scholar]
- 27.Garcia S, Chirinos J, Jimenez J, et al. Phenotypic assessment of endothelial microparticles in patients with heart failure and after heart transplantation: switch from cell activation to apoptosis. J Heart Lung Transplant 2005;24:2184-9 [DOI] [PubMed] [Google Scholar]
- 28.Wilson DO, Weissfeld JL, Balkan A, et al. Association of radiographic emphysema and airflow obstruction with lung cancer. Am J Respir Crit Care Med 2008;178:738-44 [DOI] [PMC free article] [PubMed] [Google Scholar]