

Abstract
Mu opioid receptor (MOR) selective antagonists and partial agonists have been used for the treatment of opioid abuse and addiction. Our recent efforts on the identification of MOR antagonists have provided several novel leads displaying interesting pharmacological profiles. Among them, 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-[(3'-isoquinolyl)acetamido]morphinan (NAQ) showed sub-nanomolar binding affinity to the MOR with significant selectivity over the delta opioid receptor (DOR) and the kappa opioid receptor (KOR). Its central nervous system penetration capacity together with marginal agonism in the MOR–GTPγS binding assay made it a very interesting molecule for developing novel opioid abuse and addiction therapeutic agents. Therefore, further pharmacological characterization was conducted to fully understand its biological profile. At the molecular and cellular level, NAQ not only induced no translocation of β-arrestin2 to the MOR, but also efficaciously antagonized the effect of DAMGO in MOR-βarr2eGFP-U2OS cells in the β-arrestin2 recruitment assay. At the in vivo level, NAQ displayed a potent inhibition of the analgesic effect of morphine in the tail-flick assay (ID50 = 1.19 mg/kg). NAQ (10 mg/kg) also significantly decreased the hyper-locomotion induced by acute morphine without inducing any vertical jumps. Meanwhile NAQ precipitated lesser withdrawal symptoms in morphine dependent mice than naloxone. In conclusion, NAQ may represent a new chemical entity for opioid abuse and addiction treatment.
Keywords: Mu opioid receptor, antagonist, NAQ, pharmacology, in vitro, in vivo
1. Introduction
As one of the most serious, chronic and relapsing medical disorders, heroin and prescription opioid abuse is very common and the incidence is still increasing (Fiellin et al., 2004; Manchikanti and Singh, 2008; McLellan et al., 2000; Snyder and Pasternak, 2003; SAMHSA, 2012). It has been demonstrated that for many clinically available opiates, their analgesic function and side effects (such as addiction/abuse liability) are primarily due to their interaction with the mu opioid receptor (MOR) (Contet et al., 2004; Gaveriaux-Ruff and Kieffer, 2002; Martin et al., 2003). While opioid receptor agonist methadone and partial agonist buprenorphine remain the major treatment agents for opioid abuse, opioid receptor antagonists, e.g. naltrexone and naloxone, have recently been shown to be able to block relapse and curb drug craving in opiate addicts (Chen, et al., 2010; George and Ekhtiari, 2010; Lobmaier et al., 2010; Minozzi et al., 2011; Veilleux, et al., 2010; Gold et al., 1982; Gonzalez and Brogden, 1988; Schwyzer, 1977).
Though these antagonists have shown promise alone or in combination with traditional opioid detoxification and maintenance treatments, some severe side effects have been reported. For example, patients receiving naltrexone for opioid dependence exhibited higher than expected rates of overdose and suicide (Miotto et al., 2002; Ritter, 2002). Meanwhile, high dose application of naloxone in patients treated for severe pain with an opioid may cause pulmonary edema and cardiac arrhythmias (van Dorp, 2007). While the delta opioid receptor (DOR) may be related to the mood-related behavior (Baamonde et al., 1992; Filliol et al., 2000; Tejedor-Real et al., 1998; Gaveriaux-Ruff and Kieffer, 2002; Roberts et al., 2001; Jutkiewicz et al., 2006), the kappa opioid receptor (KOR) may play an important role in the inhibition of glycinergic neurotransmission to cardiac vagal neurons (Wang et al., 2004; Lishmanov et al., 2006; Valtchanova-Matchouganska et al., 2004). More evidence will be needed to link the side effects of naltrexone and naloxone directly to their interaction with the DOR and KOR, an antagonist with high selectivity to the MOR seems more promising for opioid abuse and addiction treatment.
Thus far no optimal non-peptidic, selective, and reversible antagonist has been developed for the MOR, though some moderately potent and selective ligands, e.g. cyprodime (Schmidhammer et al., 1989), are in use. Our recent efforts on developing selective opioid receptor antagonists led to the identification of NAQ as a highly selective MOR ligand with subnanomolar affinity for the MOR and high selectivity over the DOR and KOR. NAQ showed potent antagonism in the in vivo tail-flick test (Li et al., 2009). Further characterization indicated that NAQ is a potent CNS agent (Mitra et al., 2011). Primary behavioral studies on NAQ indicated that even at a dose of ten times higher than naloxone and naltrexone, NAQ did not precipitate physical withdrawal symptoms (Yuan et al., 2011). To further characterize its pharmacological profile, a series of cellular and behavioral studies were pursued. Here we report these results to support our original hypothesis that NAQ may be potentially useful for opioid abuse/addiction treatment.
2. Material and Methods
2.1. In vitro pharmacology characterization. Confocal microscopy
Drug-induced translocation of a GFP-tagged β-arrestin2 to the MOR, DOR, and KOR was assessed using MOR-βarr2eGFP-U2OS (MBU), DOR-βarr2eGFP-U2OS (DBU), and KOR-βarr2eGFP-U2OS (KBU) cells (from Larry Barak, Duke University), respectively. Cells were plated on collagen coated glass confocal dishes (MatTek, Ashland, MA) as described in the literature (Barak et al., 1999; Béguin et al., 2012). Prior to imaging, cells were starved for 60 min in serum free MEM without phenol red (Life Technologies, Grand Island, NY). Drug was then added at 10 μM (100 μM NAQ for DBU and KBU cells) and live cell images were obtained by confocal microscopy (Leica SP5 Confocal Microscope) at 0, 5 min (25, and 20 min for NAQ in DBU and KBU, respectively).
2.2. In vivo antagonism profile characterization
2.2.1. Animals
Adult male imprinting control region (ICR) mice (25–35 g) (Harlan, Indianapolis, IN) were used for all experiments. Mice were housed in groups of four to five in standard Plexiglas containers with food and water available ad libitum. Animals were maintained in a temperature and humidity controlled colony on a 12-h light/dark cycle (lights on at 7 am). All studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health. The University of New England Institutional Animal Care and Use Committee approved all protocols involving animals.
2.2.2. Drug Solutions and Injections
Morphine sulfate and naloxone were obtained through the National Institute on Drug Abuse Drug Supply Program. NAQ was synthesized in our labs. All drugs were dissolved in distilled water for intracerebroventricular (i.c.v.) injections and physiological saline (0.9% NaCl) for intraperitoneal (i.p.) and subcutaneous (s.c.) injections. The i.c.v. injections were performed as previously described (Porreca et al., 1984). Briefly, mice were lightly anesthetized with ether, and a 5-mm incision was made along the midline of the scalp. An injection was made using a 25-μL Hamilton syringe at a point 2 mm caudal and 2 mm lateral from bregma. The injection was made using a 27-gauge needle at a depth of 3 mm in a volume of 5 μL. The i.p. and s.c. injections were administered using a 1-mL syringe with a 30-gauge needle at a volume of 10 mL/kg body weight.
2.2.3. Tail-Flick Assay
Antinociception was assessed using the 55 °C warm-water tail-flick assay. The latency to the first sign of a rapid tail-flick was used as the behavioral endpoint (Jannsen et al., 1963). Each mouse was tested for baseline latency by immersing its tail in the water bath and recording the time to response. Mice typically reacted within 1 to 2 s at this temperature, with any mice having a baseline latency of greater than 5 s eliminated from further testing. A maximal score was assigned to mice not responding in 10 s to avoid tissue damage. The percentage of antinociception was calculated as (test latency – control latency)/(10 – control latency) × 100.
2.2.4. Antinociception Studies for Determining Duration of Antagonist Effects
The duration of antagonist effects for NAQ was estimated by pretreating mice with the drug at various time points by an intravenous (i.v.) injection with the drug. Following the pretreatment time of 10, 30, or 60 min mice were injected subcutaneously (s.c.) with 18 mg/kg of morphine. Mice were tested 20 min later (time of morphine peak effect) in the tail-flick assay. A ten second cut off point was used to avoid tissue damage.
2.2.5. Antinociception Studies for Determining Antagonist Potencies
Antagonist potencies were determined by administering vehicle or various doses of the test compound i.v. 10 min prior to an A90 dose of morphine (18 mg/kg, s.c.). Mice were then tested 20 min later in the 55 °C tail-flick assay. The percentage of inhibition of the morphine effect was determined for each mouse, and an ID50 value (and 95% confidence interval) was calculated using linear regression (FlashCalc software; Dr. Michael Ossipov, University of Arizona, Tucson, AZ).
2.2.6. Locomotor Studies for Determining Onset of Antagonist Blockade
Locomotor activity was measured using an activity monitoring system (Coulbourn Instruments, Allentown, PA) and Truscan software. Each of the eight chambers consisted of 10-inch width × 10-inch length × 16-inch height arena surrounded by Plexiglas walls. The floor consisted of a removable plastic drop pan that was cleaned between sessions. A sensor ring surrounded the arena on the bottom outside edges of the four sides of the chamber and contained 16 infrared beams that transected both the length and the width of the chambers on all four sides (beam spacing = 0.6 inches, resolution of 32 × 32 squares). Measurement of the animals' position was determined every 100 ms, and the software calculated a number of parameters related to aspects of locomotor activity. The total distance traveled by each mouse was used as the primary measure of activity, with the data summed into 1-min intervals (bins). Mice were habituated to the chamber for 30 min and then received an injection of morphine sulfate (32 mg/kg s.c.) or saline (s.c.) and were immediately placed back into the chambers for an additional 30 min for a test phase. This dose of morphine was previously determined to produce robust stimulation of forward locomotion with stereotypic circling around the perimeter of the arena and Straub tail. Following this test phase mice were briefly picked up again and administered NAQ (i.v.) or saline (i.v.) and placed back into the chamber from t = 60–120 min.
2.2.7. Physical Dependence Studies
Mice received acute or chronic exposure to morphine using different protocols to produce increasingly severe levels of physical dependence. For the pellet implantation, mice were lightly anesthetized with ether, and a small opening was cut into the back (scapular region) using surgical scissors. A single morphine (25 or 75 mg) or placebo pellet was inserted s.c. toward the lumbar region of the mouse, and the wound was closed with two wound clips. The ability of the tested compound to precipitate opioid withdrawal was assessed by administering i.p. doses at various times after morphine pellet implantation (t = 4 h withdrawal for acute dependence assay, and t = 72 h for chronic dependence assay). Immediately after the injection, mice were placed into a clear Plexiglas cylinder with a filter paper bottom. Mice were videotaped for 20 min, and the tapes were analyzed by a trained observer who was blinded to all experimental treatments. Various measures were recorded for each mouse including vertical jumping, weight of urine & feces, and body weight loss (Sepulveda et al., 1999; Yano and Takemori, 1977). Each nominal sign was either present or absent, and the number of vertical jumps, and fecal boli for each animal was recorded.
2.2.8. Statistical Analysis
Dose-response curves were analyzed as described above using FlashCalc software. Most of the other antinociceptive, locomotor, and physical dependence data were analyzed using analysis of variance (ANOVA) followed by Games/Howell post hoc analysis. The nominal data were analyzed using chi square. In all cases, significance was established at the p < 0.05 level.
3. Results
3.1. In vitro cellular pharmacology
Live cell confocal imaging of MBU, DBU, and KBU cells revealed robust recruitment of β-arrestin2 to the MOR, DOR, and KOR by their agonists DAMGO, SNC80, and U69,593 respectively, as evidenced by the formation of intense green punctae after 5 min of drug exposure (Figure 1). In comparison, NAQ not only failed to induce a visible β-arrestin2 response in MBU cells (Figure 1A), but also antagonized the β-arrestin2 recruitment by DAMGO in MBU cells when pretreated prior to DAMGO treatment for 10 min (Figure 1A). Additionally, NAQ acted as a DOR and KOR partial agonist, recruiting much less βarr2-GFP than the corresponding full agonists (Figure 1B).
Fig. 1.

β-arrestin2 recruitment assay in opioid receptor expressing βarr2eGFP-U2OS cells. The cells are live imaged, and the βarr2 recruitment evoked by drug induced receptor activation is visualized as bright green punctae (white arrows) by confocal microscopy. (A) MOR expressing cells are treated with DAMGO or NAQ (both 10 μM) as indicated. DAMGO induces robust βarr2 recruitment, while NAQ does not. In addition, NAQ pretreatment completely blocks DAMGO induced βarr2 recruitment. These findings are consistent with NAQ acting as an antagonist at the MOR. (B) DOR and KOR expressing cells are treated with a control agonist (SNC80 or U69,593, 10 μM, as indicated) or 100 μM NAQ. The control agonist induces robust βarr2 recruitment, while NAQ induces fewer, dimmer punctae than the control agonist. These findings are consistent with NAQ acting as a partial agonist at the DOR and KOR.
3.2. In vivo pharmacological characterization
To further determine the potency of NAQ to antagonize the CNS mediated effect of morphine, the peak effect of its inhibition of morphine antinociception was determined. As indicated in Fig. 2A, a pretreatment time of 10 min gave the strongest antagonist effect. Therefore, based on this result, the time of NAQ antagonist peak effect was determined to be t = −10 min.
Fig. 2.
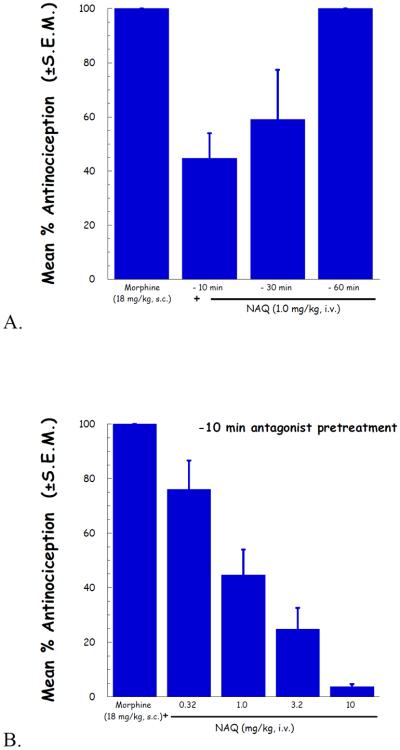
NAQ antinociception antagonism study in the tail-flick assay. (A) NAQ was pretreated as indicated for 10, 30 and 60 minutes, and followed by 18 mg/kg morphine. Maximal NAQ antagonism was seen at 10 and 30 minutes, and absent by 60 minutes.; (B) Using a 10 minute NAQ pretreatment time, an antagonist dose-response curve was generated using NAQ.
Applying the above determined NAQ pretreatment time, a complete dose–response curve for the NAQ antagonism of morphine induced antinociception was conducted. The results were depicted in Fig. 2B. The ID50 value (95% CI) of NAQ was 1.19 (0.685–2.07) mg/kg.
Then high locomotor activity was induced after a 30 min administration of morphine (32 mg/kg, s.c.), compared to saline. NAQ (1.0 mg/kg) had no effect on locomotor activity throughout the whole experiment (Figure 3A). Although 3.2 mg/kg of NAQ significantly reduced locomotion after its administration, the effect was transient (Figure 3B). In contrast, a higher dose of NAQ (10 mg/kg) produced a significant and long lasting effect in reducing morphine induced hyper-locomotion up to 1 h after its injection (Figure 3C).
Fig. 3.

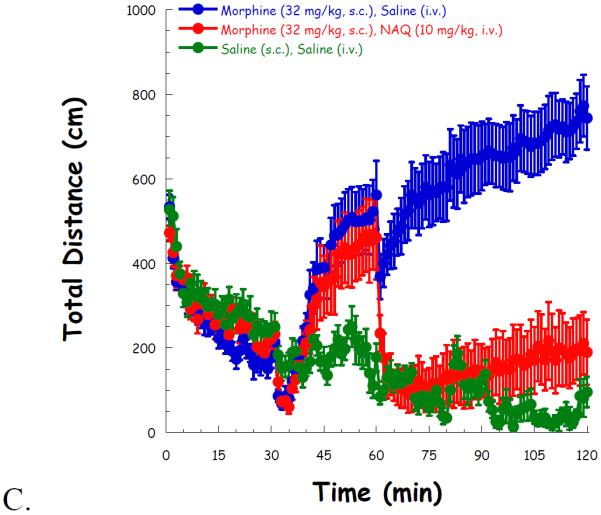
NAQ locomotor study with acute morphine (32 mg/kg, s.c.) administration at doses of 1.0 mg/kg (A), 3.2 mg/kg (B), and 10 mg/kg (C). Morphine/saline (s.c.), and NAQ/saline (i.v.) were injected at t = 30, 60 min, respectively.
Complete dose–response curves for NAQ to precipitate withdrawal jumping were generated in acute (single dose of morphine, 100 mg/kg s.c., 4 h, Figure 4) and chronic (morphine pellet, 75 mg s.c., 72 h, Figure 5) models of opioid physical dependence. Naloxone was included for comparison. As reported previously (Lowery et al., 2011), 10 mg/kg of naloxone treatment induced a significant number of vertical jumps in both models while the severity of withdrawal was significantly greater with the chronic versus acute morphine exposure (Figs 6A, 7A). In contrast, NAQ did not precipitate any jumping in the acute physical dependence assay at a dose of up to 100 mg/kg (Fig. 6A). In the chronic model of dependence, NAQ did precipitate significant jumping at a dose of 10 mg/kg and higher compared to saline, but the withdrawal inducing effect of NAQ was still lower than that of naloxone (Figure 5A).
Fig. 4.
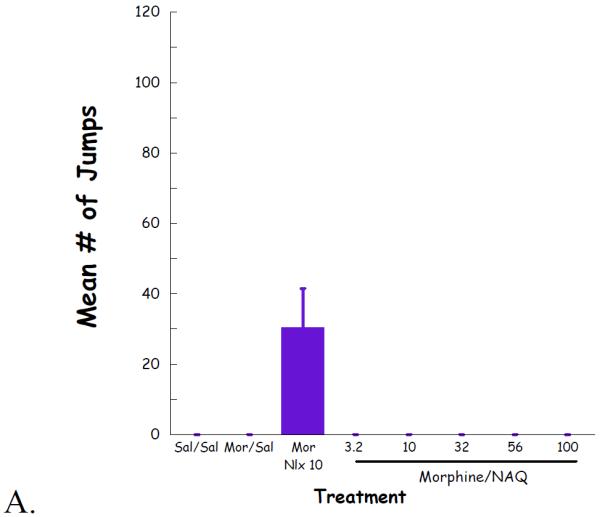

NAQ acute dependence study evaluating vertical jumps (A), weight of feces and urines (B), and weight loss (C).
Fig. 5.

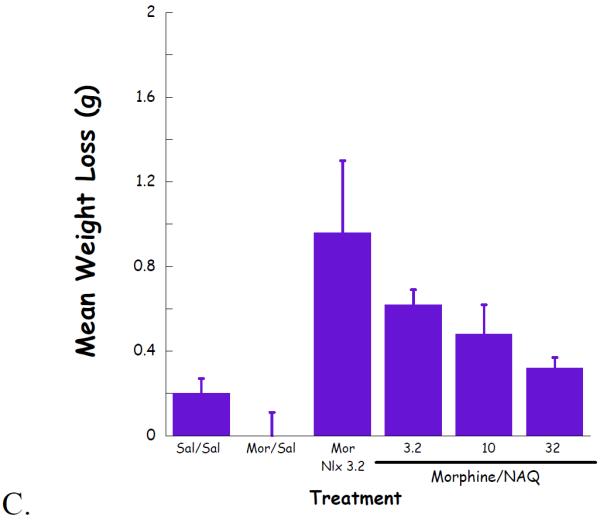
NAQ chronic dependence study evaluating vertical jumps (A), weight of feces and urines (B), and weight loss (C).
The effect of NAQ on gastrointestinal activity, weight of feces and urine, and body weight loss were also recorded. In the acute dependence group, no significant difference was observed for these two physical properties among low doses of NAQ (3.2, 10, 32 mg/kg) and 10 mg/kg of naloxone (Figure 4B & 4C). In contrast, increased doses of NAQ seemed to maintain the body weight while reducing the weight of feces and urine in the chronic dependence study. Furthermore, the same dose of NAQ showed a less significant effect on the weight of feces & urine and body weight loss than naloxone (Figure 5B & 5C).
4. Discussion
A number of the mu opioid receptor (MOR) selective antagonists and partial agonists have been applied in opioid abuse and addiction therapy (Chen, et al., 2010; George and Ekhtiari, 2010; Lobmaier et al., 2010; Minozzi et al., 2011; Veilleux, et al., 2010). NAQ, an isoquinoline substituted 6α-naltrexamine derivative, was recently reported as a highly MOR selective ligand (Li et al., 2009). Depending on the context, NAQ could act as a low efficacy MOR partial agonist or MOR antagonist (Li et al., 2009; Yuan et al., 2011). The current studies aimed to further characterize the pharmacological profile of NAQ to validate its lead compound status for future molecular design.
4.1. NAQ, opioid receptors and β-arrestin2
Arrestins regulate G protein-coupled receptor (GPCR) desensitization and internalization by binding to the agonist-activated, GPCR kinases (GRKs) phosphorylated receptor (Ferguson et al., 1996; Pierce and Lefkowitz, 2001; Shenoy and Lefkowitz, 2003). It has been postulated that opioid receptor internalization is one of the mechanisms for the development of opioid tolerance (Alvarez et al., 2001; Bohn et al., 2004; Gainetdinov et al., 2004; Groer et al., 2007). Accumulated evidence suggests that morphine activation of the MOR is primarily regulated through β-arrestin2. For example, morphine produced improved and extended antinociception in β-arrestin2 knock-out mice with limited tolerance development (Bohn et al., 1999, 2000b, 2002; Przewlocka et al., 2002). In addition, morphine-induced side effects, such as respiratory suppression and constipation, were also absent in these animals (Raehal et al., 2005). As shown in the confocal assay (Figure 3), NAQ did not promote MOR–β-arrestin2 interaction under the test conditions, although it acted as a MOR partial agonist in the [35S]GTPγS assay. While NAQ could cause β-arrestin2 interaction at the KOR and DOR, it was significantly less efficacious than the full agonists U69,593 and SNC80, respectively. Thus, it is very tempting to speculate that the development of tolerance for NAQ at the MOR would be significantly reduced since it did not recruit β-arrestin2 apparently. Further quantitative characterization of the assay is underway now and will be reported in due course.
4.2. NAQ antinociception antagonism (in vivo)
The warm water tail-flick test mainly evaluates spinal reflexes to painful thermal stimuli (Gainetdinov et al., 2004). The relative potencies of naloxone, naltrexone, and NAQ to block the antinociceptive effects of an A90 i.c.v. dose of morphine in the 55 °C tail flick assay did not differ by the injection method. The rank order of both i.c.v. and i.p. antagonist potencies was naltrexone>NAQ>naloxone. Furthermore, none of these compounds produced significant antinociception in the 55 °C tail flick assay when administered alone at doses up to 60 mg/kg i.p. (data not shown). They did not produce any antinociceptive effect in the 50 °C tail flick assay either (data not shown). Thus, NAQ can be considered as an neutral antagonist in this in vivo animal model.
4.3. NAQ locomotor and withdrawal study
NAQ decreased the hyper-locomotion induced by morphine (32 mg/kg, s.c.) in a dose–dependent manner and 10 mg/kg of NAQ completely restored the locomotor activity to the level of saline treated mice. The acute morphine dependence study revealed that at the effective dose in the locomotion assay, 10 mg/kg, NAQ did not precipitate any vertical jumps and had similar effects on the weight of feces & urine, and body weight changes, compared with 10 mg/kg naloxone. Notably, the latter produced significant jumps (~ 30) for the experiment. In contrast, although 10 mg/kg of NAQ induced half of the jumps by naloxone at the same dose in the chronic morphine dependence study, the other physical properties, such as gastrointestinal transit and body weight loss were much lower than that of naloxone and close to the parameters of the control group. Therefore, NAQ seemed to carry a more promising pharmacological profile than traditional opioid antagonists, e.g. naloxone and naltrexone, in opioid abuse and addiction treatment.
5. Conclusion
Several mu opioid receptor (MOR) selective partial agonists and antagonists have been successfully applied in opioid abuse and addiction treatment. However, the modest MOR selectivity of these ligands may compromise their therapeutic index as well as cause other side effects. NAQ, as a highly selective and potent MOR ligand has been identified through our recent effort. In principle, NAQ acted as a low efficacy partial agonist on the MOR with high selectivity over the DOR and KOR. Inspired by these preliminary results, further pharmacological characterization of NAQ was conducted and reported herein. The most profound findings are: NAQ induced no translocation of β-arrestin2 in MOR-βarr2eGFP-U2OS cells; NAQ 10 mg/kg significantly decreased the hyper-locomotion of acute morphine without inducing any vertical jumps in morphine dependent mice. In summary, NAQ may thus represent a potential lead compound to identify new entities for opioid abuse and addiction treatment.
Acknowledgement
The work was funded by PHS grants from NIH/NIDA, DA024022 (YZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdel-Salam OM. Antinociceptive and behavioral effects of ribavirin in mice. Pharmacol. Biochem. Behav. 2006;83:230–238. doi: 10.1016/j.pbb.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Altarifi S, Stevens N. Some determinants of morphine effects on intracranial self-stimulation in rats: dose, pretreatment time, repeated treatment, and rate dependence. Behavioural. Pharmacol. 2011;22:663–673. doi: 10.1097/FBP.0b013e32834aff54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez V, Arttamangkul S, Williams JT. A RAVE about opioid withdrawal. Neuron. 2001;32:761–763. doi: 10.1016/s0896-6273(01)00530-x. [DOI] [PubMed] [Google Scholar]
- Baamonde A, Daugé V, Ruiz-Gayo M, Fulga IG, Turcaud S, Fournié-Zaluski MC, Roques BP. Antidepressant-type effects of endogenous enkephalins protected by systemic RB 101 are mediated by opioid delta and dopamine D1 receptor stimulation. Eur. J. Pharmacol. 1992;216:157–166. doi: 10.1016/0014-2999(92)90356-9. [DOI] [PubMed] [Google Scholar]
- Barak LS, Ferguson SS, Zhang J, Caron MG. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 1997;272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- Béguin C, Potuzak J, Xu W, Liu-Chen LY, Streicher JM, Groer CE, Bohn LM, Carlezon WA, Cohen BM. Differential signaling properties at the kappa opioid receptor of 12-epi-salvinorin A and its analogues. Bioorg. Med. Chem. Lett. 2012;22:1023–1026. doi: 10.1016/j.bmcl.2011.11.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Caron MG. G protein-coupled receptor kinase/beta-arrestin systems and drugs of abuse: psychostimulant and opiate studies in knockout mice. Neuromol. Med. 2004;5:41–50. doi: 10.1385/NMM:5:1:041. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Caron MG. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J. Neurosci. 2002;22:10494–10500. doi: 10.1523/JNEUROSCI.22-23-10494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Chartoff EH. Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat. Protoc. 2007;2:2987–2995. doi: 10.1038/nprot.2007.441. [DOI] [PubMed] [Google Scholar]
- Charles AC, Hales TG. From inhibition to excitation: functional effects of interaction between opioid receptors. Life Sci. 2004;76:479–485. doi: 10.1016/j.lfs.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Chen H, Wu J, Zhang J, Hashimoto K. Recent topics on pharmacotherapy for amphetamine-type stimulants abuse and dependence. Curr. Drug Abuse Rev. 2010;3:222–238. doi: 10.2174/1874473711003040222. [DOI] [PubMed] [Google Scholar]
- Contet C, Kieffer BL, Befort K. Mu opioid receptor: a gateway to drug addiction. Curr. Opin. Neurobiol. 2004;14:370–378. doi: 10.1016/j.conb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Esposito RU, Perry W, Kornetsky C. Effects of d-amphetamine and naloxone on brain stimulation reward. Psychopharmacol. (Berl) 1980;69:187–191. doi: 10.1007/BF00427648. [DOI] [PubMed] [Google Scholar]
- Ferguson SSG, Barak LS, Zhang J, Caron MG. G-protein-coupled receptor regulation: role of G-protein-coupled receptor kinases and arrestins. Can. J. Physiol. Pharmacol. 1996;74:1095–1110. doi: 10.1139/cjpp-74-10-1095. [DOI] [PubMed] [Google Scholar]
- Fiellin DA, Kleber H, Trumble-Hejduk JG, McLellan AT, Kosten TR. Consensus statement in office-based treatment of opioid dependence using buprenorphine. J. Subst. Abuse Treat. 2004;27:153–159. doi: 10.1016/j.jsat.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, Befort K, Gavériaux-Ruff C, Dierich A, LeMeur M, Valverde O, Maldonado R, Kieffer BL. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat. Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gallistel CR, Freyd G. Quantitative determination of the effects of catecholaminergic agonists and antagonists on the rewarding efficacy of brain stimulation. Pharmacol. Biochem. Behav. 1987;26:731–741. doi: 10.1016/0091-3057(87)90605-8. [DOI] [PubMed] [Google Scholar]
- Gaveriaux-Ruff C, Kieffer BL. Opioid receptor genes inactivated in mice: the highlights. Neuropeptides. 2002;36:62–71. doi: 10.1054/npep.2002.0900. [DOI] [PubMed] [Google Scholar]
- George S, Ekhtiari H. Naltrexone in the treatment of opioid dependence. Br. J. Hosp. Med. (Lond) 2010;71:568–570. doi: 10.12968/hmed.2010.71.10.78943. [DOI] [PubMed] [Google Scholar]
- Gold MS, Dackis CA, Pottash AL, Sternbach HH, Annitto WJ, Martin D, Dackis MP. Naltrexone, opiate addiction, and endorphins. Med. Res. Rev. 1982;2:211–246. doi: 10.1002/med.2610020302. [DOI] [PubMed] [Google Scholar]
- Gonzalez JP, Brogden RN. Naltrexone. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of opioid dependence. Drugs. 1988;35:192–213. doi: 10.2165/00003495-198835030-00002. [DOI] [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induce mu-opioid receptor—arrestin interactions or receptor internalization. Mol. Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jannsen PAJ, Niemegeers CJE, Dorg JGH. The inhibitory effects of fentanyl and other morphine-like analgesics on the warm water-induced tail withdrawal reflex in rats. Arzneim-Forsch. 1963;13:502–505. [PubMed] [Google Scholar]
- Jutkiewicz EM. The antidepressant -like effects of delta-opioid receptor agonists. Mol. Interv. 2006;6:162–169. doi: 10.1124/mi.6.3.7. [DOI] [PubMed] [Google Scholar]
- Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715–723. doi: 10.1126/science.2903550. [DOI] [PubMed] [Google Scholar]
- Kornetsky C, Esposito RU. Euphorigenic drugs: effects on the reward pathways of the brain. Fed. Proc. 1979;38:2473–2476. [PubMed] [Google Scholar]
- Li G, Aschenbach LC, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y. Design, Synthesis and Biological Evaluation of 6α- and 6β-N-Heterocyclic Substituted Naltrexamine Derivatives as Mu Opioid Receptor Selective Antagonists. J. Med. Chem. 2009;52:1416–1427. doi: 10.1021/jm801272c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lishmanov AI, Lasukova TV, Maslov LN, Platonov AA. Role of kappa-opioid receptors in the regulation of cardiac resistance to arrhythmogenic effects of ischemia and reperfusion. Ross Fiziol Zh Im I M Sechenova. 2006;92:1419–1428. [PubMed] [Google Scholar]
- Lobmaier P, Gossop M, Waal H, Bramness J. The pharmacological treatment of opioid addiction--a clinical perspective. Eur. J. Clin. Pharmacol. 2010;66:537–545. doi: 10.1007/s00228-010-0793-6. [DOI] [PubMed] [Google Scholar]
- Lowery JJ, Raymond TJ, Giuvelis D, Bidlack JM, Polt R, Bilsky EJ. In Vivo Characterization of MMP-2200, a Mixed δ/μ Opioid Agonist, in Mice. JPET. 2011;336(3):767–778. doi: 10.1124/jpet.110.172866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchikanti L, Singh A. Therapeutic opioids: A tenyear perspective on the complexities and complications of the escalating use, abuse, and nonmedical use of opioids. Pain Phys. 2008;11:S63–S88. [PubMed] [Google Scholar]
- Martin M, Matifas A, Maldonado R, Kieffer BL. Acute antinociceptive responses in single and combinatorial opioid receptor knockout mice: distinct mu, delta and kappa tones. Eur. J. Neurosci. 2003;17:701–708. doi: 10.1046/j.1460-9568.2003.02482.x. [DOI] [PubMed] [Google Scholar]
- McLellan AT, Lewis DC, O'Brien CP, Kleber HD. Drug dependence, a chronic medical illness: Implications for treatment, insurance, and outcomes evaluation. JAMA. 2000;284:1689–1695. doi: 10.1001/jama.284.13.1689. [DOI] [PubMed] [Google Scholar]
- Minozzi S, Amato L, Vecchi S, Davoli M, Kirchmayer U, Verster A. Oral naltrexone maintenance treatment for opioid dependence. Cochrane Database Syst Rev. 2011:CD001333. doi: 10.1002/14651858.CD001333.pub2. [DOI] [PubMed] [Google Scholar]
- Miotto K, McCann M, Basch J, Rawson R, Ling W. Naltrexone and dysphoria: fact or myth? Am. J. Addict. 2002;11:151–160. doi: 10.1080/10550490290087929. [DOI] [PubMed] [Google Scholar]
- Mitra P, Venitz J, Yuan Y, Zhang Y, Gerk PM. Preclinical Disposition (in vitro) of Novel μ-Opioid Receptor Selective Antagonists. Drug Metab. Dispos. 2011;39:1589–1596. doi: 10.1124/dmd.111.038588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Selley DE, Sim-Selley LJ. Pharmacodynamic tolerance. In: Stolerman I, editor. Encyclopedia of psychopharmacology. Springer-Verlag; Berlin: 2010. pp. 994–997. [Google Scholar]
- Nestler EJ, Aghajanian G. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J. Comp. Physiol. Psychol. 1954;47:419–427. doi: 10.1037/h0058775. [DOI] [PubMed] [Google Scholar]
- Pereira Do Carmo G, Folk JE, Rice KC, Chartoff E, Carlezon WA, Negus SS. The selective non-peptidic delta opioid agonist SNC80 does not facilitate intracranial self-stimulation in rats. Eur. J. Pharmacol. 2009a;604:58–65. doi: 10.1016/j.ejphar.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira Do Carmo G, Stevenson GW, Carlezon WA, Negus SS. Effects of pain-and analgesia-related manipulations on intracranial self-stimulation in rats: further studies on pain-depressed behavior. Pain. 2009b;144:170–177. doi: 10.1016/j.pain.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat. Rev. Neurosci. 2001;2:727–733. doi: 10.1038/35094577. [DOI] [PubMed] [Google Scholar]
- Porreca F, Mosberg HI, Hurst R, Hruby VJ, Burks TF. Roles of mu, delta and kappa opioid receptors in spinal and supraspinal mediation of gastrointestinal transit effects and hot-plate analgesia in the mouse. J. Pharmacol. Exp. Ther. 1984;230:341–348. [PubMed] [Google Scholar]
- Przewlocka B, Sieja A, Starowicz K, Maj M, Bilecki W, Przewlocki R. Knockdown of spinal opioid receptors by antisense targeting beta-arrestin reduces morphine tolerance and allodynia in rat. Neurosci. Lett. 2002;325:107–110. doi: 10.1016/s0304-3940(02)00246-x. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Walker JK, Bohn LM. Morphine side effects in β-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- Reid LD. Tests involving pressing for intracranial stimulation as an early procedure for screening the likelihood of addiction of opioids and other drugs. In: Bozarth MJ, editor. Methods of assessing the reinforcing properties of abused drugs. Springer-Verlag; Berlin: 1987. pp. 391–420. [Google Scholar]
- Ritter AJ. Naltrexone in the treatment of heroin dependence: relationship with depression and risk of overdose. Aust. N. Z. J. Psychiatry. 2002;36:224–228. doi: 10.1046/j.1440-1614.2002.01012.x. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, Gold LH, Polis I, McDonald JS, Filliol D, Kieffer BL, Koob GF. Increased ethanol self-administration in delta-opioid receptor knockout mice. Alcohol Clin. Exp. Res. 2001;25:1249–1256. [PubMed] [Google Scholar]
- SAMHSA [Accessed on Jan 21, 2013];Results from the 2011 National Survey on Drug Use and Health: Summary on National findings. 2012 Available at: http://www.samhsa.gov/data/NSDUH/2011SummNatFindDetTables/Index.aspx.
- Schmidhammer H, Burkard WP, Eggstin-Aeppli L, Smith CFC. Synthesis and biological evaluation of 14-alkoxymorphinans. 2. (−)-N-(cyclopropymethyl)-4, 14-dimethoxymorphinana-6-one, a selective mu opioid receptor antagonist. J. Med. Chem. 1989;32:418–421. doi: 10.1021/jm00122a021. [DOI] [PubMed] [Google Scholar]
- Schwyzer R. ACTH: a short introductory review. Ann. NY. Acad. Sci. 1977;247:3–26. doi: 10.1111/j.1749-6632.1977.tb41843.x. [DOI] [PubMed] [Google Scholar]
- Sepulveda J, Astorga JG, Contreras E. Riluzole decreases the abstinence syndrome and physical dependence in morphine-dependent mice. Eur. J. Pharmacol. 1999;379:59–62. doi: 10.1016/s0014-2999(99)00503-8. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem. J. 2003;375:503–515. doi: 10.1042/BJ20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder SH, Pasternak GW. Historical review: Opioid Receptors. Trends Pharmacol. Sci. 2003;24:198–205. doi: 10.1016/S0165-6147(03)00066-X. [DOI] [PubMed] [Google Scholar]
- Stellar JR, Stellar E. The neurobiology of motivation and reward. Springer-Verlag; New York: 1985. [Google Scholar]
- Tejedor-Real P, Micó JA, Smadja C, Maldonado R, Roques BP, Gilbert-Rahola J. Involvement of delta-opioid receptors in the effects induced by endogenous enkephalins on learned helplessness model. Eur. J. Pharmacol. 1998;354:1–7. doi: 10.1016/s0014-2999(98)00423-3. [DOI] [PubMed] [Google Scholar]
- Valtchanova-Matchouganska A, Missankov A, Ojewole JA. Evaluation of the antidysrhythmic effects of delta- and kappa-opioid receptor agonists and antagonists on calcium chloride-, adrenaline- and ischemia/reperfusion-induced arrhythmias in rats. Methods Find Exp. Clin. Pharmacol. 2004;26:31–38. doi: 10.1358/mf.2004.26.1.793470. [DOI] [PubMed] [Google Scholar]
- Veilleux JC, Colvin PJ, Anderson J, York C, Heinz AJ. A review of opioid dependence treatment: pharmacological and psychosocial interventions to treat opioid addiction. Clin Psychol Rev. 2010;30:155–166. doi: 10.1016/j.cpr.2009.10.006. [DOI] [PubMed] [Google Scholar]
- van Dorp EL, Yassen A, Dahan A. Naloxone treatment in opioid addiction: the risks and benefits. Expert Opin. Drug Saf. 2007;6:125–132. doi: 10.1517/14740338.6.2.125. [DOI] [PubMed] [Google Scholar]
- Wang X, Dergacheva O, Griffioen KJ, Huang ZG, Evans C, Gold A, Bouairi E, Mendelowitz D. Action of kappa and Delta opioid agonists on premotor cardiac vagal neurons in the nucleus ambiguus. Neurosci. 2004;129:235–241. doi: 10.1016/j.neuroscience.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Watts VJ. Molecular mechanisms for heterologous sensitization of adenylate cyclase. J. Pharmacol. Exp. Ther. 2002;302:1–7. doi: 10.1124/jpet.302.1.1. [DOI] [PubMed] [Google Scholar]
- Wise RA. Drug-activation of brain reward pathways. Drug Alcohol. Depend. 1998;51:13–22. doi: 10.1016/s0376-8716(98)00063-5. [DOI] [PubMed] [Google Scholar]
- Yano I, Takemori AE. Inhibition by naloxone of tolerance and dependence in mice treated acutely and chronically with morphine. Res. Commun. Chem. Pathol. Pharmacol. 1977;16:721–734. [PubMed] [Google Scholar]
- Yuan Y, Li G, He H, Stevens DL, Kozak P, Scoggins KL, Mitra P, Gerk PM, Selley DE, Dewey WL, Zhang Y. Characterization of 6α- and 6β-N-Heterocyclic Substituted Naltrexamine Derivatives as Novel Leads to Development of Mu Opioid Receptor Selective Antagonists. ACS Chem. Neurosci. 2011;2:346–351. doi: 10.1021/cn2000348. [DOI] [PMC free article] [PubMed] [Google Scholar]