

ABSTRACT
The genome sequences of intestinal Bacteroidales strains reveal evidence of extensive horizontal gene transfer. In vitro studies of Bacteroides and other bacteria have addressed mechanisms of conjugative transfer and some phenotypic outcomes of these DNA acquisitions in the recipient, such as the acquisition of antibiotic resistance. However, few studies have addressed the horizontal transfer of genetic elements between bacterial species coresident in natural microbial communities, especially microbial ecosystems of humans. Here, we examine the genomes of Bacteroidales species from two human adults to identify genetic elements that were likely transferred among these Bacteroidales while they were coresident in the intestine. Using seven coresident Bacteroidales species from one individual and eight from another, we identified five large chromosomal regions, each present in a minimum of three of the coresident strains at near 100% DNA identity. These five regions are not found in any other sequenced Bacteroidetes genome at this level of identity and are likely all integrative conjugative elements (ICEs). Such highly similar and unique regions occur in only 0.4% of phylogenetically representative mock communities, providing strong evidence that these five regions were transferred between coresident strains in these subjects. In addition to the requisite proteins necessary for transfer, these elements encode proteins predicted to increase fitness, including orphan DNA methylases that may alter gene expression, fimbriae synthesis proteins that may facilitate attachment and the utilization of new substrates, putative secreted antimicrobial molecules, and a predicted type VI secretion system (T6SS), which may confer a competitive ecological advantage to these strains in their complex microbial ecosystem.
IMPORTANCE
By analyzing Bacteroidales strains coresident in the gut microbiota of two human adults, we provide strong evidence for extensive interspecies and interfamily transfer of integrative conjugative elements within the intestinal microbiota of individual humans. In the recipient strain, we show that the conjugative elements themselves can be modified by the transposition of insertion sequences and retroelements from the recipient’s genome, with subsequent transfer of these modified elements to other members of the microbiota. These data suggest that the genomes of our gut bacteria are substantially modified by other, coresident members of the ecosystem, resulting in highly personalized Bacteroidales strains likely unique to that individual. The genetic content of these ICEs suggests that their transfer from successful adapted members of an ecosystem confers beneficial properties to the recipient, increasing its fitness and allowing it to better compete within its particular personalized gut microbial ecosystem.
INTRODUCTION
The human intestine harbors a very dense microbial ecosystem containing approximately 1011 to 1012 bacteria per g of colonic content. The species within this community are diverse; however, most of the numerically dominant species are contained within two bacterial taxonomic groups, the Gram-positive phylum Firmicutes and the Gram-negative order Bacteroidales (1, 2). There are more than 25 different human gut Bacteroidales species, many colonizing this ecosystem simultaneously at high density (3, 4). Coresident gut Bacteroidales form ecological networks to utilize dietary polysaccharides (5), with mutualistic interactions likely occurring between these members. Therefore, the presence in the human intestinal microbiota of different Bacteroidales species/strains, each with different phenotypes and fitness properties, may increase the fitness of the Bacteroidales community as a whole.
Many important molecules of the gut Bacteroidales, such as those involved in microbial interactions with the host, other microbes, and dietary or abiotic substances, are not encoded by conserved genes of a species. These include the immunomodulatory polysaccharide molecule PSA of Bacteroides fragilis strain NCTC 9343, the genes for which are contained in less than one-third of B. fragilis strains (6), the B. fragilis enterotoxin (7) implicated in colon cancer (8), glycoside hydrolases and polysaccharide lyases (5) that allow these bacteria to harvest dietary and host glycans (9, 10), and secreted antimicrobial molecules (M. Chatzidaki-Livanis, M. Coyne, and L. Comstock, submitted for publication) predicted to limit local competition.
Many genes contributing to strain diversity are contained in regions likely acquired by horizontal gene transfer (HGT). The genomes of gut Bacteroidales strains show evidence of DNA acquisitions from phage (11), conjugative plasmids (12–14), and conjugative transposons (15, 16). In Bacteroides species, conjugative plasmids and conjugative transposons have been studied intensely for decades because of the importance of these mobile elements in transferring antibiotic resistance genes (12–14, 17, 18). Bacteroidales conjugative transposons fall within the classification of integrative and conjugative elements (ICEs), and as such, they encode the gene products necessary for conjugative transfer, including the mating apparatus, integrases, excisionases, and proteins that regulate transfer (reviewed in references 18 and 19). In order for conjugative transfer to occur, an ICE must excise from the chromosome and form a nonreplicative covalently closed circular intermediate. It is thought that a single strand of the element is then transferred through a mating apparatus to the recipient, with the single strands in both the donor and recipient then being replicated and the element subsequently being (re)integrated into the donor and recipient genomes. Due to the number of genes necessary for these processes, conjugative transposons are relatively large, with those described in Bacteroides averaging approximately 50 to 80 kb (18).
As mating aggregates are necessary for the transfer of conjugative elements, these processes should be favored in dense microbial ecosystems. The human gut is an ideal environment for such conjugative transfers due to its high density of related Bacteroidales species. Most studies of the transfer of mobile genetic elements (MGEs) of gut bacteria have been performed in vitro or with experimental in vivo systems (20–22). Data regarding transfer within the natural human gut ecosystem are lacking, especially regarding the extent of transfer that occurs within an individual human’s microbiota. One study provided strong evidence for the transfer of an 8.9-kb conjugative plasmid among four coresident Bacteroidales species in the gut microbiota of a human girl (23). This small plasmid contained genes and elements necessary for replication and mobilization, such as repA, mobA, mobB, and oriT, but not genes required for the mating apparatus. Due to the importance of MGEs in supplying closely related strains/species with genes that may allow them to rapidly adapt to an ecosystem (reviewed in reference 24) and to understand the nature of these genetic transfers within an individual’s microbiota and how these genomes are modified by interaction with other members of the ecosystem, we studied coresident Bacteroidales species for evidence of HGT. We provide evidence for the interspecies and interfamily transfer of large genetic elements within the gut microbial ecosystem of two healthy humans. We show that these MGEs meet the definition of ICEs or conjugative transposons and carry genes predicted to increase the fitness of the recipient.
RESULTS
Analysis of coresident Bacteroidales strains for evidence of intraecosystem DNA transfer.
Seven strains of different species cocolonizing subject CL02 and eight strains of different species cocolonizing subject CL03 were included in the analyses, with each community including both Bacteroides and Parabacteroides species (Table 1). Within the gut microbiota of each individual, these strains were each present at >108 CFU/g (3). The genomes comprising each of these communities were compared to one another at the DNA level using BLAST. To identify DNA regions with the best likelihood of intraecosystem transfer, we limited the search to identify regions that existed in at least three of the Bacteroidales strains of an individual. Moreover, these segments were required to be at least 10 kb in length and have at least 99.9% DNA identity between strains. These criteria were intentionally conservative to avoid detecting small regions coincidentally common between strains without necessarily indicating recent transfer. Each of these 15 genomes were finished to the draft level, wherein a supercontig or scaffold is assembled by linking smaller contigs, often separated by long stretches of Ns representing unassigned or ambiguous residues. As these Ns cause BLAST to split potentially contiguous hits into multiple returns, the BLAST files were parsed and the results were consolidated and counted as one region if there were gaps of ≤5,000 bp or if the coordinates overlapped. These consolidations revealed six large regions of DNA, referred to herein as regions 1 through 6, two from the CL02 community and four from the CL03 community (Table 1). In general, each of the regions was nearly 100% identical between the identified strains, with the exception of a few single-nucleotide polymorphisms (SNPs), insertion sequences (IS), and/or retroelements (RE) in some regions, as detailed below.
TABLE 1 .
Composition of natural Bacteroidales communities and identification of highly similar regions in strains coresident in a gut microbial ecosystem
| Microbial ecosystem, organisma | No. of contigs | Genome size (bp) | CL02 region (size [bp])b: |
CL03 region (size [bp])b: |
CRISPR/Cas systemc | ||||
|---|---|---|---|---|---|---|---|---|---|
| 1 (24,866) | 2 (116,095) | 3 (17,607) | 4 (60,734) | 5 (42,545) | 6 (44,124) | ||||
| CL02 | |||||||||
| B. cellulosilyticus CL02T12C19 | 25 | 7,678,000 | ✓ | ✓ | Type I | ||||
| B. dorei CL02T12C06 | 21 | 5,997,310 | ✓ | ✓ | Type I | ||||
| B. nordii CL02T12C05 | 10 | 5,707,590 | None | ||||||
| B. ovatus CL02T12C04 | 15 | 7,880,760 | None | ||||||
| B. salyersiae CL02T12C01 | 7 | 5,781,840 | ✓ | ✓ | Type I, III | ||||
| P. goldsteinii CL02T12C30 | 14 | 6,690,360 | Type I | ||||||
| P. johnsonii CL02T12C29 | 6 | 4,613,500 | ✓ | None | |||||
| CL03 | |||||||||
| B. caccae CL03T12C61 | 6 | 5,479,120 | ✓ | None | |||||
| B. dorei CL03T12C01 | 20 | 5,387,250 | Type I | ||||||
| B. fragilis CL03T12C07 | 7 | 5,214,030 | ✓ | ✓ | Type II, III | ||||
| B. ovatus CL03T12C18 | 19 | 6,972,150 | +/− | +/− | ✓ | None | |||
| B. uniformis CL03T12C37 | 14 | 4,890,740 | ✓ | ✓ | Type II | ||||
| B. xylanisolvens CL03T12C04 | 13 | 6,056,100 | ✓ | ✓ | ✓ | None | |||
| P. distasonis CL03T12C09 | 5 | 5,055,860 | ✓ | Type I, II | |||||
| P. merdae CL03T12C32 | 13 | 4,918,050 | ✓ | ✓ | None | ||||
All species belong to Bacteroides or Parabacteroides.
✓, the region is present in the organism; +/−, a large, yet partial segment of the region was identified at >99.9%.
Type(s) of CRISPR/Cas systems present in the organism.
Region 1 was detected in the CL02 community in Bacteroides cellulosilyticus, Bacteroides salyersiae, and Bacteroides dorei. There were several areas where the sequences from these three genomes diverged and were not identified as contiguous aligning segments in our initial analyses, largely due to assembler-introduced Ns. We PCR amplified and sequenced all regions containing Ns (see Table S1 in the supplemental material). These complete sequences revealed that regions 1 from B. cellulosilyticus and B. salyersiae are 100% identical over their entire 24,866-bp length (Fig. 1), whereas the B. dorei genome differed from the other two by a 12-bp insertion and 12-bp deletion and the presence of IS and RE (Fig. 1). The B. cellulosilyticus and B. salyersiae genomes contain two IS, referred to here as ISa and ISb, which are absent in B. dorei, and B. dorei contains a different IS and an RE, referred to here as ISc and REa, both of which are absent in the other two genomes (Fig. 1 and 2). Details of these IS and RE are contained in Table S2. The patterns of these IS and RE suggest that this region initially lacked these elements and was modified by preexisting copies from the genome of a recipient/donor. In fact, each of the strains containing these IS and RE have, in most cases, numerous other copies of these IS and RE in other locations in their genome (Table S2).
FIG 1 .

Comparisons of regions 1 to 5 in the three or four genomes containing these MGEs. Differences between strains for each region following sequencing to resolve Ns are shown. The remaining SNPs displayed were not tested by sequencing and represent the original genome sequence for each isolate. The positions of IS and RE in regions 1 and 2 are shown with the corresponding sizes of these elements.
FIG 2 .

Open reading frame (ORF) maps of regions 1 to 5. Regions are oriented so that the majority of the tra genes (red) read left to right. The letter above the red genes indicates the particular tra gene. An open reading frame map, excluding variable IS and RE, is shown for each region, with the locations of IS and RE indicated. Genes encoding selective orthologous proteins present in each region are color coded as indicated above. Genes comprising the type VI secretion system (T6SS) of region 2 are shown (blue). The 24,866-bp region 1 (boxed) and the 17,607-bp region 3 (boxed) are extended to show the likely extent of the MGEs that were transferred between strains.
Region 2 is very large (116,095 bp) and is present in four of the seven isolates of the CL02 community, B. cellulosilyticus, B. dorei, B. salyersiae, and Parabacteroides johnsonii. Segments containing assembler-introduced Ns were PCR amplified and sequenced (see Table S1 in the supplemental material). These data revealed that regions 2 are identical among these four strains except for an IS element (ISd), present only in P. johnsonii, and two RE, REb, present only in B. salyersiae, and REc, present in both B. salyersiae and B. dorei (Fig. 2; Table S2).
The three regions from the CL03 community contained no assembler-introduced Ns and no IS element differences between strains. The first of these (region 3) is 17,607 bp and is present in CL03 community members Bacteroides uniformis, B. dorei, and Parabacteroides merdae at 100% identity (Fig. 1 and 2).
Region 4 is 60,734 bp and is present in the genomes of CL03 members B. fragilis, Bacteroides xylanisolvens, and Parabacteroides distasonis. The sequences of these three regions agree perfectly, with the exception of one SNP. The first 44,008 bp of this sequence was also present at 100% identity in the Bacteroides ovatus CL03 genome, at the end of scaffold 1.10, and the remaining 16,726 bp was found in the middle of scaffold 1.3. The discontinuity of region 4 in this strain may be the result of an error in the assembly of this genome sequence.
Region 5 is 42,545 bp and is present in CL03 community members B. fragilis, B. xylanisolvens, and B. uniformis. The regions 5 are 100% identical between the three genomes, with the exception of two SNPs at the very end of the region in B. uniformis (Fig. 1). The second half of this region (28,967 bp) was also detected in the B. ovatus genome assembly, residing in the middle of scaffold 1.3.
Region 6 is 44,124 bp and is present in CL03 community members B. ovatus, B. xylanisolvens, and P. merdae. This region was not further analyzed due to its presence at 100% identity in numerous noncommunity members (see below).
Presence of highly identical regions in other Bacteroidales strains.
The possibility existed that these DNA segments represented very promiscuous MGEs and that their presence in these isolates was coincidental and not related to the fact that they were coresident. If so, BLAST analysis of these regions against the database of all draft and completed Bacteroidetes genomes should reveal other strains not present in these natural ecosystems that have similarly sized regions also identical at ≥99.9%. For each of these six regions, BLAST analyses were performed with each of the regions with all IS and RE removed to allow the best chance to return a similarly conserved region. The results of these BLAST analyses revealed that only one of the six regions had ≥99.9% identity to another ≥10-kb segment from other Bacteroidales strains not associated with these natural communities (Table 2). CL03 region 6, which is 44,124 bp in length, is present at 100% identity in numerous other Bacteroidales strains. In contrast, no other sequenced Bacteroidetes genomes contained regions of ≥10 kb that matched regions 1 to 5, even at 99.90% identity, whereas the identified regions in coresident strains are 99.99 to 100% identical to each other, even prior to resolving the Ns (Table 2). These data provide strong evidence that regions 1 to 5 were transferred between coresident strains of the CL02 or CL03 ecosystems, but the BLAST data do not support the intraecosystem transfer of region 6.
TABLE 2 .
BLAST output of regions 1 to 6 against the databasea
| BLAST targetb | % Identityc | Alignment length | No. of: |
Query |
Target |
Accession no. | |||
|---|---|---|---|---|---|---|---|---|---|
| MMd | Gaps | Start | End | Start | End | ||||
| Query—CL02 region 1 | |||||||||
| B. salyersiae CL02T12C01 | 100.00 | 22,005 | 0 | 0 | 1,234 | 23,238 | 1,381,606 | 1,403,610 | NZ_JH724307.1 |
| B. cellulosilyticus CL02T12C19 | 100.00 | 22,005 | 0 | 0 | 1,234 | 23,238 | 5,321 | 27,325 | NZ_JH724088.1 |
| B. dorei CL02T12C06 | 99.99 | 17,671 | 1 | 0 | 7,196 | 24,866 | 530,799 | 548,469 | NZ_JH724135.1 |
| B. eggerthii DSM 20697 | 99.78 | 24,878 | 25 | 10 | 1 | 24,866 | 622,697 | 647,557 | NZ_DS995509.1 |
| B. plebeius DSM 17135 | 97.99 | 10,412 | 200 | 9 | 10,543 | 20,946 | 174,332 | 184,742 | NZ_DS990131.1 |
| B. fragilis 3_1_12 | 95.05 | 13,605 | 615 | 45 | 11,286 | 24,866 | 1,817,695 | 1,804,125 | NZ_EQ973213.1 |
| Query—CL02 region 2 | |||||||||
| B. dorei CL02T12C06 | 100.00 | 109,844 | 2 | 1 | 6,257 | 116,095 | 1,017,432 | 907,589 | NZ_JH724134.1 |
| B. salyersiae CL02T12C01 | 100.00 | 109,844 | 4 | 1 | 6,257 | 116,095 | 567,507 | 677,350 | NZ_JH724309.1 |
| P. johnsonii CL02T12C29 | 100.00 | 55,262 | 0 | 2 | 1 | 55,262 | 109,000 | 164,259 | NZ_JH976468.1 |
| 100.00 | 53,650 | 2 | 1 | 62,451 | 116,095 | 178,109 | 231,758 | NZ_JH976468.1 | |
| B. cellulosilyticus CL02T12C19 | 100.00 | 59,402 | 2 | 0 | 1 | 59,042 | 290,506 | 231,465 | NZ_JH724088.1 |
| 99.99 | 13,339 | 1 | 0 | 59,002 | 72,340 | 231,364 | 218,026 | NZ_JH724088.1 | |
| 100.00 | 12,303 | 0 | 0 | 75,233 | 87,535 | 214,881 | 202,579 | NZ_JH724088.1 | |
| 100.00 | 28,560 | 1 | 0 | 87,536 | 116,095 | 202,478 | 173,919 | NZ_JH724088.1 | |
| B. ovatus CL02T12C04 | 99.55 | 58,086 | 263 | 27 | 58,035 | 116,095 | 5,697 | 63,752 | NZ_JH724231.1 |
| Bacteroides sp. strain 3_2_5 | 98.71 | 33,023 | 426 | 41 | 2 | 33,010 | 2,030,884 | 2,063,856 | NZ_JH636044.1 |
| 97.79 | 24,597 | 543 | 9 | 34,628 | 59,221 | 2,065,264 | 2,089,854 | NZ_JH636044.1 | |
| Query—CL03 region 3 | |||||||||
| B. uniformis CL03T12C37 | 100.00 | 17,607 | 0 | 0 | 1 | 17,607 | 96 | 17,702 | NZ_JH724271.1 |
| P. merdae CL03T12C32 | 100.00 | 17,607 | 0 | 0 | 1 | 17,607 | 142,174 | 159,780 | NZ_JH976456.1 |
| B. dorei CL03T12C01 | 100.00 | 17,607 | 0 | 0 | 1 | 17,607 | 17,607 | 1 | NZ_JH724164.1 |
| B. eggerthii 1_2_48FAA | 98.53 | 17,614 | 245 | 7 | 1 | 17,607 | 30,388 | 47,994 | NZ_AKBX01000010.1 |
| B. plebeius DSM 17135 | 98.48 | 17,615 | 250 | 13 | 2 | 17,607 | 30,516 | 48,121 | NZ_DS990120.2 |
| B. intestinalis DSM 17393 | 98.50 | 16,772 | 236 | 12 | 2 | 16,766 | 17,245 | 34,007 | NZ_ABJL02000003.1 |
| Query—CL03 region 4 | |||||||||
| B. fragilis CL03T12C07 | 100.00 | 60,734 | 0 | 0 | 1 | 60,734 | 285,831 | 346,564 | NZ_JH724182.1 |
| P. distasonis CL03T12C09 | 100.00 | 60,734 | 0 | 0 | 1 | 60,734 | 2,432,090 | 2,492,823 | NZ_JH976495.1 |
| B. xylanisolvens CL03T12C04 | 100.00 | 60,734 | 2 | 0 | 1 | 60,734 | 2,000,696 | 1,939,963 | NZ_JH724294.1 |
| B. ovatus CL03T12C18 | 100.00 | 44,008 | 2 | 0 | 1 | 44,008 | 31,399 | 75,406 | NZ_JH724250.1 |
| 100.00 | 16,726 | 0 | 0 | 44,008 | 60,733 | 215,190 | 231,915 | NZ_JH724243.1 | |
| B. fragilis NCTC 9343 | 99.20 | 38,365 | 289 | 17 | 22,378 | 60,733 | 2,040,415 | 2,078,771 | NC_003228.3 |
| 99.63 | 15,410 | 55 | 2 | 1,801 | 17,209 | 2,017,133 | 2,032,541 | NC_003228.3 | |
| B. helcogenes P 36-108 | 99.60 | 15,423 | 58 | 3 | 1,801 | 17,221 | 230,238 | 245,659 | NC_014933.1 |
| B. uniformis ATCC 8492 | 95.50 | 16,906 | 652 | 53 | 30,502 | 47,366 | 215,548 | 198,710 | NZ_DS362247.1 |
| Query—CL03 region 5 | |||||||||
| B. xylanisolvens CL03T12C04 | 100.00 | 42,545 | 0 | 0 | 1 | 42,545 | 1,171,697 | 1,214,241 | NZ_JH724294.1 |
| B. fragilis CL03T12C07 | 100.00 | 42,545 | 0 | 0 | 1 | 42,545 | 457,382 | 414,838 | NZ_JH724184.1 |
| B. uniformis CL03T12C37 | 100.00 | 42,545 | 2 | 0 | 1 | 42,545 | 725,544 | 768,088 | NZ_JH724268.1 |
| B. ovatus CL03T12C18 | 100.00 | 28,967 | 1 | 0 | 13,578 | 42,544 | 205,601 | 176,635 | NZ_JH724243.1 |
| Bacteroides sp. strain 3_1_23 | 96.50 | 18,468 | 561 | 50 | 16,314 | 34,740 | 2,449,865 | 2,431,442 | NZ_GG774949.1 |
| B. finegoldii DSM 17565 | 96.60 | 17,611 | 497 | 57 | 17,192 | 34,740 | 29,060 | 46,630 | NZ_GG688325.1 |
| B. salyersiae DSM 18765 | 97.03 | 16,978 | 442 | 35 | 17,790 | 34,740 | 554,600 | 537,659 | NZ_KB905466.1 |
| Query—CL03 region 6 | |||||||||
| B. xylanisolvens CL03T12C04 | 100.00 | 44,124 | 0 | 0 | 1 | 44,124 | 388,361 | 344,238 | NZ_JH724296.1 |
| P. merdae CL03T12C32 | 100.00 | 26,817 | 0 | 0 | 1 | 26,817 | 204,214 | 231,030 | NZ_JH976457.1 |
| 100.00 | 16,701 | 0 | 0 | 27,424 | 44,124 | 236,576 | 253,276 | NZ_JH976457.1 | |
| B. ovatus CL03T12C18 | 100.00 | 12,583 | 0 | 0 | 1 | 12,583 | 530,345 | 542,927 | NZ_JH724241.1 |
| 100.00 | 23,711 | 1 | 0 | 12,584 | 36,294 | 545,391 | 569,101 | NZ_JH724241.1 | |
| B. eggerthii DSM 20697 | 100.00 | 44,124 | 0 | 0 | 1 | 44,124 | 159,910 | 204,033 | NZ_DS995511.1 |
| P. merdae CL09T00C40 | 100.00 | 44,124 | 0 | 0 | 1 | 44,124 | 372,514 | 416,637 | NZ_JH976526.1 |
| Bacteroides sp. strain 3_1_19 | 100.00 | 44,124 | 0 | 0 | 1 | 44,124 | 180,923 | 225,046 | NZ_GG774763.1 |
| Bacteroides sp. strain D22 | 100.00 | 44,124 | 0 | 0 | 1 | 44,124 | 56,078 | 100,201 | NZ_GG774819.1 |
| Alistipes sp. strain HGB5 | 100.00 | 44,124 | 0 | 0 | 1 | 44,124 | 66,384 | 110,507 | NZ_AENZ01000040.1 |
| Alistipes onderdonkii DSM 19147 | 100.00 | 44,124 | 1 | 0 | 1 | 44,124 | 55,386 | 11,263 | NZ_KB894552.1 |
| B. intestinalis DSM 17393 | 100.00 | 44,124 | 0 | 1 | 1 | 44,124 | 456,857 | 412,735 | NZ_ABJL02000006.1 |
| B. stercoris ATCC 43183 | 100.00 | 44,124 | 2 | 0 | 1 | 44,124 | 103,168 | 59,045 | NZ_DS499672.1 |
| P. merdae ATCC 43184 | 100.00 | 44,124 | 1 | 1 | 1 | 44,124 | 73,390 | 117,512 | NZ_DS264518.1 |
| B. fragilis YCH46 DNA | 100.00 | 44,124 | 1 | 1 | 1 | 44,124 | 163,822 | 119,700 | NC_006347.1 |
All variant IS and RE were removed from query sequences. Boldface indicates strains from a natural ecosystem.
All species belong to Bacteroides or Parabacteroides, unless otherwise indicated.
% Identity was rounded to the closest hundredth of a percent.
MM, mismatches.
Analysis of highly similar regions within the genomes of mock communities of Bacteroidales.
To estimate the frequency with which one might expect to find such long and nearly identical DNA segments (i.e., ≥10 kb and ≥99.9% identity in three strains) among bacteria that were not coresident, we performed a similar BLAST search using 1,000 eight-member mock communities of Bacteroidales assembled from a set of 84 Bacteroides and Parabacteroides genome sequences of similar quality (see Materials and Methods; see Table S3 in the supplemental material). Genomes were pseudorandomly assigned to each mock community such that no collection contained two genomes of the same species and each microbiota contained at least one but not more than two Parabacteroides genomes. Each collection was further restrained by limiting it to contain no more than one genome of each of the CL02, CL03, and CL09 strains, as these groups each represent strains collected from three different subjects (3).
The mock-community BLAST analysis revealed only three unique segments of qualifying DNA that were ≥10 kb, ≥99.9% identical, and shared by 3 strains within a mock community but not by any other genomes in the BLAST comparison database (Table 3; see Table S4 in the supplemental material). The first of these regions is 12,502 bp and is contained in the same three Bacteroides strains that were present in both mock community 59 and mock community 609, the second is 13,248 bp and is present in two Bacteroides and a P. merdae genome of one mock community, and the third region is 30,598 bp and was contained in three Bacteroides genomes from one mock community.
TABLE 3 .
BLAST output of three unique regions from the mock communities against the databasea
| BLAST query (accession no.:position), targetb | % Identityc | Alignment length | No. of: |
Query |
Target |
Accession no. | |||
|---|---|---|---|---|---|---|---|---|---|
| MMd | Gaps | Start | End | Start | End | ||||
| Query—B. stercoris ATCC 43183 (NZ_DS499676.1:176961–207558) | |||||||||
| B. stercoris ATCC 43183 | 100.00 | 30,598 | 0 | 0 | 1 | 30,598 | 176,961 | 207,558 | NZ_DS499676.1 |
| B. vulgatus PC510 | 99.96 | 30,599 | 10 | 2 | 1 | 30,598 | 30,597 | 1 | NZ_ADKO01000036.1 |
| B. uniformis ATCC 8492 | 99.95 | 30,602 | 9 | 3 | 1 | 30,598 | 176,746 | 207,345 | NZ_DS362245.1 |
| B. cellulosilyticus CL02T12C19 | 99.80 | 13,767 | 23 | 3 | 1 | 13,764 | 624,314 | 610,549 | NZ_JH724089.1 |
| B. vulgatus ATCC 8482 | 99.66 | 25,496 | 76 | 10 | 1 | 25,491 | 2,046,625 | 2,021,136 | NC_009614.1 |
| P. merdae ATCC 43184 | 99.61 | 25,500 | 83 | 12 | 1 | 25,491 | 117,639 | 92,147 | NZ_DS264524.1 |
| Query—B. fragilis HMW 616 (NZ_JH815527.1:1–13248) | |||||||||
| B. fragilis HMW 616 | 100.00 | 13,248 | 0 | 0 | 1 | 13,248 | 1 | 13,248 | NZ_JH815527.1 |
| 100.00 | 13,248 | 0 | 0 | 1 | 13,248 | 80,750 | 67,503 | NZ_JH815526.1 | |
| P. merdae ATCC 43184 | 99.99 | 13,248 | 1 | 0 | 1 | 13,248 | 356,073 | 342,826 | NZ_DS264540.1 |
| Bacteroides sp. strain 4_3_47FAA | 99.99 | 13,248 | 1 | 0 | 1 | 13,248 | 561,549 | 548,302 | NZ_JH114362.1 |
| B. coprocola DSM 17136 | 89.09 | 8,440 | 800 | 87 | 4,875 | 13,248 | 8,644 | 17,028 | NZ_DS981488.1 |
| B. plebeius DSM 17135 | 89.09 | 8,440 | 800 | 87 | 4,875 | 13,248 | 241,574 | 249,958 | NZ_DS990119.1 |
| B. finegoldii CL09T03C10 | 86.88 | 5,349 | 638 | 46 | 7,935 | 13,248 | 82,421 | 77,102 | NZ_JH951901.1 |
| Query—B. faecis MAJ27 (NZ_AGDG01000049.1:1–12502) | |||||||||
| B. faecis MAJ27 | 100.00 | 12,502 | 0 | 0 | 1 | 12,502 | 1 | 12,502 | NZ_AGDG01000049.1 |
| B. plebeius DSM 17135 | 99.98 | 12,502 | 2 | 0 | 1 | 12,502 | 28,019 | 15,518 | NZ_DS990120.2 |
| B. intestinalis DSM 17393 | 99.98 | 12,502 | 2 | 1 | 1 | 12,502 | 14,748 | 2,248 | NZ_ABJL02000003.1 |
| Bacteroides sp. strain D22 | 99.87 | 12,502 | 0 | 4 | 1 | 12,502 | 35,210 | 22,725 | NZ_GG774809.1 |
| P. merdae CL03T12C32 | 98.71 | 8,731 | 112 | 1 | 603 | 9,333 | 139,124 | 130,395 | NZ_JH976456.1 |
| Bacteroides sp. strain 9_1_42FAA | 98.67 | 9,241 | 120 | 1 | 2,512 | 11,752 | 25,552 | 34,789 | NZ_EQ973174.1 |
Boldface indicates strains from a natural ecosystem.
All species belong to Bacteroides or Parabacteroides.
% Identity was rounded to the closest hundredth of a percent.
MM, mismatches.
Therefore, in the two natural communities CL02 and CL03, five unique qualifying regions were retrieved with no other matches in the database at 99.9% or greater (mean of 2.5 regions per community), whereas only four such regions (including one unique region found in two different communities) were retrieved from similar analyses of 1,000 communities of non-coresident strains (mean of 0.004 regions per community). Moreover, many of the qualifying DNA segments detected in the real communities were larger than the segments detected in the mock communities. Therefore, the likelihood of detecting such highly similar and unique regions in a set of Bacteroidales strains that are coresident is 625 times higher than the likelihood of detecting such a region among non-coresident strains, providing strong evidence that the five identified regions from the CL02 and CL03 ecosystems were transferred between strains while coresident in the gut microbiota of these humans.
Genetic content of the five transferred regions.
Conjugative transposons or ICEs contain genes encoding all the functions for their transfer, including the machinery for the conjugative mating apparatus, which in Gram-negative bacteria largely occurs by type IV secretion systems (T4SS) (19). Regions 2, 4, and 5 each contain numerous genes encoding Tra proteins of T4SS machinery, including TraD, -G, -J, -K, -L, -M, and -N. These tra genes from each region have a similar genetic architecture, displaying a modular unit of functionally related genes, characteristic of ICEs (19). Regions 1 and 3 are likely contained on larger MGEs but were truncated in our analyses due to assembly scaffold breaks in at least one of the three qualifying genomes. For region 3, the scaffold from P. merdae extended beyond the defined region, and several smaller scaffolds from both the B. uniformis and B. dorei genomes aligned at 100% identity with the larger P. merdae sequence with relatively small gaps or overlaps, indicating that the true size of the transferred element is likely ~47 kb (Fig. 3). All of the same tra genes were contained in this extended region (Fig. 2), suggesting that this MGE is also an ICE. Region 1 also continued upstream for an additional 61.5 kb at near 100% identity in two of three genomes (Fig. 3). Alignment of this extended region with the B. cellulosilyticus sequence indicated that the genome was likely misassembled in this area. However, for the two genomes that continued, the same tra genes were identified (Fig. 2). Therefore, three of the five identified regions meet the definition of an ICE, with regions 1 and 3 also likely part of a larger ICE that was truncated in our analysis due to incomplete or incorrect assembly of the genome sequences.
FIG 3 .

Likely extent of the MGEs containing regions 1 and 3. Boxed regions are the extent of regions 1 and 3 identified by the indicated BLAST criteria. (A) Expansion of region 1 in two of the three genomes. (B) Expansion of region 3 based on smaller matching scaffolds in each of the two genomes that are noncontiguous with the region from P. merdae.
These ICEs also contained other common genes, such as those encoding single-stranded-DNA-binding proteins, relaxases, ParBs, excisionases, TOPRIM-like proteins, ATPases similar to those involved in chromosomal partitioning, and proteins with DUF4133, DUF4134, and DUF4099 (Fig. 2, Table 4). Each of these regions also contains at least one gene with predicted site-specific recombinase activity, likely involved in integration of the element (Fig. 2, Table 4).
TABLE 4 .
Numbers of various products encoded by the five intracommunity-transferred regions
| Putative category | Putative assignment/function of gene products | No. of products in: |
||||
|---|---|---|---|---|---|---|
| CL02 region: |
CL03 region: |
|||||
| 1 | 2 | 3 | 4 | 5 | ||
| Conjugative transfer machinery | TraD (coupling protein) | 1 | 1 | 1 | 1 | 1 |
| TraG | 1 | 1 | 1 | 1 | ||
| TraJ | 1 | 1 | 1 | 1 | ||
| TraK | 1 | 1 | 1 | 1 | 1 | |
| TraM | 1 | 1 | 1 | 1 | 1 | |
| TraN | 1 | 1 | 1 | 1 | 1 | |
| TraO | 1 | 1 | ||||
| Recombinases | Serine site-specific recombinases | 2 | 1 | 1 | ||
| Tyrosine site-specific recombinases/integrases | 1 | 2 | 2 | |||
| Element transfer/partitioning/segregation | TOPRIM-like, DUF3991 | 2 | 1 | 1 | ||
| TOPRIM primase | 1 | 1 | 1 | |||
| Excisionase | 1 | 1 | ||||
| Single-stranded-DNA-binding protein family | 3 | 2 | ||||
| ATPases—chromosome partitioning/CobQ/CobB/MinD/ ParA nucleotide binding | 1 | 1 | 1 | 1 | 1 | |
| PRTRC system ParB family | 1 | 1 | 1 | |||
| Chromosome segregation protein SMC | 1 | 1 | ||||
| Relaxase/mobilization nuclease | 1 | 1 | 1 | |||
| Other common proteins/domains | RibD C-terminal domain, dihydrofolate reductase | 1 | 1 | 1 | ||
| DUF4099 | 1 | 1 | 1 | 1 | 1 | |
| DUF4133 | 1 | 1 | 1 | |||
| DUF4134 | 1 | 1 | 1 | |||
| DUF3408 | 1 | 1 | 1 | |||
| PH domain protein | 1 | 1 | 1 | |||
| Transcriptional regulation/DNA binding | RteC family | 1 | 1 | |||
| TetR family | 1 | 1 | 1 | |||
| Other transcriptional regulator | 2 | 1 | 1 | |||
| Other helix-turn-helix domain DNA-binding proteins | 1 | 1 | 4 | 1 | ||
| Selfish genes/element survival | Putative toxin | 1 | 1 | 1 | 2 | 1 |
| Putative antitoxin /immunity protein | 1 | 2 | 1 | 4 | 1 | |
| Anti-restriction protein | 1 | 1 | 1 | 1 | 1 | |
| DNA methylase | 1 | 3 | ||||
| Potential fitness genes | Fimbria synthesis | 2 | ||||
| MACPF domain containing | 1 | |||||
| M23 peptidase family | 2 | 3 | 1 | |||
| Type VI secretion system (T6SS) | ✓a | |||||
✓, the region is present in the organism.
As ICEs must excise from the donor genome in order to transfer to a recipient, some encode a toxin-antitoxin pair to ensure that they are not lost in the donor strain prior to replication and reintegration (25). Regions 1 to 5 each encode identifiable toxin-antitoxin or immunity proteins, likely for element maintenance (Table 4; see Table S5 in the supplemental material). In addition, each of these five regions encodes a predicted antirestriction protein, frequently contained on a conjugative element, which facilitates maintenance of the ICE in the recipient prior to its modification.
Genes that may contribute to fitness.
Each region also contains numerous genes unrelated to transfer and maintenance of the ICE. The majority of these genes encode hypothetical proteins of unknown function (see Table S5 in the supplemental material); however, many encode products with putative functions that suggest that they could contribute to fitness. Region 1 encodes genes likely involved in fimbria synthesis. Similar FimA orthologs in the oral Bacteroidales species Porphyromonas gingivalis allow this organism to attach to host cells (reviewed in reference 26). In these gut Bacteroidales, these fimbriae may expand the niche of these organisms, allowing them to attach to other host, microbial, or dietary particle surfaces in the gut.
Region 2 encodes three putative orphan DNA methyltransferases not associated with a cognate restriction enzyme. DNA methyltransferases enable genomewide epigenetic modifications which have been shown to have diverse outcomes, including transcriptional regulation, cell cycle control, and regulation of conjugal transfer (27, 28). Therefore, these newly acquired genes may have significant effects on recipient fitness.
There are also genes in these regions that may contribute to competitive ecological interactions. Regions 2 and 3 contain a total of four predicted M23 peptidases (Table 4; see Table S5 in the supplemental material) that hydrolyze peptidoglycan and have various physiological functions, including bacteriocin activity (29). In addition, region 3 encodes a protein with a membrane attack/perforin (MACPF) domain found in proteins widely distributed in Bacteroidetes species, one of which we have shown to have secreted antimicrobial activity targeting heterologous strains (M. Chatzidaki-Livanis et al., submitted for publication).
The most notable feature of these regions is a large cluster of genes in region 2 encoding characteristic type VI secretion system (T6SS) proteins (Fig. 2 and Fig. 4; see Table S6 in the supplemental material). Type VI secretion systems are widely distributed among Proteobacteria but have not previously been reported in Bacteroidetes. T6SSs translocate toxic effector proteins into neighboring cells in a contact-dependent manner, killing sensitive cells (reviewed in references 30 and 31). T6SS loci are very diverse, and certain hallmark T6SS proteins exhibit little pairwise identity in sequence-sequence comparison. Thus, the identification of these core proteins often relies on the presence of certain motifs (sequence-profile comparisons) or on remote homologies detectable by profile-profile comparisons or structural similarities.
FIG 4 .
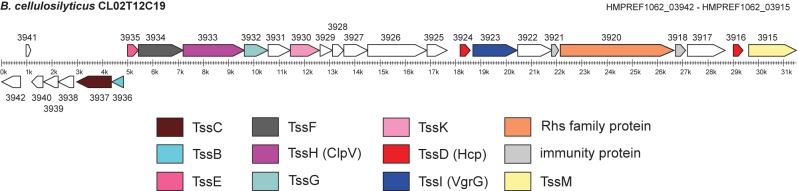
ORF map of portion of region 2, encoding a putative T6SS. Genes encoding proteins characteristic of or commonly associated with T6SS are color coded as indicated below. These designations are based on the analyses as outlined in Table S6 in the supplemental material. The putative functions of all gene products encoded by the genes shown here are included in Table S6.
Such profile-profile analyses (32, 33) reveal that this locus encodes numerous proteins encoded by T6SS loci, including TssI (VgrG) and TssD (Hcp), two proteins that comprise the T6SS cell-puncturing structure, the contractile sheath proteins TssB and TssC, the phage baseplatelike protein TssE, and the TssH (ClpV) ATPase, thought to be involved in recycling of TssB and TssC.
The locus also encodes proteins identified as TssF, TssG, and TssK, T6SS proteins whose function is less well understood, and a large transmembrane protein with both a GTP-ATP binding domain and a P-loop ATPase domain, both of which are structural features of TssM, a protein involved in anchoring the T6SS apparatus to the cell wall. Additionally, this locus encodes an Rhs protein with a deaminase domain and two putative immunity proteins, features that are also found associated with T6SS loci. As TssM (34), TssK (35), TssG, and TssF are associated with T6SS but not phage, it is unlikely that this region is an integrated phage. Although T6SS loci have been predicted to be transferred between strains by HGT, this is the first description of a putative T6SS locus likely being transferred on a conjugative element between strains within a natural human ecosystem.
DISCUSSION
By analyzing the genomes of Bacteroidales strains cocolonizing the guts of two humans, we provide evidence that as much as 140 kb of DNA has been exchanged within several strains in the microbiota of two individuals and suggest that ICE elements are likely responsible for this transfer. These transfers were not limited to Bacteroides species; they also included Parabacteroides species. Bacteroides are contained within the family Bacteroidaceae and Parabacteroides within the family Porphyromonadaceae, and as such, the Parabacteroides are more phylogenetically related to the oral pathogens Porphyromonas gingivalis and Tannerella forsythia than to the Bacteroides genus. However, the Parabacteroides have many phenotypes that are more in common with the Bacteroides than with the oral Porphyromonadaceae. A few notable phenotypes include the synthesis of multiple phase-variable capsular polysaccharides (36) and the production of the enzyme Fkp, which allows these bacteria to incorporate salvaged fucose from the gut environment into their glycans (37). The data from the current study reveal the tremendous capacity for species of these different families to share numerous phenotypes encoded by these ICEs. In fact, these data show that a Bacteroides strain and a Parabacteroides strain living together in the same human gut share many features that are not shared with other, non-coresident members of the same genus/species.
These genomic comparisons document the continued evolution of these ICEs, which are subject to continued bombardment with IS and RE elements, likely from the recipient’s genome. These modifications result in highly personalized genomes that are likely unique to each human. These data also reveal the extent to which our Bacteroidales strains are likely altered by the other members of our gut microbial community.
In this retrospective study, we cannot determine which of these strains may have been the donor of the ICE and which the recipients. However, due to the presence of particular IS or RE in an ICE of one or two strains but not all, some predictions can be made. For example, ISa and ISb are each present in the exact same locations of region 1 for both B. cellulosilyticus and B. salyersiae, but both are absent in B. dorei (Fig. 1). Therefore, it is unlikely that B. dorei received this ICE from either B. cellulosilyticus or B. salyersiae. In addition, as both B. cellulosilyticus and B. salyersiae each contain other copies of both of these IS in their genomes, these elements were likely transferred from one member’s chromosomal copy to the ICE and then transferred to the other strain. In the recipient, the IS present on the ICE then could have served as the donor for transposition into other areas of its chromosome. The data clearly demonstrate that ICEs are efficient vehicles for the transfer of IS and RE between coresident strains (38).
Although ICEs are selfish elements and contain numerous genes dedicated to their transmission and maintenance, the carriage of fitness-conferring genes would increase the chance that the recipient of an ICE is maintained in the ecosystem. Indeed, elements transferred by HGT are known to encode fitness-conferring traits (24), the most obvious being genes encoding antibiotic resistance. In this way, HGT is a means to allow for rapid adaptation of new members into specific adapted communities (39).
In analyzing the contents of these five genetic elements, we can speculate as to the influence on fitness of the transfer and acquisition of these ICEs. The predicted T6SS encoded by region 2 and the putative antimicrobial molecules encoded by regions 2 and 3 are examples of transfers/acquisitions that may be advantageous to both the donor and recipient. The recipient is now endowed with machinery that may allow it to promote antagonistic interactions to limit competition, and the donor may benefit in that the recipient can now deploy this energetically costly defensive machinery and share the burden of protecting the ecosystem from invasion. In Pseudomonas aeruginosa, a T6SS was shown to be assembled in response to mating pair formation by a T4SS of Escherichia coli, and therefore, it functions to prevent conjugal DNA transfer by killing the attempting donor strain (40). This response is postulated to block the acquisition of parasitic foreign DNA. It will be interesting to determine whether the Bacteroidales species that acquired the T6SS are now unable to receive additional T4SS-mediated DNA transfers and, if so, whether it is an advantage or disadvantage for these strains in the human gut ecosystem.
The identification of these intracommunity-transferred ICEs will allow for more in-depth analyses to address ecological interactions between these strains and other Bacteroidales strains of these natural communities that do not contain these elements. Because the majority of the genes on the five identified elements encode proteins of unknown function, there are potentially numerous advantages that these regions could confer to a recipient in its interactions with the host and other community members. As these strains represent the evolutionary winners at the time of their isolation, it is unlikely that these ICEs conferred an overall fitness disadvantage to the recipients. The isolation of additional Bacteroidales strains from these same subjects will allow us to determine whether strains containing these ICEs have been maintained over time and/or whether the ICEs have since been transferred to the remaining Bacteroidales members of these communities.
MATERIALS AND METHODS
Strains and genome sequences.
The 15 CL02 and CL03 Bacteroidales strains of this study were isolated from human feces, as described previously (3), as part of a study approved by the Partners Human Research Committee IRB that complied with all relevant federal guidelines and institutional policies. The genome sequencing of these strains was performed at the Broad Institute as part of the Human Microbiome Project (41). These sequences were deposited in GenBank and are identified by their project accession numbers, as follows: Bacteroides caccae, CL03T12C61 and PRJNA64801; B. cellulosilyticus, CL02T12C19 and PRJNA64803; B. dorei, CL02T12C06 and PRJNA64807; B. dorei, CL03T12C01 and PRJNA64809; B. fragilis, CL03T12C07 and PRJNA64813; Bacteroides nordii, CL02T12C05 and PRJNA64823; B. ovatus, CL02T12C04 and PRJNA64825; B. ovatus, CL03T12C18 and PRJNA64827; B. salyersiae, CL02T12C01 and PRJNA64829; B. uniformis, CL03T12C37 and PRJNA64835; B. xylanisolvens, CL03T12C04 and PRJNA64839; P. distasonis, CL03T12C09 and PRJNA64883; Parabacteroides goldsteinii, CL02T12C30 and PRJNA64887; P. johnsonii, CL02T12C29 and PRJNA64889; and P. merdae, CL03T12C32 and PRJNA64891.
Intracommunity genome comparisons.
The genomes comprising each of the mock or natural communities were compared to one another at the DNA level using BLAST. All hits of ≥10,000 bp that shared ≥99.9% identity were retained, with redundancy due to reciprocal hits eliminated. The BLAST files were parsed to detect instances in which a particular query scaffold returned multiple qualifying segments (≥10 kb at ≥99.9% identity) against a particular target scaffold. These results were consolidated and counted as one qualifying hit if the gaps between the query sequence coordinates were ≤5,000 bp or if the query coordinates overlapped. If the same segment of query DNA produced multiple qualifying returns from different scaffolds of the same target genome, this was also counted as one hit.
Once consolidated, the BLAST results were further parsed for contiguous query sequences producing qualifying matches against two or more target genomes within a community. The overlapping relationship between these BLAST hits was analyzed to calculate the longest contiguous stretch of query DNA present in the target genomes under examination, and the query DNA thus defined was extracted from the proper scaffolds of the query genome.
Analysis of segments found in the natural communities.
Sequences flanking the ≥10-kb, ≥99.9% identity segments present in three or more genomes of either of the two natural communities and that returned no qualifying hits from the comparison database were compared to identify areas where the sequences diverged. Once the ends of each region were established, the DNA sequences were recovered from all participating genomes and aligned using Clustal W2 (42). Areas where the multiple sequence alignment disagreed (for example, due to stretches of unaligned sequence from one or more genomes or from Ns inserted during genome sequence assembly, SNPs, etc.) were examined by PCR and/or sequencing (see Table S1 in the supplemental material). The sequencing-corrected and/or PCR-confirmed DNA sequences were realigned, and several relatively short stretches of unaligned DNA present in a subset of the genomes due to the presence of IS or RE were removed. The sequences were translated using Prodigal version 2.6 trained on the appropriate full genome (43).
Selection of genomes for mock-community analysis.
156 genomes identified by NCBI as Bacteroides or Parabacteroides species were retrieved from the RefSeq repository. Genomes from species originating from nonhuman sources (e.g., Bacteroides salanitronis, acquired from a chicken cecum, or Bacteroides helcogenes, acquired from pig feces) were eliminated from the collection. Five duplicate genomes were also removed (B. dorei CL02T00C15, B. uniformis CL03T00C23, and B. fragilis strains CL03T00C08, CL05T00C42, and CL07T00C01 each correspond to a CL0xT12Cxx strain isolated at a different time point from the same subject). The genome sequences of the T00 and the T12 isolates are nearly identical, and including them would have introduced unnecessary duplication. Individual databases prepared for each of the remaining genomes were queried via BLAST with a set of 16S ribosomal DNA sequences acquired from the Ribosomal Database Project (RDP), release 11.1 (44), representing the Bacteroides or Parabacteroides type strains. The highest-scoring segment pair resulting from each BLAST search was extracted from the target genome and examined further. Genomes with extracted segments of <1,000 bp were excluded, and the remaining segments were used as queries against the RDP database to confirm the species assigned to the genome or assign a species designation to a genome annotated only to the genus level. Genomes whose species identification by this method was ambiguous or appeared incorrect were eliminated from the local collection. Ultimately, 84 genomes representing 26 Bacteroides and Parabacteroides species were retained.
Presence of DNA regions in noncommunity members.
A collection of genomes was retrieved from NCBI to evaluate whether a qualifying DNA segment was unique to the community in which it was found. All DNAs contained in the RefSeq collection classified by NCBI as belonging to taxonomy ID 976 (phylum Bacteroidetes) that did not arise from metagenomic or environmental samples and were not also members of taxonomy ID 32644 (unclassified, e.g., unspecified or unidentified samples) were retrieved as FASTA files via the Web. This collection was further processed locally to remove entries whose sequences consisted entirely of rRNA genes and project info files. Scaffolds comprising genomes known to be duplicates were also removed.
Each qualifying segment of DNA found to exist in three or more genomes of a community was compared via BLAST to this comparison database. Only hits from outside the mock community were considered. The comparison database BLAST results were examined to enumerate the number of qualifying hits (≥10 kb at ≥99.9% identity) returned. Multiple qualifying returns originating from the same target genome were scored as a single hit.
Annotation of genes residing on regions 1 to 5.
The utilities of the HMMER suite version 3.1b1 (45) were compiled under Cygwin (version 1.7.27; http://www.cygwin.com), and hmmpress was used to convert the Pfam-A data files (version 27) (46) to binaries. Each of the protein sequences from the Prodigal-translated sequences was scanned under Cygwin for matches to the Pfam-A set of motifs using hmmscan, with the sequence and domain E value cutoffs each set to 1.0.
The position-specific score matrix (PSSM) files from NCBI’s Conserved Domain Database (CDD, version 3.10) (47) were sorted by source database (Entrez models, SMART version 6.0, TIGRFAM version 13.0, COG and KOG, and LOAD). The PSSM files corresponding to NCBI’s Protein Clusters database were further separated into curated prokaryotic and nonprokaryotic groups based on the naming convention of the PSSM files (48). Each of these groupings of PSSM files was compiled separately into RPS-BLAST databases using the NCBI makeprofiledb utility with default settings. Protein sequences derived from the conserved sequences were scanned for conserved motifs using the NCBI rpsblast utility. The results of these motif scans and those of the Pfam-A scans were collected for each protein and used to inform the annotation (see Table S5 in the supplemental material).
The segment encoding the predicted T6SS detected in region 2 was more extensively analyzed using the HHpred server (http://toolkit.tuebingen.mpg.de/hhpred) (32). The use of HMM-HMM profile comparisons and comparisons to structured proteins contained in the Protein Data Bank (PDB; http://www.rcsb.org/pdb) (49) allowed the detection of remote homologs not detectable by sequence-sequence or sequence-profile analyses.
SUPPLEMENTAL MATERIAL
PCR primers used to elucidate DNA sequences of segments containing Ns.
Characteristics of insertion sequences (IS) and retroelements (RE) present in regions 1 to 5. Each IS or RE element listed is different and shares little if any DNA homology to the other IS or RE listed. A pound sign (#) in a genome refers to additional copies not including the copy in the specified region.
Human gut Bacteroidales strains used to compile the mock communities.
Presence in 1,000 mock Bacteroidales communities of highly similar sequences with no or few other highly similar matches in the database.
Putative annotations and motifs of the proteins encoded within regions 1 to 5. The genes with colored text correspond to the color coding shown in Fig. 2
Assignments of proteins encoded within a putative type VI secretion system of region 2 based on motif and homology/structure analyses (HHpred).
ACKNOWLEDGMENTS
The authors declare no competing financial interests.
We acknowledge NIH for funding the sequencing of CL0 strains with grant U54-HG004969 to the Broad Institute. This project has been funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract no. HHSN272200900018C and grants AI081843 and AI093771.
Footnotes
Citation Coyne MJ, Zitomersky NL, McGuire AM, Earl AM, Comstock LE. 2014. Evidence of extensive DNA transfer between Bacteroidales species within the human gut. mBio 5(3):e01305-14. doi:10.1128/mBio.01305-14.
REFERENCES
- 1. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. 2005. Diversity of the human intestinal microbial flora. Science 308:1635–1638. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Doré J, Meta HITC, Antolin M, Artiguenave F, Blottiere HM, Almeida M, Brechot C, Cara C, Chervaux C, Cultrone A, Delorme C, Denariaz G, Dervyn R, Foerstner KU, Friss C, van de Guchte M, Guedon E, Haimet F, Huber W, van Hylckama-Vlieg J, Jamet A, Juste C, Kaci G, Knol J, Lakhdari O, Layec S, Le Roux K, Maguin E, Mérieux A, Melo Minardi R, M’rini C, Muller J, Oozeer R, Parkhill J, Renault P, Rescigno M, Sanchez N, Sunagawa S, Torrejon A, Turner K, Vandemeulebrouck G, Varela E, Winogradsky Y, Zeller G, Weissenbach J, Ehrlich SD, Bork P, Merieux A, Melo Minardi R, M'Rini C, Muller J, Oozeer R, Parkhill J, Renault P, Rescigno M, Sanchez N, Sunagawa S, Torrejon A, Turner K, Vandemeulebrouck G, Varela E, Winogradsky Y, Zeller G, Weissenbach J, Ehrlich SD, Bork P. 2011. Enterotypes of the human gut microbiome. Nature 473:174–180. 10.1038/nature09944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zitomersky NL, Coyne MJ, Comstock LE. 2011. Longitudinal analysis of the prevalence, maintenance, and IgA response to species of the order Bacteroidales in the human gut. Infect. Immun. 79:2012–2020. 10.1128/IAI.01348-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, Rosenbaum M, Gordon JI. 2013. The long-term stability of the human gut microbiota. Science 341:1237439. 10.1126/science.1237439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rakoff-Nahoum S, Coyne MJ, Comstock LE. 2014. An ecological network of polysaccharide utilization among human intestinal symbionts. Curr. Biol. 24:40–49. 10.1016/j.cub.2013.10.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coyne MJ, Tzianabos AO, Mallory BC, Carey VJ, Kasper DL, Comstock LE. 2001. Polysaccharide biosynthesis locus required for virulence of Bacteroides fragilis. Infect. Immun. 69:4342–4350. 10.1128/IAI.69.7.4342-4350.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Franco AA. 2004. The Bacteroides fragilis pathogenicity island is contained in a putative novel conjugative transposon. J. Bacteriol. 186:6077–6092. 10.1128/JB.186.18.6077-6092.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL. 2009. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15:1016–1022. 10.1038/nm.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martens EC, Koropatkin NM, Smith TJ, Gordon JI. 2009. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J. Biol. Chem. 284:24673–24677. 10.1074/jbc.R109.022848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koropatkin NM, Cameron EA, Martens EC. 2012. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 10:323–335. 10.1038/nrmicro2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ogilvie LA, Caplin J, Dedi C, Diston D, Cheek E, Bowler L, Taylor H, Ebdon J, Jones BV. 2012. Comparative (meta)genomic analysis and ecological profiling of human gut-specific bacteriophage phiB124-14. PLoS One 7:e35053. 10.1371/journal.pone.0035053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hecht DW, Jagielo TJ, Malamy MH. 1991. Conjugal transfer of antibiotic resistance factors in Bacteroides fragilis: the btgA and btgB genes of plasmid pBFTM10 are required for its transfer from Bacteroides fragilis and for its mobilization by IncP beta plasmid R751 in Escherichia coli. J. Bacteriol. 173:7471–7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith CJ, Macrina FL. 1984. Large transmissible clindamycin resistance plasmid in Bacteroides ovatus. J. Bacteriol. 158:739–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Welch RA, Jones KR, Macrina FL. 1979. Transferable lincosamide-macrolide resistance in Bacteroides. Plasmid 2:261–268. 10.1016/0147-619X(79)90044-1 [DOI] [PubMed] [Google Scholar]
- 15. Hecht DW, Malamy MH. 1989. Tn4399, a conjugal mobilizing transposon of Bacteroides fragilis. J. Bacteriol. 171:3603–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salyers AA, Shoemaker NB, Stevens AM, Li LY. 1995. Conjugative transposons: an unusual and diverse set of integrated gene transfer elements. Microbiol. Rev. 59:579–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salyers AA, Gupta A, Wang Y. 2004. Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol. 12:412–416. 10.1016/j.tim.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 18. Waters JL, Salyers AA. 2013. Regulation of CTnDOT conjugative transfer is a complex and highly coordinated series of events. mBio 4(6):e00569-13. 10.1128/mBio.00569-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wozniak RA, Waldor MK. 2010. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8:552–563. 10.1038/nrmicro2382 [DOI] [PubMed] [Google Scholar]
- 20. Duval-Iflah Y, Raibaud P, Tancrede C, Rousseau M. 1980. R-plasmic transfer from Serratia liquefaciens to Escherichia coli in vitro and in vivo in the digestive tract of gnotobiotic mice associated with human fecal flora. Infect. Immun. 28:981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feld L, Schjørring S, Hammer K, Licht TR, Danielsen M, Krogfelt K, Wilcks A. 2008. Selective pressure affects transfer and establishment of a Lactobacillus plantarum resistance plasmid in the gastrointestinal environment. J. Antimicrob. Chemother. 61:845–852. 10.1093/jac/dkn033 [DOI] [PubMed] [Google Scholar]
- 22. Trobos M, Lester CH, Olsen JE, Frimodt-Møller N, Hammerum AM. 2009. Natural transfer of sulphonamide and ampicillin resistance between Escherichia coli residing in the human intestine. J. Antimicrob. Chemother. 63:80–86. 10.1093/jac/dkn437 [DOI] [PubMed] [Google Scholar]
- 23. Shkoporov AN, Khokhlova EV, Kulagina EV, Smeianov VV, Kuchmiy AA, Kafarskaya LI, Efimov BA. 2013. Analysis of a novel 8.9kb cryptic plasmid from Bacteroides uniformis, its long-term stability and spread within human microbiota. Plasmid 69:146–159. 10.1016/j.plasmid.2012.11.002 [DOI] [PubMed] [Google Scholar]
- 24. Rankin DJ, Rocha EP, Brown SP. 2011. What traits are carried on mobile genetic elements, and why? Heredity 106:1–10. 10.1038/hdy.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wozniak RA, Waldor MK. 2009. A toxin-antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet. 5:e1000439. 10.1371/journal.pgen.1000439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Amano A, Nakagawa I, Okahashi N, Hamada N. 2004. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J. Periodontal Res. 39:136–142. 10.1111/j.1600-0765.2004.00719.x [DOI] [PubMed] [Google Scholar]
- 27. Wion D, Casadesús J. 2006. N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 4:183–192. 10.1038/nrmicro1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marinus MG, Casadesus J. 2009. Roles of DNA adenine methylation in host-pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol. Rev. 33:488–503. 10.1111/j.1574-6976.2008.00159.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baba T, Schneewind O. 1996. Target cell specificity of a bacteriocin molecule: a C-terminal signal directs lysostaphin to the cell wall of Staphylococcus aureus. EMBO J. 15:4789–4797 [PMC free article] [PubMed] [Google Scholar]
- 30. Ho BT, Dong TG, Mekalanos JJ. 2014. A view to a kill: the bacterial type VI secretion system. Cell Host Microbe 15:9–21. 10.1016/j.chom.2013.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Russell AB, Peterson SB, Mougous JD. 2014. Type VI secretion system effectors: poisons with a purpose. Nat. Rev. Microbiol. 12:137–148. 10.1038/nrmicro3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Biegert A, Mayer C, Remmert M, Söding J, Lupas AN. 2006. The MPI Bioinformatics toolkit for protein sequence analysis. Nucleic Acids Res. 34:W335–W339. 10.1093/nar/gkl217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hildebrand A, Remmert M, Biegert A, Söding J. 2009. Fast and accurate automatic structure prediction with HHpred. Proteins 77(Suppl 9):128–132. 10.1002/prot.22499 [DOI] [PubMed] [Google Scholar]
- 34. Ma LS, Narberhaus F, Lai EM. 2012. IcmF family protein TssM exhibits ATPase activity and energizes type VI secretion. J. Biol. Chem. 287:15610–15621. 10.1074/jbc.M111.301630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zoued A, Durand E, Bebeacua C, Brunet YR, Douzi B, Cambillau C, Cascales E, Journet L. 2013. TssK is a trimeric cytoplasmic protein interacting with components of both phage-like and membrane anchoring complexes of the type VI secretion system. J. Biol. Chem. 288:27031–27041. 10.1074/jbc.M113.499772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coyne MJ, Comstock LE. 2008. Niche-specific features of the intestinal Bacteroidales. J. Bacteriol. 190:736–742. 10.1128/JB.01559-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coyne MJ, Reinap B, Lee MM, Comstock LE. 2005. Human symbionts use a host-like pathway for surface fucosylation. Science 307:1778–1781. 10.1126/science.1106469 [DOI] [PubMed] [Google Scholar]
- 38. Toleman MA, Walsh TR. 2011. Combinatorial events of insertion sequences and ICE in gram-negative bacteria. FEMS Microbiol. Rev. 35:912–935. 10.1111/j.1574-6976.2011.00294.x [DOI] [PubMed] [Google Scholar]
- 39. Polz MF, Alm EJ, Hanage WP. 2013. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet. 29:170–175. 10.1016/j.tig.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ho BT, Basler M, Mekalanos JJ. 2013. Type 6 secretion system-mediated immunity to type 4 secretion system-mediated gene transfer. Science 342:250–253. 10.1126/science.1243745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. NIH HMP Working Group, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, Peavy H, Pontzer C, Portnoy M, Sayre MH, Starke-Reed P, Zakhari S, Read J, Watson B, Guyer M. 2009. The NIH Human Microbiome Project. Genome Res 19:2317–2323. 10.1101/gr.096651.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 43. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. 2014. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42:D633–D642. 10.1093/nar/gkt1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39:W29–W37. 10.1093/nar/gkr367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res. 40:D290–D301. 10.1093/nar/gkr1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. 2013. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 41:D348–D352. 10.1093/nar/gks1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. ONeill K, Klimke W, Tatusova T. 2007. Protein clusters: a collection of proteins grouped by sequence similarity and function. National Center for Biotechnology Information, Bethesda, MD. http://www.ncbi.nlm.nih.gov/books/NBK3797 [Google Scholar]
- 49. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PCR primers used to elucidate DNA sequences of segments containing Ns.
Characteristics of insertion sequences (IS) and retroelements (RE) present in regions 1 to 5. Each IS or RE element listed is different and shares little if any DNA homology to the other IS or RE listed. A pound sign (#) in a genome refers to additional copies not including the copy in the specified region.
Human gut Bacteroidales strains used to compile the mock communities.
Presence in 1,000 mock Bacteroidales communities of highly similar sequences with no or few other highly similar matches in the database.
Putative annotations and motifs of the proteins encoded within regions 1 to 5. The genes with colored text correspond to the color coding shown in Fig. 2
Assignments of proteins encoded within a putative type VI secretion system of region 2 based on motif and homology/structure analyses (HHpred).