

Abstract
Cigarette smoke exposure is known to induce obstructive lung disease and several cardiovascular disease states in humans and also in animal models. Smoking leads to oxidative stress and inflammation that are important in triggering pulmonary and cardiovascular disease. The objective of the current study was to quantify differences in expression levels of plasma proteins of cigarette smoke exposed and control mice, at the time of disease onset, and identify these proteins for use as potential biomarkers of the onset of smoking-induced disease. We utilized two dimensional difference in-gel electrophoresis/ mass spectroscopy (2D-DIGE/MS) to characterize these proteomic changes. 2D-DIGE of plasma samples identified 11 differentially expressed proteins in cigarette smoke exposed mice. From these 11 proteins, 9 were down-regulated and two were up-regulated. The proteins identified are involved in vascular function, coagulation, metabolism, and immune function. Among these, the alterations in fibrinogen (2.2 fold decrease), alpha-1-antitrypsin (1.8 fold increase) and arginase (4.5 fold decrease) are of particular interest since these have been directly linked to cardiovascular and lung pathology. Differences in expression levels of these proteins were also confirmed by immunoblotting. Thus, we observe that chronic cigarette smoke exposure in mice leads to prominent changes in the protein expression profile of blood plasma and these changes in turn can potentially serve as markers predictive of the onset and progression of cardiovascular and pulmonary disease.
Keywords: proteomics, cigarette smoking, mass spectrometry, inflammation, heart disease, lung disease, biomarkers
1. Introduction
Cigarette smoking and second hand smoke have been linked to development of several chronic disorders. Exposure to tobacco smoke in humans is associated with the onset of coronary disease [1], atherosclerosis [2], and ischemic heart disease [3]. While over the last 2 decades, there has been increasing concern regarding the role of cigarette smoking in cardiovascular (CV) and pulmonary disease, many questions remain regarding the precise mechanism by which smoking induces disease. Cigarette smoking is a major risk factor for cardiovascular events [4]. The exact mechanism by which smoking causes these events is not fully understood; however, this is known to involve oxidative stress [5] which leads to induction of endothelial dysfunction [6, 7]. It is believed that cigarette smoke (CS) exposure leads to absorption of tobacco combustion products including a variety of poly aromatic hydrocarbons and particulates into the systemic circulation that stimulate systemic inflammation which injures the arterial endothelium leading to atherogenesis [8]. In the lungs this exposure is well known to induce chronic obstructive pulmonary disease.
As depicted in Figure 1, with an exposure to CS, free-radical mediated oxidative stress and its influence on protein expression can be a mechanism leading to atherosclerosis. Excess free radicals are generated in vivo with CS-exposure from: a) smoke components; b) by activation of endogenous sources of reactive oxygen species like uncoupled eNOS (endothelial nitric oxide synthase); xanthine oxidase and the mitochondrial electron transport chain [9]. An increase in oxidative stress with free radical generation leads to activation of neutrophils and monocytes triggering endothelial adhesion and transmigration with tissue injury [10]. Oxidative stress also induces platelet activation and aggregation leading to a prothrombotic state [11, 12]. Further increase in oxidative stress decreases NO availability and its protective action, due to reaction of superoxide and NO leading to the formation of peroxynitrite [13, 14]. Also, increase in oxidative stress affects the expression profile of certain groups of proteins which ultimately cause the proatherogenic and prothrombotic events leading to initiation and progression of disease [15].
Figure 1. Diagram illustrating mechanisms of cigarette smoke-exposure mediated cardiovascular dysfunction.

H2O2 = hydrogen peroxide; O2 .− = superoxide; ONOO− = peroxynitrite; METC = mitochondrial electron transport chain; NADPH = nicotinamide adenine dinucleotide phosphate reduced form; NOS = nitric oxide synthase
Despite the association between CS exposure and pulmonary and cardiovascular disease (CVD) states, many questions remain regarding disease onset and the precise mechanism by which smoking induces disease. This may be, in part, due to the lack of a well-characterized controlled animal model and a sensitive and reliable method that can detect CS-induced biochemical and molecular changes.
Proteomic techniques have been applied to achieve greater understanding of CS-induced pulmonary and CV disease. Yet, most studies to date have used invasive methods for sampling. Only a few reports have used blood plasma, although it is a more readily available clinical sample. Samples from human bronchoalveolar lavage fluid (BALF) have shown increased levels of neutrophil defensins, calgranulin A, and calgranulin B in smokers with COPD compared to smokers with no COPD [16]. Another human BALF study reported differential expression of a number of proteins in asymptomatic smokers compared to healthy non-smokers [17]. A biomarker discovery study on plasma samples of smokers with COPD has suggested differential expression of candidate proteins including, retinal-binding protein (RETB), fibrinogen, apolipoprotein E, inter-α-trypsin inhibitor heavy chain H4 and glutathione peroxidase [18].
Based on previous observations in the literature, we hypothesize that exposure to toxic components of CS and the secondary oxidative stress-mediated inflammation leads to changes in expression levels of certain circulating proteins, in blood plasma and these protein alterations could be markers of and possible mediators of disease pathology and progression. Proteomic studies related to protein alterations caused by cigarette smoke exposure can provide new insights into specific markers, which could enable development of new methods for early detection and molecular characterization of cardiovascular and pulmonary disease.
Previously, proteomics studies of differential protein expression have utilized separation of proteins by 2D gel electrophoresis [19, 20] along with protein identification through mass spectrometry [21-23]; however, technical problems limit sensitivity and reliability for detection of quantitative differences in protein expression levels. Differential In-gel Electrophoresis (DIGE) provides more accurate quantitative data on differential protein expression. DIGE was originally developed by Minden and colleagues [24, 25].
The technique involves two sets of proteins which are covalently labeled with Cy3 and Cy5 fluorescent dyes. The labeled proteins are pooled together, and separated in the same 2D gel. Since minimal labeling with Cy dyes does not change molecular mass and charge of the proteins, the pooled proteins existing in both sample sets will move to the same locations in 2D gels, therefore enhancing the reproducibility. Quantitation of protein expression levels can be accurately measured based on the fluorescent intensity of individual protein spots. This technique has been applied in many studies including identification of esophageal squamous cell cancer markers [25], markers of liver toxicity [26], cancer-specific proteins [27], and markers for bladder cancer [28].
We have recently developed and characterized a mouse model of smoking induced cardiovascular disease in a widely available background mouse strain in order to facilitate delineation of the mechanisms of disease allowing for the use of specific knockout or overexpressed genetic models [29]. In these C57BL/6J mice that are not otherwise predisposed to disease, we observed that after 32-weeks of CS-exposure, less body-weight gain was seen and markedly higher blood pressure developed. Furthermore, at 32-weeks CS-exposure: acetylcholine-induced vasorelaxation was significantly impaired; left ventricular mass was significantly larger along with increased heart to body-weight ratio; and in vitro cardiac function was mildly impaired [29, 30]. At this time these CS-exposed mice also first exhibit significant increases in lung volumes indicative of early COPD. Thus in this mouse model with 32 weeks of smoking exposure, hypertension, endothelial dysfunction, mild cardiac hypertrophy, and COPD occur. Of note, at 16 weeks of exposure these abnormalities were not seen and 32 weeks exposure corresponded to the first time of significant disease onset. This model has been proposed as a useful tool to enable further elucidation of the molecular and cellular mechanisms of smoking-induced cardiovascular diseases.
In the current study, we applied DIGE to investigate changes in the abundance of differentially expressed proteins in plasma samples of CS exposed mice compared to non-exposed control mice, at the time of disease onset. We aim to identify differences in expression levels of plasma proteins induced by CS exposure that could serve as potential markers for smoking-induced oxidative stress and the onset of inflammatory cardiovascular and pulmonary disease.
2. Materials and methods
2.1. Animal model and exposure
Twenty (10 control and 10 CS exposed) male C57BL/6J mice, 8-9 weeks of age (Harlan Sprague Dawley, USA), were housed at a 23 ± 2°C, at 55% relative humidity, with 12h day-night cycle, and maintained on standard rodent chow and tap water ad libitum. After a week of acclimatization, mice were exposed to whole body mainstream, and side stream CS using a SCIREQ “InExpose” smoking system (SCIREQ Inc., Montreal, Canada) and 3R4F reference research cigarettes (University of Kentucky) that deliver 9.4 mg tar/0.726 mg nicotine per cigarette under the standard Cambridge filter smoking condition. The smoking machine was programmed according to the following standard parameters (ISO 1991): One 35 ml puff of 2-sec duration followed by 58-sec of fresh air at a rate of 6 ml/sec. The CS was directed in a 5 liter volume exposure chamber at a smoke to air ratio of 1:10. Mice were exposed to CS for 48-min/day, 5 days/week, for 32 consecutive weeks, and were sacrificed 24-hr after the last CS exposure. Agematched, air exposed mice served as controls. Carboxyhemoglobin (CO-Hb) levels in whole blood were measured spectrophotometrically using an Agilent 8453 Diode Array UV/VIS spectrophotometer (Agilent Technologies, USA). CO-Hb level immediately after the exposure was ~12% in CS-exposed mice and ~1% in air-exposed controls. All animal procedures were performed in accordance with the regulations of the Institutional Animal Care and Use Committee at The Ohio State University, and conformed to the Guide for the Care and Use of Laboratory Animals.
2.2 Materials
Multiple affinity removal system was purchased from Agilent Technologies. All chemicals used for 2D DIGE labeling and gels (urea, thiourea, DTT, 3-[(3-cholamidopropyl) dimethyl-amonio]-1-propanesulfonate (CHAPS), Cy dyes) were purchased from GE Healthcare.
2.3 Sample preparation for 2D-DIGE
Blood plasma samples were collected 24 hours after the last exposure time in individual heparinized tubes from both CS-exposed as well as control mice. Plasma samples were immediately centrifuged at 3000 g for 15 minutes at 4°C and then frozen in liquid nitrogen and stored at -80°C for further analysis. Plasma samples were depleted of most abundant proteins (albumin, IgG and transferrin) using the Multiple Affinity Removal System (Agilent Technologies) as per manufacturer’s instructions. Depleted samples were cleaned with 2D Clean Up kit (GE) according to manufacturer’s protocol and re-suspended in lysis buffer (7M urea, 2 M thiourea, 4% CHAPS, 30mM Tris pH 8.5). Bradford assay was used for sample quantification with Bovine gamma globulin as a standard (Bio-Rad).
Equal aliquot of all samples (4 control and 4 treated) were combined together to make Internal standard (IS). Then 30 μg each of control, treated samples and internal standard were labeled with fluorescent Cy dyes. Samples were labeled with Cy 3 and Cy 5 dyes in random manner to avoid dye specific bias. For labeling we used 8 pmol of Cy dye per 1 μg of protein. Labeling reaction was performed on ice for 30 min. 1 μl of 10 mM Lysine (Sigma) was added to stop the reaction. Then labeled samples were mixed together, vortexed, and diluted with sample buffer containing 7 M urea, 2 M thiourea, 2% CHAPS, 130 mM DTT and 0.5% IPG buffer (GE) up to a volume of 460 μl. This solution was used for IPG strip rehydration.
For preparative gels, we combined all samples into a total volume of ~ 200 μl with ~ 500 μg protein that was diluted to 460 μl with sample buffer and applied onto 24 cm IPG strip (pH 4-7) for 18 hour rehydration.
2.4 2D-DIGE and image analysis
A Multiphor II system from Amersham was used for the isoelectric focusing protocol. Protein sample (90 μg) was applied on pre-cast IPG strips (24 cm; pH 4-7) by overnight rehydration, and first-dimensional separation was performed until the focusing time reached to 40 kV-h. After focusing, the IPG strips were treated with equilibration buffer 6 M urea, 30% glycerol, 2% SDS, 75 mM Tris (pH 8.8) and 0.5% DTT for 15 min. Strips were again incubated with equilibration buffer containing 4.5% iodoacetamide instead of DTT. The strips were rinsed in 2X SDS running buffer (25 mM Tris, 192 mM glycine, 0.2% w/v SDS), placed on the gels and overlaid with overlay solution (0.5% w/v agarose in running buffer and trace amount of bromophenol blue). Second dimension was run on 12% SDS-PAGE (26×20 cm) at constant 2 W per gel for 45 min followed by constant 15 W per gel for 4 hr and 10 min. The Cy2-, Cy3- and Cy5-labeled gel images were acquired using a laser scanner (Typhoon 9400, GE healthcare) for image analysis. To quantify the differences in protein expression levels, statistical analysis was done by using Decyder software (Amersham Biosciences, Inc.). The gel spots with more than 1.5-fold changes in volume after normalization between two sets of samples were considered as differentially expressed. Preparative gels were stained with Lava Purple (Fluortechniques) and matched to the master gel.
2.5 Mass spectrometry analysis
The altered protein spots were excised from the preparative gel and digested with sequencing grade trypsin (Promega, Madison, WI) using the Multiscreen Solvinert Filter Plates from Millipore (Bedford, MA).
Ettan Spot Handling Workstation (Amersham Biosciences) was used to core protein spots of interest using a pick list and placed in a 96 well plate. The 96 well plates were placed inside the robot for wash and digestion. Briefly, gel pieces were washed in 100 μL solution of 50% methanol/5% acetic acid for 30 minutes. The gel pieces were washed 3 times and placed in a storage solution of 50 μL of 50% methanol/5% acetic acid until digestion. Digestion was carried out by incubating samples with 100 μL water for 10 minutes followed by 100 μL acetonitrile for 10 minutes. Following removal of water and acetonitrile, gel pieces were rehydrated and incubated first with DTT (5 mg/ml in 100 mM ammonium bicarbonate) for 30 minutes and then with iodoacetamide (15 mg/ml in 100 mM ammonium bicarbonate solution) for 30 minutes. After the removal of iodoacetamide, the gel pieces were washed with cycles of acetonitrile and 100 mM ammonium bicarbonate in 10 and 5 minute increments, dried for 10 minutes and rehydrated with 25 μL of 5 μg/mL trypsin in 50 mM ammonium bicarbonate for 3 hours at 37° C. Peptides were extracted 3 times with 50 µL of extraction solution (50% acetonitrile and 5% formic acid). All extracted fractions were pooled together, dried for 15 minutes and removed immediately to prevent complete drying.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis was carried out on Thermo Finnigan LTQ mass spectrometer. This instrument has nanospray ion source (working in positive mode) and linear ion trap analyzer. UltiMate™ Plus system from Dionex (Sunnyvale, CA) was used for LC separation of digested peptides. Two solvents, A (50 mM acetic acid in water) and B (acetonitrile) and two columns were used for LC separations. At first, samples were applied on to the trapping column (LC-Packings A Dionex Co, Sunnyvale, CA), and washed with solvent A. Then peptides were eluted onto second column, a 5 cm ProteoPep II C18 column (New Objective, Inc. Woburn, MA). A gradient of solvent B (2-80%) was used to elute the peptides from the second column into LTQ. A flow rate of 300 nL/min was set with total run time of 60 minutes for each sample. The nanospray source was operated at 3 KV.
The parameters for the MS/MS acquisition were set as follows: scan range 350–2000 Da and MS/MS consecutive scans of the 10 most abundant peaks in the spectrum were used for reconstructing amino acid sequences of the fragments. Fragmentation CID energy (35%), Dynamic exclusion was used with a repeat count of 30 seconds, an exclusion duration of 350 seconds with the low and high mass width as 0.5 and 1.5 Da, respectively. After mass spectra acquisition was complete, the raw data files were converted into merged file (mgf) format, and searched against SwissProt database version 56.5 (402482 sequences; 145232059 residues) by using Mascot Daemon search mechanism (Matrix Science version 2.2.1, Boston, MA). Search parameters were used as: 2 Da for the precursor mass accuracy; 0.5 Da for the fragment mass accuracy; Carbamidomethylation of Cys and oxidation of Met for variable modifications; number of allowed miscleavages as two. Peptides with score of 20 or lower were excluded from the analysis. Protein identifications were accepted only if they made by at least two peptides and mascot score of 50.
2.6 SDS-PAGE and Immunoblot
Plasma protein samples were depleted for abundant proteins as described before in section 2.3, and separated on 1D criterion precast (Bio-Rad) 8-16% gradient gel. In order to make an equal protein load on western blot, all protein samples were measured before and after immunodepletion by using Coomassie Protein Assay reagent Kit (Pierce Biotech, Rockford, IL) with BSA as standard. An equal amount of protein (5μg) was used for each sample. Following SDS-PAGE, Proteins were transferred onto nitrocellulose membrane (0.45um, Bio-Rad). For immunoblotting, primary antibodies were used as follows: rabbit polyclonal anti-alpha-1 antitrypsin (1:500, Abbiotec, LLC, San Diego, CA), rabbit polyclonal anti-ariginase 1 (1:1000, GenWay Biotech, Inc., San Diego, CA), rabbit polyclonal anti-fibrinogen (1:10000, Abcam Inc., Cambridge, MA). Goat anti-rabbit Alexa 488-conjugated antibody (1:2000, Invitrogen, Carlsbad, CA) was used as secondary antibody. Immunopositive protein bands were visualized by scanning the blot on a laser scanner (Pharos-FX, Bio-Rad). Densitometric analysis of protein bands was done by using ImageJ software (NIH).
2.7 Statistical analysis
Differences in protein expression levels in 2D gel spots were carried out using DeCyder software. Statistical analysis was done on 12 analytical images (three images from each gel). All images were matched to master gel image. The pixel volume of each spot was quantified depending on the spot intensity and area and then normalized with the total pixel volume of all the spots in gel image. The pixel volume of each spot was used to compare the change in protein expression.
Set of volume ratio values of Cy3/Cy2 and Cy5/Cy2 were calculated, and normalized for each gel separately. This value is called standardized abundance and its log 10 is used for statistical analysis.
The log of standardized abundance values are used in an order that the data points approach a normal distribution around zero and therefore are suitable for statistical analysis. Students t test used to evaluate difference in protein expression between control and treated experimental groups. The null hypothesis is that there are no changes in protein abundance between experimental groups (control vs. CS-exposed) and average ratio between two groups is 1. The t-test p values represent the probability of data having the same protein abundance. We considered protein expression to be significantly different when p < 0.05 and fold changes > 1.5.
All experimental details are illustrated in Figure 2.
Figure 2. Flow chart of DIGE analysis of plasma samples obtained from control and CS-exposed mice.

CS, Cigarette Smoke; IS, internal standard; ID, identification.
3 Results
3.1 2D Gel Separation of Proteins
The acquired 2D-DIGE gel images for each representative gel were overlayed to match the protein spots, and for further analysis, as explained in experimental details. The master gel overlay image with Cy dye colors is shown in Fig 3A. Altogether about 1100 to 1400 proteins were separated on each gel. The largest numbers of protein spots were on the master gel (1439). The differentially expressed protein spots on 2D gels with their respective numbers, as shown in Fig 3B, were used for gel excision and MS analysis.
Figure 3. DIGE gel images of differential expression of proteins.
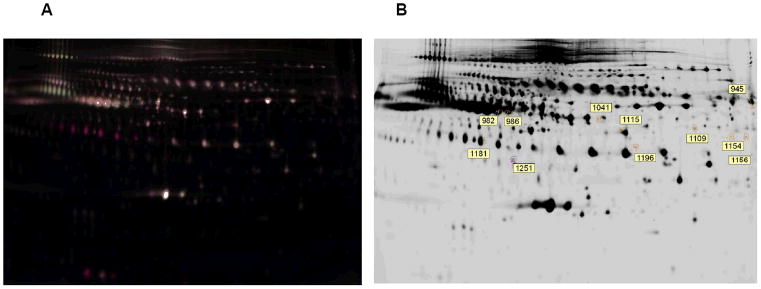
A, Overlay image of the master gel containing both Cy3 and Cy5- labeled proteins of plasma samples. B, lava purple stained master gel image. The labeled control and CS-exposed samples were mixed and separated on gel. The same gel stained by lava purple (B) gave the combined protein profile of both control and smoke-exposed plasma samples.
3.2 Differential Expression of Proteins between Cy3- and Cy5-imaged Gels
Figure 4 illustrates the 3-D view corresponding to the pixel volume distribution for the Cy5/Cy3 ratios of proteins which are differentially expressed between plasma samples of CS-exposed and control mice. The protein peak under consideration is being outlined with pink line around it. Most protein spots showed decreased expression in smoke-exposed samples while few showed an increase in expression level.
Figure 4. 3D view of differential expressed protein spots.

The volume of each spot was calculated using Decyder software and is graphically represented. The spot pairs of down-regulated (A) and up-regulated proteins (B) in plasma samples were shown. The amount of the protein is proportional to the volume of the peak
3.3 Quantitation and identification of differential protein expression
Comparisons of protein expression in control and CS-exposed samples are calculated based on the relative change of sample with respect to its in-gel internal standard. Comparison is done by student t-test and for calculations we used log standardized protein abundance. The degree of difference between protein groups is expressed as average ratio (Table 1). The average ratio between two sets of values for a given protein was represented by lines marked with a cross (Fig. 5).
Table 1.
Differentially Expressed Proteins in CS-Exposed versus Control plasma.
| Spot No. | p-value | Av ratio | Protein name | Swiss-Prot accession | Mascot score | MW (Da) | Peptide matches | pI |
|---|---|---|---|---|---|---|---|---|
| 945 | 0.048 | -2.2 | Fibrinogen beta chain | Q8K0E8 | 98 | 55402 | 2 | 6.68 |
| 1041 | 0.021 | -2.25 | S-adenosylmethionine synthetase | Q91X83 | 389 | 44051 | 20 | 5.51 |
| 1109 | 0.025 | -3.91 | S-adenosylhomocysteinase | P50247 | 884 | 48170 | 46 | 6.08 |
| 1115 | 0.0018 | -2.09 | Adenosine kinase | P55264 | 149 | 40446 | 4 | 5.84 |
| 1154 | 0.012 | -4.51 | Arginase-1 | Q61176 | 286 | 34957 | 7 | 6.51 |
| 1156 | 0.031 | -3.24 | Sorbitol dehydrogenase | Q64442 | 585 | 38795 | 23 | 6.56 |
| 1181 | 0.035 | -3.93 | H-2 class 1 histocompatibility antigen | P01898 | 84 | 37455 | 6 | 5.13 |
| 1196 | 0.018 | -4.88 | Fructose-1,6-bisphosphatase1 | Q9QXD6 | 983 | 37288 | 34 | 6.15 |
| 1251 | 0.022 | -3.17 | Regucalcin | Q64374 | 863 | 33899 | 35 | 5.15 |
| 982 | 0.036 | 1.65 | Alpha-1-1 antitrypsin 1-3 | Q00896 | 667 | 45966 | 26 | 5.25 |
| 986 | 0.047 | 1.77 | Alpha-1-1 antitrypsin 1-1 | P07758 | 485 | 46145 | 18 | 5.44 |
Figure 5. The average ratio of expression for protein spots for control and sample (smoke-exposed) by computational analysis (DeCyder software).

The average ratio was presented by lines marked with a cross. A, Down-regulated protein spots; B, Up-regulated protein spots.
All 11 Proteins spots (9 under-expressed and 2 over-expressed) identified by 2D-DIGE as significantly different among CS-exposed versus control samples, were excised from lava purple stained gels. The excised gel spots were subjected to in-gel digestion and LC-MS/MS analysis. The molecular masses of eluting peptides and their fragments were determined by mass spectrometry and matched with protein sequences available in the SwissProt data base. The protein identification was achieved with the help of MASCOT. Description of spots and the identified proteins differentially expressed between smoke exposed and control samples are shown in Table 1 with identification numbers matched to those of the 2D-DIGE spots shown in gel images (Fig. 3B). All identified proteins had high mascot score leading to high confidence in their IDs. Negative values for the average ratio indicate decreased protein expression.
Differential expression studies show that alpha1-antitrypsin levels were increased (1.8-fold) after 32 weeks of smoke-exposure.
Differential expression studies also demonstrated the under-expression of 9 distinct plasma proteins (fibrinogen beta chain: -2.2, S-adenosylmethionine synthetase: -2.25, adenosylhomocysteinase: -3.9, adenosine kinase: -2.1, arginase-1: -4.5, sorbitol dehydrogenase: -3.2, H-2 class 1 histocompatibility antigen: -3.9, fructose-1, 6 bisphosphatase 1: -4.9, and regucalcin: -3.2) in smoke-exposed mice as compared to controls. These proteins fall into three categories: 1) proteins involved in vascular function and coagulation; 2) proteins involved in metabolism; 3) proteins involved in immune function.
A decrease in body weight was observed in CS-exposed mice. By 32 weeks significantly higher values of diastolic blood pressure (BP) were observed with decreased pulse pressure suggesting decreased vascular compliance. There was a significant increase in lung volumes seen in CS-exposed mice, indicative of emphysematous changes and early COPD. At 32 weeks exposure, a mild increase in LV mass was seen with increased wall thickening, stroke volume and cardiac output. It is noteworthy that the observed changes in protein expression coincide with the onset of cardiovascular and metabolic disease states. For instance, these mice developed hypertension, endothelial dysfunction, impairment of cardiac function and lung emphysema at 32 weeks of CS exposure [29]. Moreover, hypoglycemia and ketosis were observed in these CS-exposed mice (Fig. 6). Consistent with this, CS-exposed mice all show low body weight and fat contents [31].
Figure 6. Metabolic changes following CS exposure.

Mice were exposed to CS for 32 weeks; blood samples were taken 24h following the last exposure for detection of blood glucose (A) and ketone (B) levels. Results are shown as average ± SD; n= 4-6. Statistical significance was determined by t-test, where p-values are * <0.05
3.4 Immunoblot analysis of plasma proteins
Immunoblot analysis was performed to confirm the proteomics results for 3 of the differentially expressed proteins in plasma samples of smoke-exposed and control mice. The expression levels of these proteins were clearly altered due to CS-exposure (Fig. 7). The expression levels of alpha-1 antitrypsin were increased significantly (approximately three times) in smoke-exposed mice as compared to controls. In accordance with the 2D-DIGE results, decreases in expression of arginase 1 and fibrinogen were seen with about two-fold and three-fold decrease in expression levels of arginase 1 and fibrinogen, respectively (Fig. 7A & 7B). Overall, these results confirm the 2D-DIGE findings regarding increased or decreased protein expression levels.
Figure 7. Detection and quantification of Alpha-1 antitrypsin, Arginase 1, and Fibrinogen by western blotting.

Western blots of depleted mice plasma for alpha-1 antitrypsin (AT-1), arginase-1 (ARG-1), and fibrinogen (A). Protein band density was calculated by software (B). Results are shown as average ± SD; n=3. Statistical significance was determined by t-test, where p-values are * <0.05: Control, non-exposed; CS-exposed, CigaretteSmoke-exposed, AU, Arbitrary Units.
4 Discussion
In the current study, mice were chronically exposed to CS for 32 weeks, and plasma samples were obtained to assess the difference in protein profile. This study demonstrates a unique application of 2D-DIGE coupled with mass spectrometry for quantitative protein analysis in plasma of a well characterized mouse model of chronic CS-induced CV pathology. We provide a promising approach for the identification of smoke-exposed inflammatory biomarkers which could be relevant to cardiovascular and pulmonary pathology. The uniqueness of our study is first that we selected to analyze plasma as the protein sample of choice because it contains many important secreted and extracellular proteins, and can readily be used for screening of proteomic alterations as biomarkers of smoking induced disease. Moreover, the use of plasma samples would reduce the hurdles in applying the powerful technology of 2D-DIGE to patients. Second, by using the mouse, the preferred animal model for genetic manipulation in cardiovascular research [32-34], our study offers the opportunity to analyze the presence and importance of protein changes during various stages of CVD states under well defined laboratory conditions and in animals with identical genetic backgrounds, thereby facilitating proteomic comparisons by limiting biological variation. Third, the proteomic changes reported in this study are in good agreement with the onset of disease states observed.
Proteomic analysis with the use of DIGE for differential expression of proteins provides some advantages. As the two sets of protein samples were resolved in the same gel, the reproducibility of protein separation is better compared to conventional 2D gel. Also, it provides more accurate differences in 2D gel images, based on the fluorescence of the labeled Cy3 and Cy5 dyes, which can be used for accurate measurement of differential expression of proteins. In DIGE technique, proteins spots are visualized and quantified based on fluorescence of individual spots with a dynamic range of four orders of magnitude, therefore it is more useful for comparing the differences in protein expression as compared to standard 2D gel based methods.
We observed that chronic CS exposure induces changes in the levels of proteins present in plasma samples and DIGE/MS was used to identify differentially expressed proteins. In the 2D-DIGE based differential expression study, nine distinct proteins were observed to be greatly under-expressed in smoking-exposed samples as compared to controls and two were over-expressed. Changes were seen in expression levels of fibrinogen and alpha antitrypsin (A1AT) which are inflammation-sensitive plasma proteins (ISPs). Fibrinogen influences thrombogenesis and there by can affect vascular patency, blood flow, blood viscosity and platelet aggregation. CS exposure is known to cause functional changes in platelets and is the source of fibrin that is required for thrombus formation. Changes in architecture and amount of fibrin fibers affect the dynamics of clot formation and this can be linked to increased cardiovascular mortality [35, 36]. Fibrinogen levels have been identified as an independent risk factor for cardiovascular disease. The decrease in fibrinogen could be due to increased consumption or decreased hepatic production.
ISPs have been reported to be associated with increased incidence of COPD and heart failure [37, 38, 39]. Literature data [40] suggest that elevated levels of ISPs are associated with increased risk for development of peripheral arterial disease (PAD). We observed that fibrinogen levels were also markedly decreased, while A1AT levels were increased. A1AT protects lung tissue from damage caused by neutrophil elastase which is released during infection or inflammation and destroys lung air sacs causing emphysema. It is believed that CS exposure mediated oxidative stress and inflammation causes oxidative damage to A1AT [41]. This important protein, involved in preventing lung disease and chronic obstructive pulmonary disease (COPD), has also identified as being subject to HNE modification and tyrosine nitration [42]. The increase in its expression level could be a compensatory response to replace modified or inactivated protein. Since highly significant differences were seen in the expression levels of these proteins, and they are involved in critical functions that are or may be altered with smoking, they appear to be highly promising biomarkers of smoking induced disease. The alterations in fibrinogen, alpha-1-antitrypsin and arginase are of particular interest since alterations in the level of these proteins are directly associated with cardiovascular and lung pathology.
The most altered protein levels included arginase-1 that was 4.5-fold decreased. It is constitutively expressed in the airways, particularly in the bronchial epithelium and in fibroblasts. Arginase-1 controls L-arginine levels and through this, NO synthase function [43, 44]. Arginase-1 represents a new therapeutic target for the treatment of vasculoproliferative disorders. A decrease in arginase-1 would be expected to result in elevated L-arginine levels, which is the key substrate for nitric oxide (NO) production from NO synthase. We measured the level of L-arginine in plasma samples and found it to be 2-fold higher in CS-exposed mice as compared to controls (data not shown). This is consistent with the decrease of arginase expression level in DIGE and western blot results. The decrease in the arginase level could be a compensation for reduced levels of active endothelial NO synthase (eNOS). It is important to note that expression of some proteins may change as a result of tissue damage or acute stress instead of CS-induced disease. Earlier time proteomic and lung histopathology data at 16 weeks did not show abnormalities (data not shown); suggesting that the differential protein expression observed in our study at 32 weeks is a marker of CS-induced disease.
Fructose-1, 6-bisphosphatase 1, plays an important role in carbohydrate metabolism and gluconeogenesis, and was also 4.9-fold decreased. The deficiency of Fructose-1, 6-bisphosphatase 1 has been reported to cause hypoglycaemia and marked elevation of plasma fatty acids concentrations that may accompany a rise in blood acetoacetate and 3-hydroxybutyrate levels. It has been reported that smokers develop hypoglycemia that conditions the body to crave a cigarette to get an immediate rise in blood sugar levels. Following this short lived rise, prolonged CS-induced hypoglycemia causes the body to burn fat as a source for energy [45, 46]. We observed that chronic CS exposure causes low blood glucose levels, slight elevation in blood ketone levels, low body weight and low body fat/body weight ratio.
Both S-adenosylmethionine synthetase (AdoMetS) and S-adenosylhomocysteinase are key enzymes involved in methionine metabolism. AdoMetS catalyzes formation of S-adenosylmethionine (AdoMet) from methionine and ATP. AdoMet is the main methyl group donor and plays a central role in cellular functions, transmethylation reactions, and the transsulphuration pathway. AdoMet is involved in biosynthesis of polyamines. S-adenosylhomocysteinase catalyzes the hydrolysis of S-adenosylhomocysteine to adenosine and homocysteine. A decrease in the expression level of both enzymes suggests reduced activity in the methyl-cycle which could alter not only the cellular methylation process but also change the homocysteine pool in blood plasma of CS-exposed mice. CS has been associated with increased homocysteine levels [47, 48]. A high plasma homocysteine level in smokers has been reported to be associated with a 12-fold increase in the risk of CVD compared with non-smokers with normal plasma homocysteine [49]. Furthermore, increased homocysteine concentrations provoke endothelial dysfunction, possibly mediated by oxidative stress or interference with nitric oxide function [50, 51]. Based on the above reports, a decrease of S-adenosylmethionine synthetase and of S-adenosylhomocysteinase could be due to a compensatory response with chronic CS-exposure to limit the increase of the homocysteine pool.
A decrease in expression levels of H-2 class 1 histocompatibility antigen represents the effects of CS exposure on the decline of immune function. Studies with tobacco extracts [52] have shown a loss or decrease in MHC (Major Histocompatibility Complex) class 1 protein expression. This CS exposure induced change may lead to a defect in antigen processing and decreased immune response.
In summary, we report the feasibility of using 2D-DIGE and related novel proteomics technology, as a tool for measurement of differential protein expression in blood plasma with identification of oxidative stress related markers from the plasma protein profile of smoke-exposed mice. This study in the mouse model demonstrates that smoking exposure results in decreases or increases in the levels of certain plasma proteins that are involved in vascular function, metabolism and immune function. Overall our data show that cigarette smoke exposure induces changes in the plasma protein expression profile with alterations in several ISPs. As we have previously reported, these changes closely correlate with the initial onset of physiological changes of cardiovascular and pulmonary disease [53, 54]. Some physiological and structural alterations were observed in the cardiovascular and pulmonary systems of mice with chronic smoking exposure that are similar to smoking induced disease in humans. These changes are consistent with early hypertension associated cardiac alterations. Thus, the first physiological or structural signs of disease were observed after 32 weeks of CS exposure.
The changes in ISPs and other proteins could be of critical importance in the pathogenesis of smoking induced disease. We conclude that the differentially expressed proteins which we identify in the mouse model could serve as potential biomarkers of smoking induced disease. Further studies in human smokers will be needed to evaluate the potential use of these proteomic changes as a predictive measure of disease onset.
Acknowledgments
We are thankful to Kari B. Green-Church for her help on 2D-DIGE analysis and mass spectrometry and to Wesley Johnson for technical support with the mouse smoking exposure model, and Tamer M. Abdelghany for other technical assistance. We also thank Dr. M. A. Hassan Talukder for helpful comments on the manuscript and related support. This work and the personnel who performed it were supported by NIH grants HL63744, HL65608, HL38324 and the biomarker initiative grant from Lorillard Tobacco Company.
Abbreviations
- Cy3
1-(5-carboxypentyl)-1’- propylindocarbocyanine halide N-hydroxysuccinimidyl ester
- Cy5
1-(5-carboxypentyl)-1’-methylindodi-carbocyanine halide N-hydroxysuccinimidyl ester
- DTT
dithiothreitol
- CS
cigarette smoke
- CV
cardiovascular
- ISPs
inflammation sensitive proteins
- COPD
Chronic obstructive pulmonary disease
- 2D-DIGE
two-dimensional difference in-gel electrophoresis
Footnotes
Conflict of Interest Statement
There are no financial or commercial conflicts of interest.
References
- 1.Price JF, Mowbray PI, Lee AJ, Rumley A, et al. Relationship between smoking and cardiovascular risk factors in the development of peripheral arterial disease and coronary artery disease: Edinburgh Artery Study. Eur Heart J. 1999;20:344–353. doi: 10.1053/euhj.1998.1194. [DOI] [PubMed] [Google Scholar]
- 2.Chen L, Chester M, Kaski JC. Clinical factors and angiographic features associated with premature coronary artery disease. Chest. 1995;108:364–369. doi: 10.1378/chest.108.2.364. [DOI] [PubMed] [Google Scholar]
- 3.Njolstad I, Arnesen E, Lund-Larsen PG. Smoking, serum lipid, blood pressure, and sex differences in myocardial infarction. A 12-year follow-up of the Finnmark study. Circulation. 1996;93:450–456. doi: 10.1161/01.cir.93.3.450. [DOI] [PubMed] [Google Scholar]
- 4.Lavi S, Prasad A, Yang EH, Mathew V, et al. Smoking is associated with epicardial coronary endothelial dysfunction and elevated white blood cell count in patients with chest pain and early coronary artery disease. Circulation. 2007;115:1–7. doi: 10.1161/CIRCULATIONAHA.106.641654. [DOI] [PubMed] [Google Scholar]
- 5.Morrow JD, Frei B, Longmire AW, Gaziano JM, et al. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers: smoking as a cause of oxidative damage. N Engl J Med. 1995;332:1198–1203. doi: 10.1056/NEJM199505043321804. [DOI] [PubMed] [Google Scholar]
- 6.Celermajer D, Sorensen K, Georgakopoulos D, Bull C, et al. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation. 1993;88:2149–2155. doi: 10.1161/01.cir.88.5.2149. [DOI] [PubMed] [Google Scholar]
- 7.Zeiher AM, Schachinger V, Minners J. Long-term cigarette smoking impairs endothelium-dependent coronary arterial vasodilator function. Circulation. 1995;92:1094–1100. doi: 10.1161/01.cir.92.5.1094. [DOI] [PubMed] [Google Scholar]
- 8.Powell JT. Vascular damage from smoking: disease mechanisms at the arterial wall. Vasc Med. 1998;3(1):21–28. doi: 10.1177/1358836X9800300105. [DOI] [PubMed] [Google Scholar]
- 9.Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovas Res. 2006;70(2):181–190. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 10.Fraticelli A, Serrano CV, Jr, Bochner BS, Capogrossi MC, et al. Hydrogen peroxide and superoxide modulate leukocyte adhesion molecule expression and leukocyte endothelial adhesion. BBA. 1996;310(3):251–259. doi: 10.1016/0167-4889(95)00169-7. [DOI] [PubMed] [Google Scholar]
- 11.Rival J, Riddle JM, Stein PD. Effects of chronic smoking on platelet function. Thromb Res. 1987;45:75–85. doi: 10.1016/0049-3848(87)90258-1. [DOI] [PubMed] [Google Scholar]
- 12.Fusegawa Y, Goto S, Handa S, Kawada T, et al. Platelet spontaneous aggregation in platelet-rich plasma is increased in habitual smokers. Thromb Res. 1999;93:271–278. doi: 10.1016/s0049-3848(98)00184-4. [DOI] [PubMed] [Google Scholar]
- 13.Kojda G, Harrison D. Interaction between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res. 1999;43:562–571. doi: 10.1016/s0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 14.Wang P, Zweier JL. Measurement of nitric oxide and peroxynitrite generation in the postischemic heart: EVIDENCE FOR PEROXINITRITE-MEDIATED REPERFUSION INJURY. J Biol Chem. 1996;271(46):29223–29230. doi: 10.1074/jbc.271.46.29223. [DOI] [PubMed] [Google Scholar]
- 15.Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;19:1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 16.Merkel D, Rist W, Seither P, Weith A, et al. Proteomic study of human bronchoalveolar lavage fluids from smokers with chronic obstructive pulmonary disease by combining surface-enhanced laser desorption/ionization-mass spectrometry profiling with mass spectrometry protein identification. Proteomics. 2005;5:2972–2980. doi: 10.1002/pmic.200401180. [DOI] [PubMed] [Google Scholar]
- 17.Plymoth A, Lofdahl CG, Ekberg-Jansson A, Dahlback M, et al. Human bronchoalveolar lavage: Biofluid analysis with special emphasis on sample preparation. Proteomics. 2003;3:962–972. doi: 10.1002/pmic.200300387. [DOI] [PubMed] [Google Scholar]
- 18.Bandow JE, Baker JD, Berth M, Painter C, et al. Improved image analysis workflow for 2-D gels enables large-scale 2-D gel-based proteomics studies – COPD biomarker discovery study. Proteomics. 2008;8:3030–3041. doi: 10.1002/pmic.200701184. [DOI] [PubMed] [Google Scholar]
- 19.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 20.Gorg A, Postel W, Gunther S. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis. 1988;9:531–546. doi: 10.1002/elps.1150090913. [DOI] [PubMed] [Google Scholar]
- 21.Dongre AR, Eng JK, Yates JR. III Emerging tandem-mass spectrometry techniques for the rapid identification of proteins. Trends Biotechnol. 1997;15:418–425. doi: 10.1016/S0167-7799(97)01110-4. [DOI] [PubMed] [Google Scholar]
- 22.Gygi SP, Han DK, Gingras AC, Sonenberg N, Aebersold R. Protein analysis by mass spectrometry and sequence database searching: tools for cancer research in the post-genomic era. Electrophoresis. 1999;20:310–319. doi: 10.1002/(SICI)1522-2683(19990201)20:2<310::AID-ELPS310>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 23.Rout MP, Aitchison JD, Suprapto A, Hjertaas K, et al. The yeast nuclear pore complex: composition, architecture, and transport mechanism. J Cell Biol. 2000;148:635–651. doi: 10.1083/jcb.148.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 25.Zhou G, Li H, DeCamp D, Chen S, et al. 2D differential in-gel electrophoresis for the identification of Esophageal scans cell cancer-specific protein markers. Mol Cell Prot. 2002:117–123. doi: 10.1074/mcp.m100015-mcp200. [DOI] [PubMed] [Google Scholar]
- 26.Kleno TG, Leonardsen LR, Kjeldal HO, et al. Mechanisms of hydrazine toxicity in rat liver investigated by proteomics and multivariate data analysis. Proteomics. 2004;4:868–880. doi: 10.1002/pmic.200300663. [DOI] [PubMed] [Google Scholar]
- 27.Friedman DB, Hill S, Keller JW, et al. Proteomic analysis of human colon cancer by two-dimensional difference gel electrophoresis and mass spectrometry. Proteomics. 2004;4:793–811. doi: 10.1002/pmic.200300635. [DOI] [PubMed] [Google Scholar]
- 28.Orenes-Pinero E, Corton M, Gonzalez-Peramato P, Algaba F, et al. Searching urinary tumor markers for bladder cancer using a two-dimensional differential gel electrophoresis (2D-DIGE) approach. J Prot Res. 2007;6:4440–4448. doi: 10.1021/pr070368w. [DOI] [PubMed] [Google Scholar]
- 29.Talukder MAH, Johnson WM, Varadharaj S, Lian J, et al. Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice. AJP. doi: 10.1152/ajpheart.00868.2010. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, El-Sherbiny GA, Collard E, Huang X, et al. Application of carbon fiber composite minielectrodes for measurement of kinetic constants of nitric oxide decay in solution. Nitric Oxide. 2010;23:311–318. doi: 10.1016/j.niox.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson D, Lian J, El-Mahdy MA, Zweier JL. Effects of Smoking on Mouse Adipose Tissue Volumes Measured by IDEAL at 11.7T. 18th Annual Proceedings of the International Society of Magnetic Resonance in Medicine 2010; Stockholm, Sweden. May 1, 2010; Abstract 5267. [Google Scholar]
- 32.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, et al. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci U S A. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plump AS, Smith JD, Hayek T, Aalto-Setala K, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 34.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 35.Undas A, Szudrzynski K, Stepien E, Zalewski J, Godlewski J, et al. Reduced clot permeability and susceptibility to lysis in patients with acute coronary syndrome: effects of inflammation and oxidative stress. Atherosclerosis. 2008;196:551–557. doi: 10.1016/j.atherosclerosis.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 36.Barua RS, Sy F, Srikanth S, Huang G, et al. Effects of cigarette smoke exposure on clot dynamics and fibrin structure. Arterioscler Thromb Vasc Biol. 2010;30:75–79. doi: 10.1161/ATVBAHA.109.195024. [DOI] [PubMed] [Google Scholar]
- 37.Chhetri SK. Alpha-1-antitrypsin deficiency and increased lung cancer risk. Thorax. 2008;63:950. [Google Scholar]
- 38.Yang P, Sun Z, Krowka MJ, et al. Alpha1-antitrypsin deficiency carriers, tobacco smoke, chronic obstructive pulmonary disease, and lung cancer risk. Arch Intern Med. 2008;168:1097–103. doi: 10.1001/archinte.168.10.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engstrom G, Hedblad B, Tyden P, Lindgarde F. Inflammation- sensitive plasma proteins are associated with increased incidence of heart failure: A population-based cohort study. Atherosclerosis. 2009;202:617–622. doi: 10.1016/j.atherosclerosis.2008.05.038. [DOI] [PubMed] [Google Scholar]
- 40.Fowkes FG. Fibrinogen and peripheral arterial disease. Eur Heart J. 1995;16:36–41. doi: 10.1093/eurheartj/16.suppl_a.36. [DOI] [PubMed] [Google Scholar]
- 41.Evans MD, Pryor WA. Cigarette smoking, emphysema, and damage to alpha 1-proteinase inhibitor. Am J Physiol Lung Cell Mol Physiol. 1994;266:L593–L611. doi: 10.1152/ajplung.1994.266.6.L593. [DOI] [PubMed] [Google Scholar]
- 42.Tewari AK, El-Mahdy MA, Zweier JL. Proteomic analysis of biomarkers for smoking induced diseases in a mouse model. 63rd Tobacco Science Research Conference; September 27-30 2009; Florida, USA. p. 61. Program Booklet and Abstracts. [Google Scholar]
- 43.Masrsingh H, Zuidhof AB, Bos ST, Duin M, et al. Arginase inhibition protects against allergen-induced airways obstruction, hyperresponsiveness, and inflammation. Am J Respir Crit Care Med. 2008;178:565–573. doi: 10.1164/rccm.200710-1588OC. [DOI] [PubMed] [Google Scholar]
- 44.Cardounel AJ, Cui H, Samouilov A, Johnson W, et al. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J Biol Chem. 2007;282(2):879–887. doi: 10.1074/jbc.M603606200. [DOI] [PubMed] [Google Scholar]
- 45.Van den Berghe G. Disorders of gluconeogenesis. J Inher Metab Dis. 1996;19:470–477. doi: 10.1007/BF01799108. [DOI] [PubMed] [Google Scholar]
- 46.Hemingway DC. Smoking and hypoglycemia. Journal of Orthomolecular Medicine. 1989;4(3):136–138. [Google Scholar]
- 47.Nygard O, Vollset SE, Refsum H, Stensvold I, et al. Total plasma homocysteine and cardiovascular risk profile: The Hordaland Homocysteine Study. J Am Med Assoc. 1995;274:1526–1533. doi: 10.1001/jama.1995.03530190040032. [DOI] [PubMed] [Google Scholar]
- 48.Stein JH, Bushara M, Bushara K, McBride PE, et al. Smoking Cessation, but not smoking reduction, reduces plasma homocysteine levels. Clin Cardiol. 2002;25:23–26. doi: 10.1002/clc.4950250107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Callaghan P, Meleady R, Fitzgerald T, Graham I, et al. Smoking and plasma homocysteine. European Heart Journal. 2002;23:1580–1586. doi: 10.1053/euhj.2002.3172. [DOI] [PubMed] [Google Scholar]
- 50.Jacobsen DW, Catanescu O, Dibello PM, Barbato JC. Molecular targeting by homocysteine: a mechanism for vascular pathogenesis. Clin Chem Lab Med. 2005;43:1076–1083. doi: 10.1515/CCLM.2005.188. [DOI] [PubMed] [Google Scholar]
- 51.Splaver A, Lamas GA, Hennekens CH. Homocysteine and cardiovascular disease: biological mechanisms, observational epidemiology, and the need for randomized trials. Am Heart J. 2004;148:34–40. doi: 10.1016/j.ahj.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 52.Fine CI, Han CD, Sun X, Liu Y, et al. Tobacco reduces membrane HLA class 1 that is restored by transfection with transporter associated with antigen processing 1 cDNA. J Immunol. 2002;169:6012–6019. doi: 10.4049/jimmunol.169.10.6012. [DOI] [PubMed] [Google Scholar]
- 53.Zweier JL, Talukder MA, Johnson W, Varadharaj S, et al. Cigarette smoke exposure results in hypertension, leukocyte-specific reactive oxygen species (ROS) generation, endothelial dysfunction, and cardiac hyperthrophy in mice. 63rd Tobacco Science Research Conference; September 27-30 2009; Florida, USA. p. 59. Program Booklet and Abstracts. [Google Scholar]
- 54.El-Mahdy MA, Johnson W, Hemann C, Tewari AK, et al. Development of in-vivo mouse model for smoking-induced cardiovascular disease. 63rd Tobacco Science Research Conference; September 27-30 2009; Florida, USA. p. 19. Program Booklet and Abstracts. [Google Scholar]