

Abstract
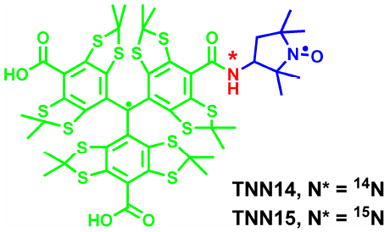
Simultaneous evaluation redox status and oxygenation in biological systems is of great importance for the understanding of biological functions. Electron paramagnetic resonance spectroscopy coupled with the use of the nitroxide radicals have been an indispensable technique for this application but are still limited by its low oxygen sensitivity, and low EPR resolution in part due to the moderately broad EPR triplet and spin quenching through bioreduction. In this study, we showed that these drawbacks can be overcome through the use of trityl-nitroxide biradicals allowing for the simultaneous measurement of redox status and oxygenation. A new trityl-nitroxide biradical TNN14 composed of a pyrrolidinyl-nitroxide and a trityl, and its isotopically labeled 15N analogue TNN15 were synthesized and characterized. Both biradicals exhibited much stronger spin-spin interaction with J > 400 G than the previous synthesized trityl-nitroxide biradicals TN1 (~160 G) and TN2 (~52 G) with longer linker chain length. The enhanced stability of TNN14 was evaluated using ascorbate as reductant and the effect of different types of cyclodextrins on its stability in the presence of ascorbate was also investigated. Both biradicals are sensitive to redox status, and their corresponding trityl-hydroxylamines resulting from the reduction of the biradicals by ascorbate share the same oxygen sensitivity. Of note is that the 15N-labeled TNN15-H with an EPR doublet exhibits improved EPR signal amplitude as compared to TNN14-H with an EPR triplet. In addition, cyclic voltammetric studies verify the characteristic electrochemical behaviors of the trityl-nitroxide biradicals.
1. Introduction
Noninvasive measurement and monitoring of molecular oxygen (O2) or “oximetry” and redox status are of importance for the understanding of biological functions, and the pathogenesis of various diseases. Nuclear magnetic resonance (NMR)- and low field electron paramagnetic resonance (EPR)-based techniques, coupled with the use of exogenous paramagnetic probes, are the most appropriate approaches for the noninvasive in vivo O2 measurement and redox assessment due to the reasonable depth of magnetic field penetration in animal tissues.1-4 In principle, EPR methods (both spectroscopic and imaging) show better sensitivity to O2 and redox measurement compared to NMR methods since the former have a much higher intrinsic sensitivity to exogeneous probe concentration. EPR imaging has been widely used to measure oxygenation and redox status in living tissue and in vivo mice.5-8 Recently, a new MRI-based technique, commonly called Overhauser-enhanced MRI (OMRI) that combines the sensitivity of EPR with the advantage of MRI, has been employed for the measurement of tissue oxygenation and redox status.9-11
Among the commonly used paramagnetic probes, nitroxide radicals are the most popular but their application in vivo were greatly limited by their rapid bioreduction and their relatively low sensitivity to O2. Although the information on the redox status was extracted from the bioreduction of the nitroxides in the investigated systems by EPR imaging or OMRI, this spin quenching mechanism greatly limits the imaging resolution. Recent development of extraordinary stable trityl radicals has tremendous benefits in EPR imaging and OMRI.12-21 To date, trityl radicals have been utilized to measure extracellular22 and intracellular17,23 oxygen level, superoxide radical anion24 and pH.18,25 However, their application as redox probes has been a challenge due to their inertness to biological reductants such as ascorbate and glutathione.16,24
Recently, we described a novel strategy using trityl-nitroxide biradicals to simultaneously measure redox status and oxygenation through EPR spectrocopy.26 As redox and O2 probes, these biradicals possess several advantages compared to nitroxide radicals: 1) their bioreduction results in the formation of the trityl-hydroxylamine monoradical whose signal intensity is enhanced therefore effectively increasing the EPR signal and secondary sensitivity. In contrast, the EPR signals of simple nitroxides are completely quenched by bioreduction; 2) the resulting trityl radical from bioreduction has narrow singlet EPR line and high stability which can allow to further increase quality and resolution of EPR imaging in less time; 3) introduction of an amide group into the trityl molecule affords a partial overlapping sharp triplet EPR signal that exhibits a higher O2 sensitivity as compared to the usual trityl radical with a singlet EPR signal or the nitroxide radicals. As part of our continuing efforts in trityl-nitroxide biradical development, we herein report the synthesis of a new trityl-nitroxide biradical TNN14 and its isotopically labeled analogue TNN15 (Chart. 1) with a short linker group. These two biradicals exhibit higher sensitivity for simultaneous measurement redox status and oxygenation as compared to the prior trityl-nitroxide biradicals with longer linker group. In addition, their electrochemical properties were also investigated.
Chart 1.
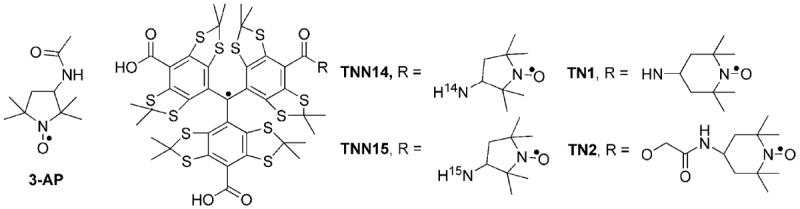
Molecular strutures of biradicals and nitroxide 3-AP.
2. Results and discussion
The synthesis of the biradical TNN14 and its analogue TNN15 is shown in Scheme 1. While the nitroxide radical 14NN (also called as 3-amino-PROXYL) is commercially available, its isotopical analogue 15NN was synthesized using 3-carboxy-proxyl (3-CP) as a starting material according to the previous procedure.27 3-CaP was obtained by conjugation of 3-CP with HOSu, followed by amidation using 15NH4Cl. Subsequent treatment of 3-CaP with sodium hypobromite at 0-70°C for 3h affords the 15N-labelled nitroxide, 15NN, with a high yield of 52% from 3-CP. The trityl radical CT-03 was synthesized via the previously reported 4-step method15 and linked with 14NN or 15NN, to afford the biradicals TNN14 or TNN15 which were characterized by HRMS and IR. The purity of both biradicals was determined by HPLC (>98%) and EPR method17 (97±1% for TNN14 and 95±1% for TNN15) (See Supporting Information (SI)). Interestingly, two close peaks were observed in their HPLC chromatograms indicating two components which were further determined as two isomers by MS analysis (See SI). These two isomers are possibly diastereomers associated with the inherent chirality of the trityl moiety.
Scheme 1.
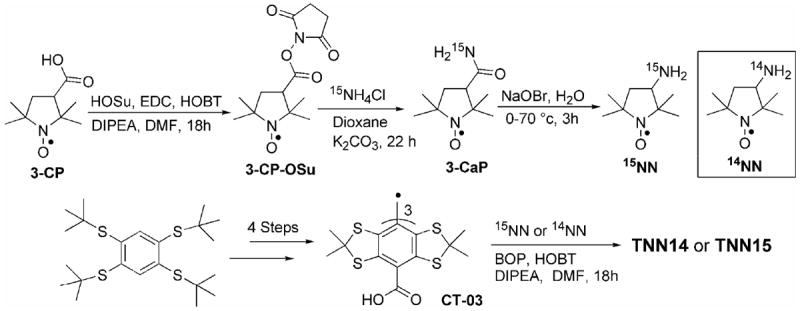
Synthesis of biradicals TNN14 and TNN15
Fig. 1 shows the room-temperature EPR spectra of TNN14 and TNN15. Completely identical spectral profiles for both biradicals were observed indicating that the isotopic labeling in the amide group did not affect the magnitude of spin-spin interaction between the two radical moieties. EPR spectra for both biradicals present a well-resolved triplet with the lines spaced by ~7.9 G, slightly less than half the 14N hyperfine splitting of the five-membered ring nitroxide 3-AP (16.1 G, Chart 1) in aqueous solution. Interestingly, the EPR triplet lines have almost the same linewidth and amplitude owing to the strong intramolecular spin-spin interaction. EPR spectral profile of TNN14 is not affected at a concentration range of 10-500 μM (see Fig. S3 in the SI), further confirming the intramolecular nature of the spin interaction. Computer simulation28 of these spectra gave a J-coupling value of > 400 G for TNN14 and TNN15 at room temperature which is much higher than the values of the previously reported biradicals, TN1 (~160 G) and TN2 (~52 G).26 The high J-coupling values for TNN14 and TNN15 are most likely due to the short distance between the NO moiety and the central carbon of the trityl radical compared to those of TN1 and TN2.
Figure 1.
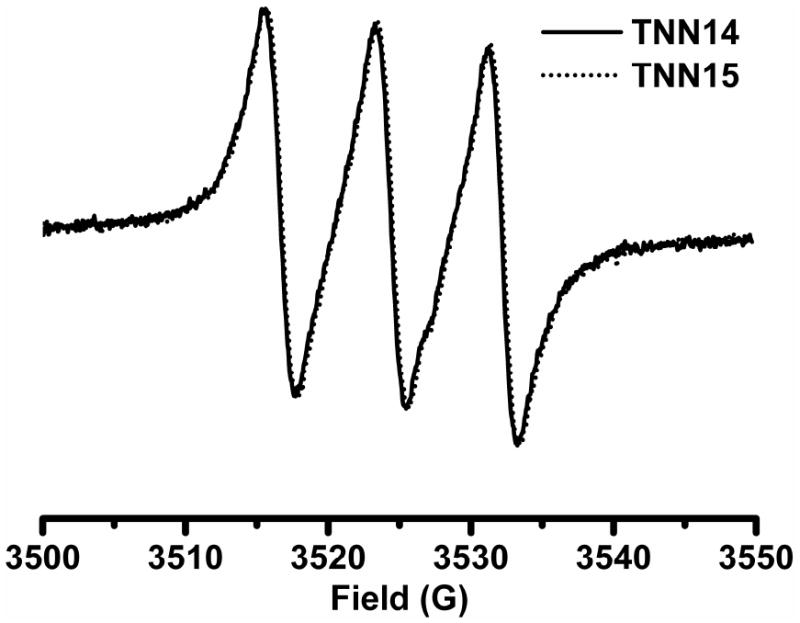
Room-temperature X-band EPR spectra of TNN14 and TNN15 in PBS (pH 7.4, 50 mM). Spectra were recorded with 10 mW microwave power and 1 G modulation amplitude.
The potential application of the new biradicals as redox probes was then evaluated using ascorbate, which is the main reductant of nitroxides in biological systems. The reduction of TNN14 by ascorbate resulted in the formation of the monoradical trityl-hydroxylamine (TNN14-H) and affords an intense partially-overlapped EPR triplet signal (Fig. 2A and inset). The formation of the signal was accompanied by a relatively slow decrease of the biradical signal due to the transformation of TNN14 into TNN14-H. The unchanged UV absorbance around 469 nm proved that the trityl moiety is stable towards ascorbate (See SI). Addition of the oxidant K3Fe(CN)6 resulted in the recovery of the signal due to TNN14 from TNN14-H (See SI), suggesting the reversibility of the reduction of the nitroxide moiety. Fig. 2B shows the effect of the ascorbate concentration on the production of TNN14-H. Higher concentrations of ascorbate led to a faster increase in the signal intensity of the singlet peak and vice versa. According to the data shown in Fig. 2B, the second order rate constant (k2) for the TNN14 reduction by ascorbate was determined to be 0.44 ± 0.07 M-1s-1. A similar k2 value was observed for the nitroxide 3-AP (Chart. 1) (0.36 ± 0.06 M-1s-1, see SI) which indicates that linkage of the nitroxide to the trityl does not significantly affect the reactivity of the nitroxide moiety. Moreover, TNN14 has much smaller k2 compared to the piperidinyl nitroxide-trityl biradicals TN1 (4.14 ± 0.14 M-1s-1) and TN2 (3.48 ± 0.09 M-1s-1).9 This result is consistent with the previous work showing the lower rates of reduction for the pyrrolidinyl nitroxide compared to the piperidinyl nitroxide due to the conformational flexibility of the latter compared to the former.29
Figure 2.
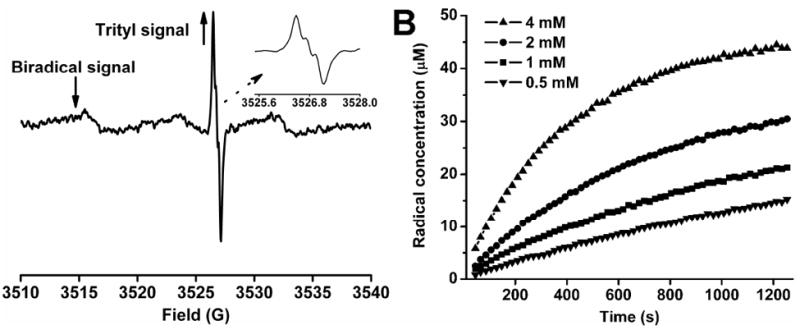
(A) EPR spectrum obtained from the reaction mixture containing TNN14 (50 μM) and ascorbate (4 mM) in PBS (pH 7.4, 50 mM) after 45 s at room temperature; (↑) indicates the increasing signal of the trityl monoradical TNN14-H which resulted from the reduction of the biradical TNN14, (↓) shows the decreasing signal of the biradical TNN14; the inset shows an expanded portion of the spectrum of the trityl monoradical in order to better visualize its triplet hyperfine structure. In order to detect both signals from the biradical and the resulting trityl radical without any distortion, spectra were recorded with 1 mW microwave power and 0.08 G modulation amplitude. These low values of microwave power and modulation amplitude are required to prevent broadening or saturation of the trityl radical spectrum. (B) Plots of the TNN14-H concentration as a function of time at various ascorbate concentrations at room temperature.
Although the nitroxide bioreduction provides important information on the redox state in a given biological system, the bioreduction is often too fast to be monitored. In this study, the use of five-membered ring nitroxide for the construction of the trityl-nitroxide biradicals effectively slows their reduction by ascorbate as mentioned above. In order to further stabilize the biradicals and enhance their in vivo application potential, we investigated the effect of three different types of cyclodextrins (CDs) on the stability of the biradicals on the basis of the knowledge that CDs can effectively protect nitroxide radicals30-32 and nitrone spin adducts33-35 from bioreduction. As shown in Fig. 3, β-CD and its methylated analogue (M-β-CD) effectively decrease the reduction of TNN14 by ascorbate as evidenced by the slower formation of the trityl signal intensity in the solution of TNN14 containing ascorbate. Comparatively, γ-CD enhances the reaction between TNN14 and ascorbate with 148% higher signal intensity in the presence of γ-CD than in the absence of γ-CD. The larger cavity size of γ-CD compared to β-CD could accommodate both the biradical, (or at least the nitroxide moiety), and ascorbate, therefore facilitating their reaction. Although high concentrations of β-CDs were used to provide protection to biradicals in this study, the covalent linkage of biradicals with methyl-β-CDs may offer better strategy for more stable biradicals with longer half-lives in biological milleiu.
Figure 3.
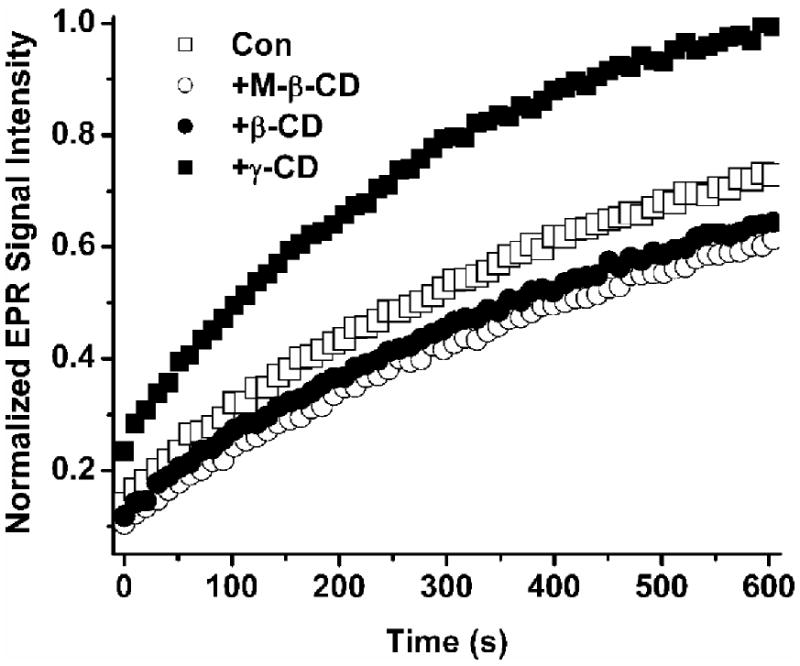
Effect of various cyclodextrins (2 mM) on the reduction of TNN14 (50 μM) and ascorbate (4 mM) in PBS.
Under anaerobic condition, the partially-overlapped EPR triplet signal of TNN14-H (Fig. 2A) becomes more pronounced (Fig. 4, top) with a hyperfine splitting constant (hfc) of 0.23 G due to reduced Heisenberg exchange between TNN14-H and paramagnetic O2. Comparatively, the trityl-hydroxylamine TNN15-H obtained through reduction of TNN15 by ascorbate gave a well-resolved doublet with a hfc of 0.33 G under anaerobic condition (Fig. 4). These additional hyperfine splittings observed from the trityl-hydroxylamines should be due to the amide-N (I =1 for 14N and I = ½ for 15N) of the linker group, verifying our previous theoretical and experimental results showing long range radical coupling.26 Apart from the hyperfine interactions with the main nitrogen isotope (i.e., 14N for TNN14-H and 15N for TNN15-H), the interactions with the 13C (natural abundance, 1.11%) mainly from the carbons of three aromatic groups and the other nitrogen isotope (natural abundance of 15N (0.37%) for TNN14-H and 14N (1%) for TNN15-H in our experiment) can be also observed, giving the weak flanking lines as shown in Fig. 4.36 Interestingly, the direct linkage of the amide group with the trityl moiety can further split these signals from CT-03 into doublet for TNN15-H or triplet for TNN14-H (see the enlarged signals in Fig. 4).
Figure 4.
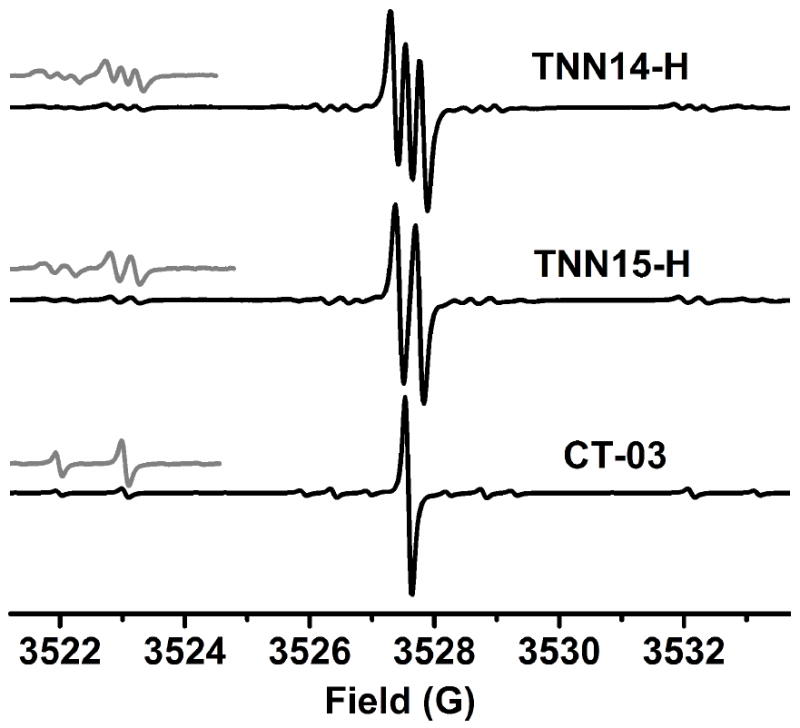
X-band EPR spectra of TNN14-H, TNN15-H and CT03 in PBS (pH 7.4, 50 mM) under anaerobic conditions. Gray lines show the enlarged (5×) portion of the spectrum. Spectra were recorded with 0.5 mW microwave power and 0.03 G modulation amplitude. These low values were required to prevent distortion of the very sharp trityl radical spectra detected under anaerobic conditions.
It has been demonstrated that the partially overlapped EPR peaks of the trityl radicals is sensitive to O2 and the spectral ratio (Iin/Iout) exhibits enhanced sensitivity towards O2, especially at low pO2, relative to the linewidth.21,26 It is therefore reasonable to use the ratio Iin/Iout to evaluate the oxygen sensitivity of TNN14-H and TNN15- H. Fig. 5A shows the EPR spectra of TNN14-H and TNN15-H which are denoted by the spectral intensities, Iin and Iout. As shown in Fig. 5B, the spectral ratio Iin/Iout is highly sensitive to O2 and both radicals have almost identical O2 sensitivities with values of 1.55×10-3 /%O2 for TNN14-H and 1.53×10-3/%O2 for TNN15-H. Although the isotopic 15N labeling does not improve the oxygen sensitivity of TNN15-H, TNN15-H has 3/2=1.5 times larger EPR signal amplitude than TNN14-H at the same concentrations due to the reduced number of hyperfine lines (Fig. 5A). Therefore, the isotopic 15N labeling of the biradical can greatly improve its sensitivity and resolution for simultaneous measurement of redox status and oxygenation as compared to nitroxides, trityl radicals alone as well as the non-labeled trityl-nitroxide biradicals, TN1, TN2 and TNN14.
Figure 5.
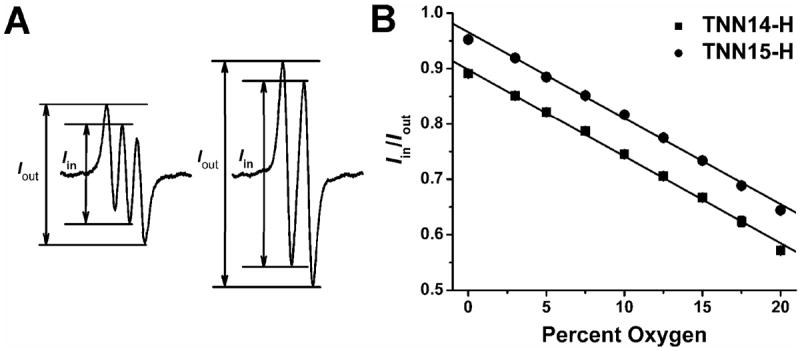
(A) EPR spectra of TNN14-H and TNN15-H denoting the spectral ratios Iin and Iout in the presence of 10%O2-90%N2. Spectra were recorded with 0.5 mW microwave power and 0.03 G modulation amplitude. (B) Plot of Iin/Iout as a function of percent oxygen. The trityl-hydroxylamines were generated by mixing ascorbate (4 mM) with the corresponding biradical (50 μM).
It has been demonstrated that as neutral radicals, both trityl and nitroxide radicals can be either reduced or oxidized via one-electron process. It is thus interesting to investigate the electrochemical behavior of the trityl-nitroxide biradicals. In doing so, the cyclic voltammeric studies were carried out on the biradical TNN14. As shown in Fig. 6, TNN14 undergoes two one-electron reversible oxidation of TNN14 to the corresponding trityl cation and oxammonium at E½ (ox) = 0.515 V vs Ag/AgCl (ΔEp = 64 mV) and 0.686 V vs Ag/AgCl (ΔEp = 61 mV), respectively. A quasi-reversible one-electron reduction at E½ (red) = -0.581 V vs Ag/AgCl (ΔEp = 113 mV) was also observed which was assigned to the reduction of the trityl moiety to the corresponding anion. Under our experimental conditions, the reduction of the nitroxide moiety was not observed although the above result has showed that the biradical TNN14 can undergo one-electron reduction by ascorbate to the corresponding trityl-hydroxylamine. Compared to the redox potential of the parent trityl CT-03 [E½ (ox) = 0.45 V vs Ag/AgCl and E½ (red) = -0.63 V vs Ag/AgCl],16 the trityl moiety in TNN14 is more difficultly oxidized [E½ (ox) = 0.515 V vs Ag/AgCl] but easier reduced [E½ (red) = -0.581 V vs Ag/AgCl] because one of carboxylate groups in CT-03 is replaced by a stronger electron-withdrawing amide group in the case of TNN14. CV studies were also carried out on the biradical TN1. The replacement of the pyrrolidinyl nitroxide in TNN14 with a piperidinyl nitroxide makes the first oxidation (E1/2 = 0.531 V) and reduction (E1/2 = -0.587 V) slightly more difficult but easier for the second oxidation (E1/2 = 0.655 V).
Figure 6.
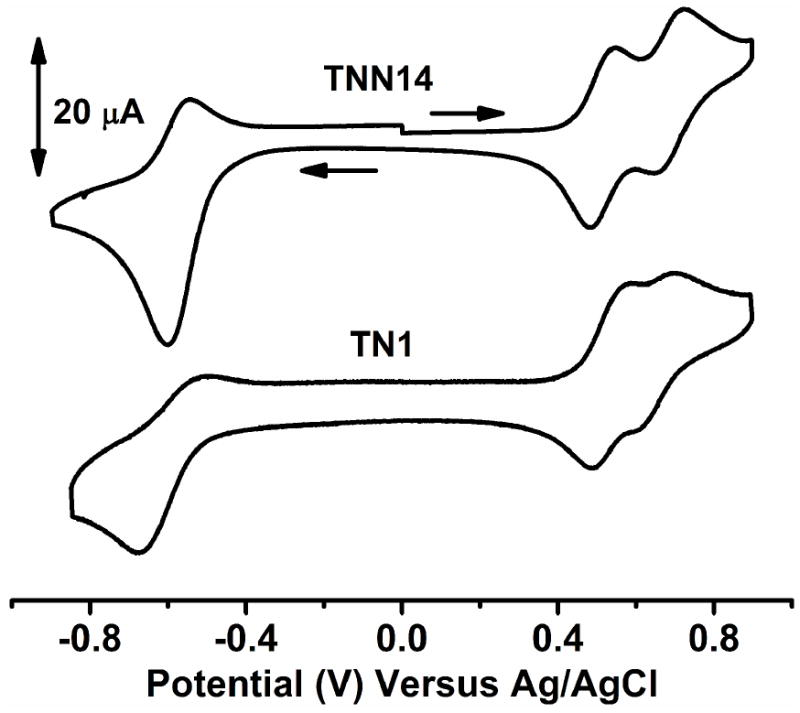
Cyclic voltammograms of TNN14 (1 mM) and TN1 (1 mM) in PBS (20 mM, pH 7.4) containing 0.15M NaCl. Scan rate: 100 mV/s.
In summary, the newly synthesized trityl-nitroxide biradicals TNN14 and its 15N-labeled analogue TNN15 possess strong intramolecular spin-spin interaction (J > 400 G) due to the short distance between two radical centers. Both biradicals are sensitive to the variation of the redox status. The use of five-membered ring nitroxide for the construction of the trityl-nitroxide biradicals as well as exogenous addition of β-CD and its methylated analogue M-β-CD effectively increases the stability of these two biradicals towards the reductant ascorbate, therefore enhancing their suitability for in vivo application. While both trityl-hydroxylamines TNN14-H and TNN15-H resulting from the reduction of the corresponding biradicals show high oxygen sensitivity, the 15N-labeled TNN15-H with its doublet EPR signal exhibits improved EPR signal resolution compared to TNN14-H with its EPR triplet. Therefore the new probes also exhibit enhanced sensitivity for simultaneous measurement of redox status and oxygenation compared to either nitroxide or trityl radicals alone.
Experimental sections
EPR measurements and simulations
EPR measurements were carried out on a Bruker EMX-X band with an HS resonator at room temperature. General instrument settings were as follows: modulation frequency, 100 kHz; microwave frequency, 9.87 GHz; microwave power, 10 mW for biradicals and nitroxide radicals and 0.5-2 mW for the trityl-hydroxylamines; microwave frequency, 9.87 GHz; modulation amplitude, 1.0 G for biradicals and nitroxide radicals and 0.03-0.08 G for the trityl-hydroxylamines; receiver gain, (1-10.00) × 104; time constant, 10.24-40.96 ms; sweep time, 10.49-41.94 s. Measurements were performed using 50 μL capillary tubes.
Simulations were carried out using a very developed EPR simulation program (ROKI\EPR).28 The fitting routine to determine the J values of the trityl-nitroxide biradicals was similar to the method described in our previous study.26 Since we could not find any improvement about the quadratic error between experimental and calculated spectra when J was larger than 400 G, the J values for TNN14 and TNN15 were suggested to be > 400 G.
Oxygen sensitivity
Oxygen sensitivities of the trityl-hydroxylamine TNN14-H and TNN15-H were evaluated according to our previous method. In brief, ascorbic acid (4 mM) was added to a solution of the biradicals (50 μM) in PBS buffer. After 60 min, the solution was transferred into a gas-permeable Teflon tube (i.d. = 0.8 mm) and was sealed at both ends. The sealed sample was placed inside a quartz EPR tube with open ends. Nitrogen or N2/O2 gas mixture with varying concentrations of O2 was allowed to bleed into the EPR tube and after about 4 min, was changed into another gas mixture. EPR spectra were recorded using a model of incremental sweep. According to the resulting spectra, the spectral ratio (Iin/Iout) was calculated.
Cyclic Voltammetry
Cyclic voltammetry was performed on a potentiostat and computer-controlled electroanalytical system. Electrochemical measurements were carried out in a 10 mL cell equipped with a glassy carbon working electrode (7.07 mm2), a platinum-wire auxiliary electrode and a Ag/AgCl reference electrode. Solutions of biradicals (1 mM) were degassed by bubbling with the nitrogen gas before the detection. The redox potentials were calculated according to the relation E = (Eap+Ecp)/2.
Reaction kinetics of TNN14 with ascorbate
Various concentrations of ascorbic acid (0.5, 1, 2 and 4 mM) were added to the solution of TNN14 (50 μM) in PBS (50 mM, pH 7.4). Incremental EPR spectra were recorded 45 s after mixing. The concentration of the trityl-hydroxylamine TNN14-H at each time point was obtained by comparing their double integrated signal intensities relative to CT03 as standard. Since the ascorbic acid concentration (0.5, 1, 2 and 4 mM) used was in greater excess than the biradical concentration (50 μM), the reaction kinetics of the biradical with ascorbic acid is a pseudo first-order reaction. The resulting curves in Fig.2B were fitted with the equation: ln[(C0-Ct)/C0] = -kobst, where C0 is the initial concentration of TNN14, Ct the concentration of the trityl-hydroxylamine TNN14-H at each time point, and kobs the observed pseudo first-order rate constant. Considering kobs = k2[Asc], the approximated second-order rate constant k2 was finally calculated from the slope of the plot of kobs versus [Asc].
Synthesis
1-Oxyl-2,2,5,5-tetramethylpyrrolidinyl-3-carboxylic acid N-hydroxysuccinimide ester (3-CP-OSu)
To a solution of 3-carboxy-proxyl (0.53 g, 2.86 mmol), HOBT (1.61 g, 8.60 mmol), DIPEA (2.49 mL, 14.3 mmol) and N-hydroxysuccinimide (0.66 g, 5.73 mmol) in DMF (20 mL) was added a solution of EDCI (1.10 g, 5.73 mmol) in DMF (5 mL) at 0°C. After addition, the reaction mixture was stirred for 18h at room temperature. Solvents were removed under vacuum and the residue was purified by flash column chromatography using the solvents ethyl acetate/ petroleum ether (1:3, 1:2, and then 1:1, v/v) as eluents. A light yellowish solid (0.71 g) was obtained. Yield: 88%. IR (cm-1, neat): 2978, 2937, 1810, 1783, 1735, 1464, 1427, 1366, 1304, 1245, 1200, 1092, 1065, 1047, 994, 958, 920, 899, 813, 735, 647; GC-MS: retention time, 5.90 min; [M-C4H4NO3+H]+, 186.11(calculated), 186.00 (measured); [M-C9H15NO3+H]+, 115.03 (calculated), 114.94 (measured).
3-Carbamoyl (15N)-2,2,5,5-tetramethylpyrrolidin-1-yloxy (3-CaP)
To a solution of 3-CP-OSu (0.5 g, 1.77 mmol) and K2CO3 (0.3 g) in dioxane (5 mL) and water (1 mL) was added solid 15NH4Cl (99% isotopic purity, 0.106 g, 1.94 mmol). The reaction mixture was stirred for 22h at room temperature, filtered and evaporated to dryness under vacuum. The residue was redissolved in 10 mL of ethyl acetate. After filtration, the filtrate was concentrated and separated by flash column chromatography using 2% methanol in dichlomethane as an eluent. The yellow solid was obtained and recrystallized from hexane and ethyl acetate to give the pure 3-Cap (0.31g, 94%). IR (cm-1, neat): 3346, 3195, 2977, 2934, 1668, 1462, 1426, 1365, 1321, 1283, 1244, 1171, 1154, 1102, 830, 739; GC-MS: retention time, 7.22 min; [M]+, 186.13 (calculated), 185.99 (measured).
3-Amino (15N)-2,2,5,5-tetramethyl-1-pyrrolidin-1-yloxy (15NN)
To a solution of sodium hypobromite prepared from 0.36g NaOH (9 mmol), 4 mL of water and 0.34 g of bromide (2.13 mmol) was added 0.28 g of 3-CaP (1.50 mmol) at 0°C. The reaction mixture was stirred for 2h at 0°C and then heated to 70 °C and kept this temperature for 1h. The reaction mixture was cooled with the ice bath and treated with 6 g KOH, and extracted with ether. The extract was dried on anhydrous Na2SO4, concentrated and separated by flash column chromatography using CH2Cl2:CH3OH:NH4OH =10:1:0.05 as an eluent. A yellow solid (0.15 g) was obtained. Yield: 63%. IR (cm-1, neat): 3483, 3363, 3291, 3198, 2972, 2933, 1610, 1463, 1363, 1315, 1254, 1228, 1195, 1164, 1102, 1059, 1024, 895, 834, 763, 673; GC-MS: retention time, 5.13 min; [M]+, 158.13 (calculated), 157.99 (measured).
TNN14
To the solution of CT-03 (100 mg, 0.1 mmol), HOBt (40.5 mg, 0.3 mmol) and BOP (46.4 mg, 0.105 mmol) in dry DMF (10 ml) was added DIPEA (90 μL) under N2. The reaction mixture was stirred at room temperature for 20 min and then 14NN (16.5 mg, 0.105 mmol) in 5 mL of DMF was added dropwise. The resulting mixture was continuously stirred for 18 h at room temperature. Solvent was removed under vacuum, and the residue was dissolved in phosphate buffer (0.1 M, pH 7.4) and purified by column chromatography on reverse phase C-18 using water followed by 0-15% acetonitrile in water as eluants to give the biradical TNN14 as a green solid (75 mg, 66 %). Purity: >98% by HPLC (See SI) and 97 ± 1% versus TEMPOL determined as previously reported.17 IR (cm-1, neat): 3425.1, 2971, 2933, 1646, 1580, 1454, 1367, 1312, 1237, 1168, 1150, 1113, 886, 820, 725, 697; MS ([M+H]+, m/z): 1139.038 (measured), 1139.071 (calculated); ([M+Na]+, m/z): 1161.000 (measured), 1161.052 (calculated).
TNN15
To the solution of CT-03 (63.3 mg, 63.3 μmol), HOBt (25.6 mg, 189.9 μmol) and BOP (29.4 mg, 66.5 μmol) in dry DMF (8 ml) was added DIPEA (60 μL) under N2. The reaction mixture was stirred at room temperature for 20 min and then 15NN (10 mg, 63.3 μmol) in 5 mL of DMF was added dropwise. The resulting mixture was continuously stirred for 18 h at room temperature. Solvent was removed under vacuum, and the residue was dissolved in phosphate buffer (0.1 M, pH 7.4) and purified by column chromatography on reverse phase C-18 using water followed by 0-15% acetonitrile in water as eluants to give the biradical TNN15 as a green solid (45 mg, 62 %). Purity: >98% by HPLC (See SI) and 95 ± 1% versus TEMPOL determined as previously reported.17 IR (cm-1, neat): 3423, 2973, 2918, 1646, 1579, 1455, 1367, 1238, 1150, 883, 727. MS ([M+H]+, m/z):1140.044 (measured), 1140.068 (calculated); MS ([M+K]+, m/z): 1177.985 (measured), 1178.024 (calculated).
Supplementary Material
Acknowledgments
This work was supported by supported by NIH grants HL38324, EB0890, EB4900 (J. L. Z.) and HL81248 (F. A. V.).
Footnotes
Supporting Information Available: GC-MS, IR, HRMS spectra and HPLC chromatograms, kinetic studies of the nitroxide radical 3-AP with ascorbate. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Berliner LJ, Fujii H, Wan XM, Lukiewicz SJ. Magn Reson Med. 1987;4:380–384. doi: 10.1002/mrm.1910040410. [DOI] [PubMed] [Google Scholar]
- 2.Zweier JL, Kuppusamy P. Proc Natl Acad Sci U S A. 1988;85:5703–5707. doi: 10.1073/pnas.85.15.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grucker D. Prog Nucl Magn Reson Spectrosc. 2000;36:241–270. [Google Scholar]
- 4.Hyodo F, Soule BP, Matsumoto KI, Matusmoto S, Cook JA, Hyodo E, Sowers AL, Krishna MC, Mitchell JB. J Pharm Pharmacol. 2008;60:1049–1060. doi: 10.1211/jpp.60.8.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He GL, Shankar RA, Chzhan M, Samouilov A, Kuppusamy P, Zweier JL. Proc Natl Acad Sci U S A. 1999;96:4586–4591. doi: 10.1073/pnas.96.8.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuppusamy P, Li HQ, Ilangovan G, Cardounel AJ, Zweier JL, Yamada K, Krishna MC, Mitchell JB. Cancer Res. 2002;62:307–312. [PubMed] [Google Scholar]
- 7.Elas M, Williams BB, Parasca A, Mailer C, Pelizzari CA, Lewis MA, River JN, Karczmar GS, Barth ED, Halpern HJ. Magn Reson Med. 2003;49:682–691. doi: 10.1002/mrm.10408. [DOI] [PubMed] [Google Scholar]
- 8.Gallez B, Swartz HM. NMR Biomed. 2004;17:223–225. doi: 10.1002/nbm.913. [DOI] [PubMed] [Google Scholar]
- 9.Li HH, Deng YM, He GL, Kuppusamy P, Lurie DJ, Zweier JL. Magn Reson Med. 2002;48:530–534. doi: 10.1002/mrm.10222. [DOI] [PubMed] [Google Scholar]
- 10.Li HH, He GL, Deng YM, Kuppusamy P, Zweier JL. Magn Reson Med. 2006;55:669–675. doi: 10.1002/mrm.20804. [DOI] [PubMed] [Google Scholar]
- 11.Utsumi H, Yamada K, Ichikawa K, Sakai K, Kinoshita Y, Matsumoto S, Nagai M. Proc Natl Acad Sci U S A. 2006;103:1463–1468. doi: 10.1073/pnas.0510670103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ardenkjaer-Larsen JH, Laursen I, Leunbach I, Ehnholm G, Wistrand LG, Petersson JS, Golman K. J Magn Reson. 1998;133:1–12. doi: 10.1006/jmre.1998.1438. [DOI] [PubMed] [Google Scholar]
- 13.Reddy TJ, Iwama T, Halpern HJ, Rawal VH. J Org Chem. 2002;67:4635–4639. doi: 10.1021/jo011068f. [DOI] [PubMed] [Google Scholar]
- 14.Xia SJ, Villamena FA, Hadad CM, Kuppusamy P, Li YB, Zhu H, Zweier JL. J Org Chem. 2006;71:7268–7279. doi: 10.1021/jo0610560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhimitruka I, Velayutham M, Bobko AA, Khramtsov VV, Villamena FA, Hadad CM, Zweier JL. Bioorg Med Chem Lett. 2007;17:6801–6805. doi: 10.1016/j.bmcl.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Villamena FA, Zweier JL. Chem Commun. 2008:4336–4338. doi: 10.1039/b807406b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Villamena FA, Sun J, Xu Y, Dhimitruka I, Zweier JL. J Org Chem. 2008;73:1490–1497. doi: 10.1021/jo7022747. [DOI] [PubMed] [Google Scholar]
- 18.Dhimitruka I, Bobko AA, Hadad CM, Zweier JL, Khramtsov VV. J Am Chem Soc. 2008;130:10780–10787. doi: 10.1021/ja803083z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Driesschaert B, Charlier N, Gallez B, Marchand-Brynaert J. Bioorg Med Chem Lett. 2008;18:4291–4293. doi: 10.1016/j.bmcl.2008.06.100. [DOI] [PubMed] [Google Scholar]
- 20.Dhimitruka I, Grigorieva O, Zweier JL, Khramtsov VV. Bioorg Med Chem Lett. 2010;132:3946–3949. doi: 10.1016/j.bmcl.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bobko AA, Dhimitruka I, Eubank TD, Marsh CB, Zweier JL, Khramtsov VV. Free Radic Biol Med. 2009;47:654–658. doi: 10.1016/j.freeradbiomed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kutala VK, Parinandi NL, Pandian RP, Kuppusamy P. Antioxid Redox Signal. 2004;6:597–603. doi: 10.1089/152308604773934350. [DOI] [PubMed] [Google Scholar]
- 23.Liu YP, Villamena FA, Sun J, Wang TY, Zweier JL. Free Radic Biol Med. 2009;46:876–883. doi: 10.1016/j.freeradbiomed.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rizzi C, Samouilov A, Kutala VK, Parinandi NL, Zweier JL, Kuppusamy P. Free Radic Biol Med. 2003;35:1608–1618. doi: 10.1016/j.freeradbiomed.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 25.Bobko AA, Dhimitruka I, Zweier JL, Khramtsov VV. J Am Chem Soc. 2007;129:7240–+. doi: 10.1021/ja071515u. [DOI] [PubMed] [Google Scholar]
- 26.Liu YP, Villamena FA, Rockenbauer A, Zweier JL. Chem Commun. 2010;46:628–630. doi: 10.1039/b919279d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rozantsev EG. Free Nitroxide Radicals. Plenum Press; 1970. [Google Scholar]
- 28.Rockenbauer A, Korecz L. Appl Magn Reson. 1996;10:29–43. [Google Scholar]
- 29.Couet WR, Brasch RC, Sosnovsky G, Lukszo J, Prakash I, Gnewuch CT, Tozer TN. Tetrahedron. 1985;41:1165–1172. [Google Scholar]
- 30.Ohara M, Hettler H, Gauss D, Cramer F. Bioorganic Chem. 1979;8:211–217. [Google Scholar]
- 31.Ebel C, Ingold KU, Michon J, Rassat A. Tetrahedron Lett. 1985;26:741–744. [Google Scholar]
- 32.Franchi P, Fani M, Mezzina E, Lucarini M. Org Lett. 2008;10:1901–1904. doi: 10.1021/ol800405b. [DOI] [PubMed] [Google Scholar]
- 33.Karoui H, Rockenbauer A, Pietri S, Tordo P. Chem Commun. 2002:3030–3031. doi: 10.1039/b209787g. [DOI] [PubMed] [Google Scholar]
- 34.Han YB, Tuccio B, Lauricella R, Villamena FA. J Org Chem. 2008;73:7108–7117. doi: 10.1021/jo8007176. [DOI] [PubMed] [Google Scholar]
- 35.Han YB, Liu YP, Rockenbauer A, Zweier JL, Durand G, Villamena FA. J Org Chem. 2009;74:5369–5380. doi: 10.1021/jo900856x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowman MK, Mailer C, Halpern HJ. J Magn Reson. 2005;172:254–267. doi: 10.1016/j.jmr.2004.10.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.