

Abstract
Background
Breast cancer frequently metastasizes to the brain, colonizing a neuro-inflammatory microenvironment. The molecular pathways facilitating this colonization remain poorly understood.
Methods
Expression profiling of 23 matched sets of human resected brain metastases and primary breast tumors by two-sided paired t test was performed to identify brain metastasis–specific genes. The implicated DNA repair genes BARD1 and RAD51 were modulated in human (MDA-MB-231-BR) and murine (4T1-BR) brain-tropic breast cancer cell lines by lentiviral transduction of cDNA or short hairpin RNA (shRNA) coding sequences. Their functional contribution to brain metastasis development was evaluated in mouse xenograft models (n = 10 mice per group).
Results
Human brain metastases overexpressed BARD1 and RAD51 compared with either matched primary tumors (1.74-fold, P < .001; 1.46-fold, P < .001, respectively) or unlinked systemic metastases (1.49-fold, P = .01; 1.44-fold, P = .008, respectively). Overexpression of either gene in MDA-MB-231-BR cells increased brain metastases by threefold to fourfold after intracardiac injections, but not lung metastases upon tail-vein injections. In 4T1-BR cells, shRNA-mediated RAD51 knockdown reduced brain metastases by 2.5-fold without affecting lung metastasis development. In vitro, BARD1- and RAD51-overexpressing cells showed reduced genomic instability but only exhibited growth and colonization phenotypes upon DNA damage induction. Reactive oxygen species were present in tumor cells and elevated in the metastatic neuro-inflammatory microenvironment and could provide an endogenous source of genotoxic stress. Tempol, a brain-permeable oxygen radical scavenger suppressed brain metastasis promotion induced by BARD1 and RAD51 overexpression.
Conclusions
BARD1 and RAD51 are frequently overexpressed in brain metastases from breast cancer and may constitute a mechanism to overcome reactive oxygen species–mediated genotoxic stress in the metastatic brain.
Brain metastases represent one of the most devastating consequences of breast cancer (1,2). Approximately 35% of patients with metastatic HER2 (human epidermal growth factor receptor 2)–positive or triple negative (TN; hormone receptor negative, HER2 normal) tumors develop brain metastases (3,4), likely the result of systemic responses to chemotherapy at extracranial sites allowing more time for brain relapse, the inability of most anticancer agents to cross the blood–tumor barrier in effective concentrations, and increased diagnostic imaging. Brain metastases are also common in lung cancer and melanoma and will likely increase in other cancer types as effective systemic therapies emerge. Brain metastasis patients experience seizures, impaired motor function, and cognitive losses; brain metastases increasingly contribute to patient deaths (5).
The brain is an unusual environment for cancer cells, and unique molecular pathways are believed to regulate brain colonization. The brain is protected by the blood–brain barrier, which may remain partially patent in metastases (6,7). The brain metastatic microenvironment is also modified by a neuro-inflammatory response containing activated microglia and astrocytes (8,9), and colonization is punctuated by periods of dormancy (10,11). To date, only ST6GALNAC5 has been reported to confer brain-specific metastasis (12).
To better understand the pathways that promote human brain metastasis, we performed cancer-specific gene expression profiling on rare sets of primary breast tumors and resected brain metastases from the same patients. Overexpression of DNA double-strand break (DSB) repair genes, including BARD1 and RAD51, were observed in the matched brain metastases. DNA DSB, generated by genotoxic insults or DNA replication fork collapse from attempted traversal of various lesions, can be repaired nonfaithfully by nonhomologous end joining or faithfully by homologous recombination (HR). RAD51 is a critical component of the HR process, which, by forming a nucleofilament on the 3’ ends of single-stranded DNA at the break site, along with various comediators, facilitates invasion and annealing to the homologous DNA template on the sister chromatid [reviewed in (13)]. In addition, RAD51 has important other HR-related (14–19) and non-HR functions (16,20–23). Cancer phenotypic responses of altered RAD51 expression have been reported in models of chemotherapy (24–26) and radiation (27,28). To the best of our knowledge, no in vivo phenotypes have been reported for RAD51 overexpression (29).
BRCA1-associated RING domain protein 1 (BARD1) is a heterodimeric binding partner of the breast cancer type 1 susceptibility protein (BRCA1), with which it interacts through their N-terminal RING finger domains (30,31). This interaction is essential for the E3 ubiquitin ligase activity (32,33) and tumor suppressor functions of BRCA1 (34). Specifically, BARD1 facilitates BRCA1 nuclear translocation for DNA repair (35), enhances BRCA1 DNA binding (36), and may be required for BRCA1 function in homology-dependent DNA repair and maintaining genomic integrity (37). During S and G2 cell-cycle phase and upon DNA damage induction, the BARD1/BRCA1 heterodimer assembles into nuclear foci in protein complexes that also contain BRCA2 and the pivotal HR protein RAD51 (38,39). At the sites of DNA damage, this supercomplex directs HR through the BRCA1–BRCA2 linker protein PALB2 (40–42). BRCA1-independent functions of BARD1 have also been reported (43,44). Functional in vivo phenotypes of BARD1 overexpression have not been described.
We examined whether the unexpected overexpression of BARD1 or RAD51 in human brain metastases functionally promotes experimental brain or lung metastasis in mouse xenograft models. Further, we address our hypothesis that BARD1 and RAD51 overexpression enhances brain metastasis development, specifically by providing resistance against the genotoxic effects of reactive oxygen species (ROS) produced in the inflammatory metastatic brain.
Methods
Gene Expression Analysis
Archived, anonymized, formalin-fixed, paraffin-embedded sample pairs of brain metastases and primary tumors, each from patients treated between 1996 and 2006 in six Polish institutions were evaluated. This study was approved by the Institutional Review Board of the coordinating centers (Medical University of Gdansk, Poland; Indiana University). Gene expression of 23 matched sets of primary breast tumors and brain metastases were evaluated using cDNA-mediated annealing, selection, extension, and ligation (DASL) analysis according to the manufacturer’s instructions (Sentrix Universal Array; Illumina, San Diego, CA). More details appear in the Supplementary Methods (available online). This study used archival formalin-fixed, paraffin-embedded blocks, and all personal data were made anonymous and coded; therefore patient consent was not sought.
Cell Lines
The human MDA-MB-231 brain-tropic cell line (MDA-MB-231-BR) and murine 4T1-BR brain-tropic line were described previously (6,45). Human BARD1 and RAD51 cDNAs were subcloned into lentiviral vectors pCDH-CMV-MCS-EF1 (Systems Biosciences, Mountain View, CA); empty vector served as control. More details about overexpression and knockdown protocols appear in the Supplementary Methods (available online).
In Vitro Assays
All in vitro assays were performed using standard protocols and, where applicable, following the recommendations of product manufacturers. Detailed description of assay procedures, reagents, and antibodies appear in the Supplementary Methods (available online).
Mouse Models
Experiments were carried out under an approved Animal Use Agreement with the National Cancer Institute. Intracardiac-injected brain metastasis models were performed, and metastases were quantified as previously described (45,46). For these brain metastasis models, female NCr nu/nu mice aged 5 to 7 weeks (Charles River, Frederick, MD) were inoculated in the left cardiac ventricle with 175000 human MDA-MB-231-BR cells or 50000 murine 4T1-BR cells under isoflurane/oxygen anesthesia. Ten mice per group were injected. Mice were killed under carbon dioxide asphyxiation at onset of signs of tumor-induced morbidity, such as weight loss, ataxia, and/or paralysis.
For lung metastasis models, female NCr nu/nu mice or BALB/c mice aged 5 to 7 weeks (Charles River, Frederick, MD) were tail-vein inoculated with 50000 MDA-MB-231-BR or 10000 4T1-BR cells, respectively. Ten mice per group were injected. Upon signs of tumor-induced morbidity, mice were killed under carbon dioxide asphyxiation. Lungs were preserved in Bouin’s solution, and lung surface metastases were counted.
Additional details on brain metastasis and lung metastasis assays, as well as tempol feeding study, appear in the Supplementary Methods (available online).
Statistical Analysis
BARD1 and RAD51 expression changes between primary tumors and brain metastases by DASL were compared by paired t test of log2-tranformed, normalized expression values. BARD1 and RAD51 expression between brain metastases and systemic metastases was compared by unpaired t test on RMA-normalized, log2-transformed expression values. The P values in Table 1 were adjusted for multiple comparisons of 42 DNA repair–related genes examined in the DASL array using Bonferroni–Holm correction for multiple comparisons.
Table 1.
Fold changes (FC) and P-values (P) for DNA repair genes overexpressed in the brain metastases compared with the primary breast tumors of the same patients in 23 matched sample sets by cDNA-mediated annealing, selection, extension, and ligation (DASL) array analysis*
| DNA repair process | Symbol | FCall† | P all‡ | P all§ | FCTN | P TN | FCHER2+ | P HER2+ | FCER+ | P ER+ |
|---|---|---|---|---|---|---|---|---|---|---|
| Homologous recombination | RAD51 | 1.46 | <.001 | .004 | 1.67 | .09 | 1.49 | <.001 | 1.11 | .43 |
| Homologous recombination | BARD1 | 1.74 | <.001 | .02 | 2.40 | .04 | 1.64 | .007 | 1.20 | .58 |
| Homologous recombination | BRCA1 | 1.31 | .004 | .15 | 1.31 | .39 | 1.34 | .01 | 1.23 | .28 |
| Homologous recombination | BRCA2 | 1.50 | .004 | .14 | 1.78 | .08 | 1.53 | .03 | 1.10 | .76 |
| Homologous recombination | RAD54B | 1.59 | <.001 | .004 | 1.65 | .04 | 1.71 | .004 | 1.26 | .25 |
| Homologous recombination | RAD54L | 1.26 | .02 | .48 | 1.28 | .23 | 1.38 | .02 | 0.96 | .52 |
| Homologous recombination | XRCC2 | 1.51 | <.001 | .001 | 1.74 | .01 | 1.54 | .002 | 1.19 | .58 |
| Base excision repair | LIG3 | 1.42 | .04 | .84 | 1.92 | .14 | 1.34 | .12 | 1.03 | .88 |
| Base excision repair | XRCC1 | 1.30 | .02 | .62 | 1.57 | .16 | 1.26 | .10 | 1.07 | .71 |
| Mismatch excision repair | MSH2 | 1.46 | .03 | .76 | 1.83 | .34 | 1.42 | .02 | 1.10 | .86 |
| Mismatch excision repair | MSH6 | 1.14 | .03 | .68 | 1.20 | .11 | 1.17 | .06 | 1.01 | .91 |
| Mismatch excision repair | PMS1 | 1.69 | .002 | .06 | 2.60 | .03 | 1.49 | .03 | 1.06 | .33 |
| Nonhomologous end-joining | XRCC4 | 1.24 | .03 | .76 | 1.44 | .12 | 1.24 | .13 | 1.01 | .96 |
| Nonhomologous end-joining | XRCC5 | 1.23 | .05 | .82 | 1.28 | .19 | 1.28 | .11 | 1.03 | .98 |
| Nucleotide excision repair | CCNH | 1.23 | .02 | .61 | 1.40 | .02 | 1.26 | .12 | 0.94 | .28 |
| Nucleotide excision repair | CDK7 | 1.28 | .008 | .26 | 1.38 | .02 | 1.28 | .11 | 1.15 | .56 |
| Nucleotide excision repair | ERCC4 | 1.15 | .03 | .75 | 1.20 | .08 | 1.22 | .03 | 0.89 | .06 |
| Nucleotide excision repair | ERCC5 | 1.75 | .04 | .88 | 3.23 | .07 | 1.31 | .22 | 1.05 | .86 |
| Nucleotide excision repair | LIG1 | 1.79 | .004 | .14 | 2.79 | .07 | 1.52 | .03 | 1.23 | .58 |
| Nucleotide excision repair | XPA | 1.47 | .01 | .36 | 2.21 | .11 | 1.30 | .02 | 1.00 | .90 |
| Repair of DNA crosslinks and adducts | FANCA | 1.62 | .03 | .72 | 1.85 | .65 | 1.50 | .01 | 1.63 | .34 |
| Repair of DNA crosslinks and adducts | FANCG | 1.25 | <.001 | .02 | 1.44 | .04 | 1.26 | .005 | 1.02 | .85 |
| DNA damage response | CHEK1 | 1.53 | .03 | .79 | 1.64 | .47 | 1.52 | .08 | 1.43 | .33 |
| DNA polymerases (catalytic subunits) | PCNA | 1.29 | .01 | .37 | 1.18 | .28 | 1.38 | .04 | 1.19 | .45 |
| Disease sensitivity to DNA damage | BLM | 1.55 | .009 | .28 | 1.42 | .52 | 1.75 | .01 | 1.25 | .37 |
* Analyses are shown for all 23 matched sets (all), and by subtypes: triple negative (TN), human epidermal growth factor receptor 2 overexpressing/amplified (HER2+), and estrogen receptor positive (ER+) primary breast tumors. Bold rows indicate the genes functionally evaluated in the manuscript.
† Fold change (FC) comparing genes expression in brain metastases with matched primary tumors.
‡ P values for gene expression comparisons between brain metastases and matched primary tumors. P values were calculated using two-sided, paired t test of log2-tranformed, normalized expression values.
§ P values adjusted for multiple comparisons of 42 DNA repair–related genes examined in the cDNA-mediated annealing, selection, extension, and ligation array array using Bonferroni–Holm correction for multiple comparisons.
Distributions of continuous variables were examined for normality using Shapiro–Wilk tests. If normal, bicategorical data were compared with unpaired t tests with equal variances or corrected for unequal variances by Welch correction. Multicategorical data were globally examined by analysis of variance, and each experimental group was compared with unified control group using Dunnett’s multiple comparisons test. Otherwise, bicategorical data were compared by Mann–Whitney test, and multicategorical data were globally examined by Kruskal–Wallis test, comparing individual experimental groups with a unified control group using Dunnett’s multiple comparisons test. For clonogenic survival assays, multiple unpaired t tests were performed using the Holm–Sidak method (alpha = 0.05). All tests were two-sided, and P values less than .05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism version 6.0 software (GraphPad, La Jolla, CA).
Results
BARD1 and RAD51 Expression in Human Brain Metastases of Breast Cancer
Twenty-three pairs of resected primary tumors and brain metastases from the same patients were profiled for the mRNA expression of 502 cancer-related genes (Supplementary Table 1, available online). A general DNA repair phenotype was apparent for brain metastases of breast cancer. Of 42 DNA repair genes assayed, 25 had statistically significantly higher expression in brain metastases compared with primary tumors, with HR pathway genes exhibiting the most robust signals (Table 1). Our functional studies focused on BARD1 and RAD51 because both proteins coexist in a critical HR DNA repair supercomplex (47,48) and physically interact with several other HR pathway genes overexpressed in brain metastases in this dataset, such as BRCA1, BRCA2, and RAD54B. Furthermore, both proteins have previously been implicated in brain metastatic progression as part of a 13-gene signature predictive of rapid brain relapse in HER2-positive metastatic breast cancer patients (49).
Analysis of all 23 sample pairs showed consistent overexpression of both BARD1 (1.74-fold; P < .001) and RAD51 (1.46-fold; P < .001) in brain metastases compared with matched primary tumors (Figure 1, A and B). Trends of differential BARD1 and RAD51 expression were observed in matched sets of TN tumors (2.40-fold, P = .04; 1.67-fold, P = .09, respectively) and HER2-positive tumors (1.64-fold, P = .008; 1.49-fold, P < 0.001, respectively) (Figure 1, C and D). Comparable expression levels of both genes were observed in estrogen receptor–positive tumor sample sets. Quantitative real-time polymerase chain reaction was performed to assess RAD51 expression in 13 of the 23 sample pairs for which sufficient RNA was available. This analysis confirmed RAD51 overexpression in brain metastases samples compared with primary tumors, with a median fold change of 2.1 (P = .02) (Supplementary Figure 1A, available online) and with general agreement between the two mRNA quantification methodologies (Supplementary Figure 1B, available online).
Figure 1.
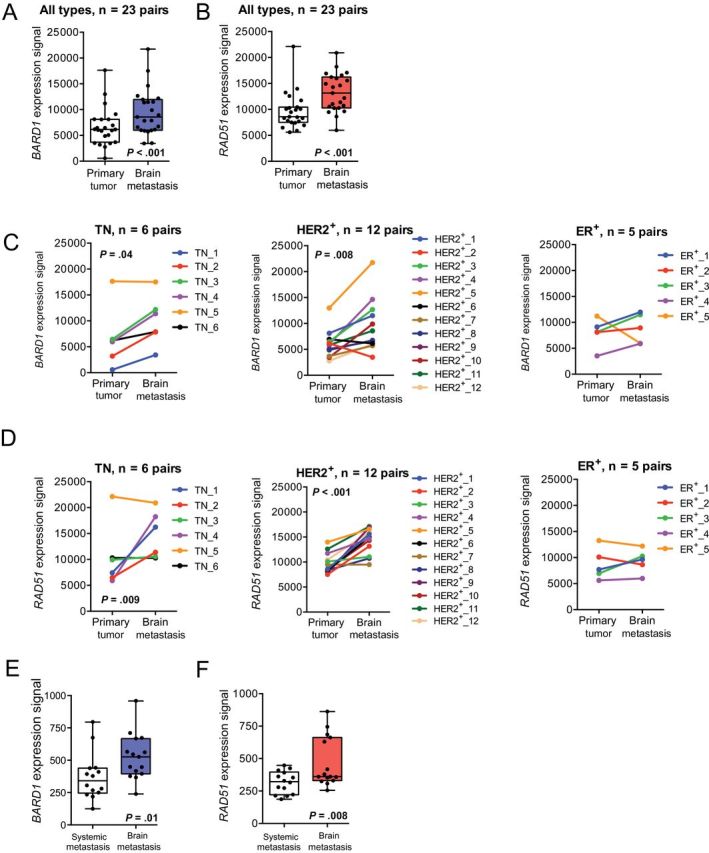
Expression of the DNA repair genes BARD1 and RAD51 in brain metastases from breast cancer. Box and whisker plots show expression of BARD1 (A) and RAD51 (B) in a set of 23 resected brain metastases compared with their matched primary tumors, as detected by cDNA-mediated annealing, selection, extension, and ligation assay. The Box represents the 25th to 75th percentile, the whiskers represent the minimum and maximum values, and the dots represent individual patient data points. Subset analyses of BARD1 (C) and RAD51 (D) expression are presented by tumor subtype. Differential expression in primary tumors vs brain metastases was assessed separately for patients with triple-negative (TN), human epidermal growth factor receptor 2–positive (HER2+), and estrogen receptor-positive (ER+) tumors. The line graphs show expression changes between primary tumors and brain metastases for individual matched pairs. For (A–D), P values were determined by two-sided, paired t tests. E and F) In a publically available microarray dataset (Gene Expression Ominbus accession number GSE14017) of distant breast cancer metastases (n = 15 brain, 4 lung, 10 bone), both BARD1 (E) and RAD51 (F) expression in brain metastases was compared with systemic (lung/bone) metastases; box-and-whisker plots with median are shown. The Box represents the 25th to the 75th percentile, the whiskers represent the minimum and maximum values, and the dots represent individual patient data points. P values were determined by two-sided, unpaired t tests.
To investigate whether overexpression of BARD1 and RAD51 is a specific feature of breast cancer brain metastases or is generally associated with breast cancer spread, we compared BARD1 and RAD51 expression in breast cancer metastases at different organ sites (brain, lung, and bone) in a publically available microarray dataset (GEO No. GSE14017) (50). BARD1 and RAD51 were overexpressed (1.49-fold, P = .01; 1.44-fold, P = .008, respectively) in brain metastases compared with systemic (lung/bone) metastases (Figure 1, E and F).
Effect of BARD1 and RAD51 Overexpression on DNA Repair Function and Chemo-Resistance In Vitro
Given the clinical observation that most breast cancer patients who develop brain metastases do so after initial chemotherapy and extracranial metastatic relapse (51), at least two distinct roles for BARD1 and RAD51 overexpression could be envisioned. Their overexpression could alter the natural history of breast cancer to specifically promote brain metastasis or, alternatively, maintain tumor cell survival through rounds of chemotherapy, indirectly facilitating brain metastasis. To test a direct effect of BARD1 and RAD51 overexpression, we generated stable wild-type human BARD1- and RAD51-overexpressing polyclonal populations and empty vector-expressing controls in the brain-tropic TN human breast carcinoma cell line MDA-MB-231-BR (Figure 2, A and B). In vitro, overexpression of both genes increased genomic stability under normal culture conditions, evidenced by reductions in markers of unrepaired DNA DSBs. As such, BARD1- and RAD51-overexpressing transfectants exhibited fewer γ-H2AX DNA DSB foci than vector controls (P = .006 and P < .001, respectively) (Figure 2C), which were enumerated in Geminin-positive cells as a readout of S to early M cell-cycle phase, when sister chromatid availability enables DNA repair by HR (representative images of γ-H2AX costaining with Geminin are shown in Supplementary Figure 2D, available online). Similarly, both BARD1- and RAD51-overexpressing transfectants had fewer DNA DSBs, as shown by neutral comet assay (P < .001 and P < .001, respectively) (Figure 2D). These reductions in residual (unrepaired) DNA DSB loads are consistent with increases in DSB repair; consequently, higher rates of HR were observed in BARD1 and RAD51 overexpressers compared with vector controls (P < .001 and P < .001, respectively) (Figure 2E). Thus, the expression levels of BARD1 and RAD51, were limiting for HR in vitro.
Figure 2.

In vitro phenotypes of BARD1 and RAD51 overexpression in brain-metastatic MDA-MB-231-BR cells. Western blot was done to confirm overexpression of BARD1 (A) and RAD51 (B) in brain metastatic MDA-MB-231-BR cells. Alpha-Tubulin was used as loading control. Numbers indicate the expression fold changes relative to tubulin. C and D) DNA repair proficiency in BARD1 and RAD51 transfectants of MDA-MB-231-BR cells was assessed. Means with 95% confidence intervals (CIs) are shown. C) DNA double-strand breaks (DSBs) were measured by the mean number of γ-H2AX foci observed in S-M phase (Geminin-positive = GMNN+). D) Frequency of DNA DSBs were quantified by a neutral comet assay, shown normalized to vector transfectants. E) Homologous recombination (HR) proficiency was assessed by quantitative real-time polymerase chain reaction assay to detect reconstituted LacZ from two mutated plasmids. Relative HR frequency was calculated by delta-delta CT method with reference to primers detecting mutated plasmids to adjust for transformation efficiency and normalized to recombinant LacZ detection in vector controls. The means of six triplicate measurements with 95% confidence intervals are shown. Two-sided, multiplicity adjusted P values shown in (C–E) are based on one-way analysis of variance with Dunnett’s correction for multiple comparisons. F) Resistance to DNA damage in BARD1 and RAD1 transfectants was measured by clonogenic survival assays after treatment with 5nM and 10nM doxorubicin. The mean percentage surviving colonies relative to untreated ± standard deviation are shown. P values are based on two-sided, unpaired t tests corrected for multiple comparisons by the Holm–Sidak method.
Another role of RAD51 is in restarting stalled replication forks, which, if unresolved, can disintegrate into DSBs (21). RAD51-overexpressing MDA-MB-231-BR cells exhibited lower frequencies of replication fork stalling after a 2-hour hydroxyurea block compared with vector controls (P = .01) (Supplementary Figure 2A, available online), confirming that RAD51 is also limiting for this function in vitro.
Despite evidence of increased function in HR, BARD1- and RAD51-overexpressing MDA-MB-231-BR cells failed to exhibit any distinct phenotypes in proliferation, invasion, or cell-cycle distribution under normal culture conditions (Supplementary Figure 2, B, C, and F, available online). However, when challenged with a DNA DSB-inducing agent such as doxorubicin, a clonogenic phenotype emerged for both BARD1- and RAD51-overexpressing MDA-MB-231-BR cells (P < .01 for both genes and doses) (Figure 2F). A similar, although less robust protection against doxorubicin-induced DNA damage, was noted in a 3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide survival assay (Supplementary Figure 2E, available online). Together, these in vitro data indicate that BARD1 and RAD51 overexpression in MDA-MB-231-BR cells resulted in a functional enhancement of HR and DNA repair activity in response to both endogenous and chemotherapy-induced DNA damage, but only generated growth and survival phenotypes in the latter.
Next, we investigated how BARD1 and RAD51 overexpression could functionally enhance DNA DSB repair. We hypothesized that their participation in a DNA repair supercomplex was fundamental, particularly with regard to BARD1 potentiation of endogenous RAD51 function. Figure 3A shows a proposed schematic of a DNA DSB repair supercomplex containing BRCA1, which, along with its binding partner BARD1, directs BRCA2/RAD51 to sites of DNA damage through the mediator protein PALB2 (41). The overall expression of these supercomplex proteins was generally unaffected by overexpression of either BARD1 or RAD51 under normal culture conditions or upon DNA damage induction (Figure 3B). One noteworthy exception was a slight increase in PALB2 after BARD1 and RAD51 overexpression under doxorubicin treatment (Figure 3B). RAD51 foci formation was absent or infrequent in vector-expressing MDA-MB-231-BR cells, but efficient assembly of RAD51 foci was observed in both BARD1 and RAD51 overexpressers (Figure 3, C and D). For the BARD1 overexpressers, this was accompanied by upstream assembly of both BARD1 and BRCA1 foci. In RAD51 and vector transfectants, however, BARD1 and BRCA1 foci were largely absent. These patterns are similar in the absence and presence of doxorubicin treatment and suggest that BARD1 overexpression in MDA-MB-231-BR cells exerts its enhancement of HR and DNA repair phenotypes by facilitating assembly of endogenous RAD51 into active repair foci (representative images of the BARD1, BRCA1, and RAD51 foci assemblage quantified in Figure 3, C and D, are shown in Supplementary Figure 3, A and B, available online).
Figure 3.
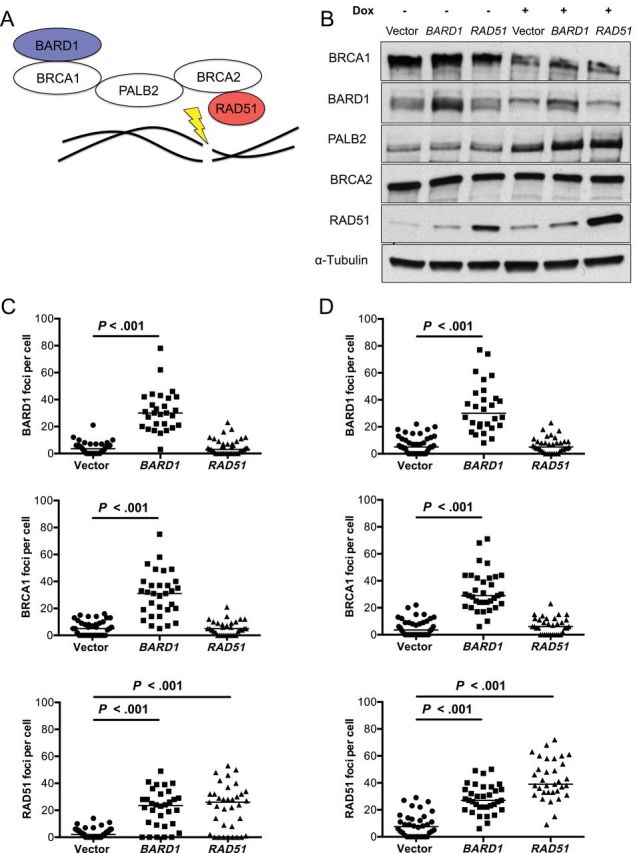
Effect of BARD1 and RAD51 overexpression on DNA double-strand break (DSB) repair foci assemblage. A) Schematic representation of BRCA1-containing supercomplex, in which BARD1/BRCA1 heterodimer directs BRCA2 and RAD51 to sites of DNA DSBs by linker protein PALB2. B) Western blot for five BRCA1 supercomplex components (BCRA1, BARD1, PALB2, BRCA2, and RAD51) are shown in the absence and presence of the DNA-damaging agent doxorubicin (Dox, 100nM). Alpha-Tubulin was used as the loading control. Quantification of BARD1, BRCA1, and RAD51 foci is shown, as detected by immunofluorescence in the absence (C) and presence (D) of 100nM doxorubicin. Individual foci counts per cell with medians are indicated. P values are two-sided and adjusted for multiplicity based on Kruskal–Wallice test with Dunn’s correction for multiple comparisons.
Effect of BARD1 and RAD51 Overexpression on Brain Metastasis Development In Vivo
A direct contribution of BARD1 and RAD51 overexpression to brain metastasis development was investigated using an intracardiac-injected hematogenous brain metastasis model. Surprisingly, both BARD1 and RAD51 overexpression resulted in threefold to fourfold increase in brain micrometastases (P = .002 and P = .008, respectively) and large brain metastases (P = .01 and P = .02, respectively), the latter corresponding to the minimal unidimensional size of a lesion detectable by magnetic resonance imaging in a human brain (Figure 4, A and B) (46). No difference in the multiplicity of lung surface metastases was apparent upon BARD1 or RAD51 overexpression using a tail-vein experimental metastasis assay (Figure 4C). Additionally, the increase in brain metastases after overexpression of both genes was not associated with increased proliferation of frankly metastatic cells in vivo, as measured by Ki67 proliferation index (Figure 4D).
Figure 4.
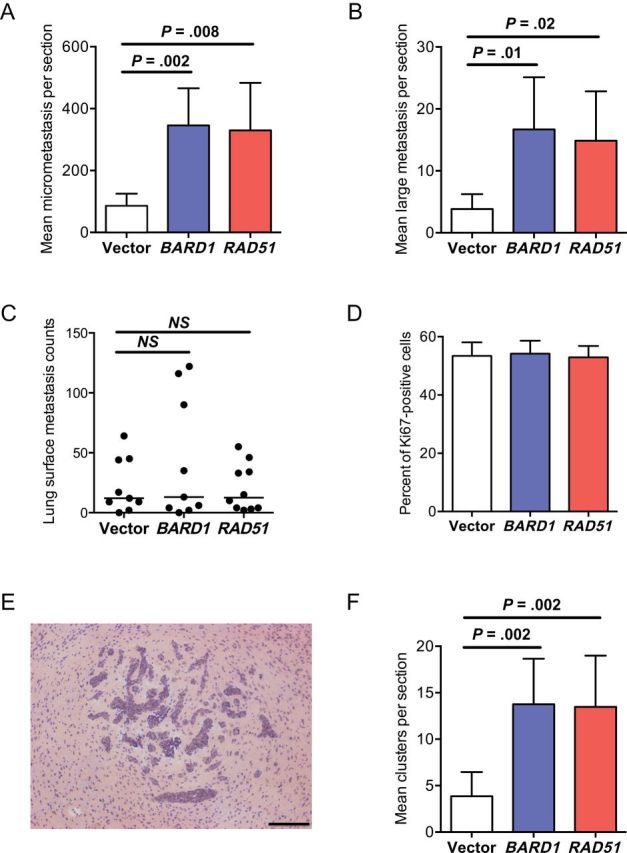
Effect of BARD1 and RAD51 overexpression in MDA-MB-231-BR cells on brain metastasis development. In an intracardiac-injected brain metastasis assay, the effect of BARD1 and RAD51 overexpression on brain metastasis multiplicity was assessed separately for micrometastases (P = .002 and P = .008, respectively) (A) and large brain metastases (P = .02 and P = .01, respectively) (B). Mean counts per section with 95% confidence intervals (CIs) are shown. C) An experimental lung metastasis assay of MDA-MB-231-BR after tail-vein injection of vector-, BARD1-, and RAD51-transfected tumor cells showed no effect on multiplicity of lung surface metastases; individual lung counts with medians are shown. NS = not significant. D) No differences are evident in proliferation indices of brain metastases between BARD1, RAD51, and vector transfectants, as assessed by Ki67 staining of brain metastases; mean percentage Ki67-positive cells with 95% confidence intervals are presented. E) A representative image of a brain metastasis cluster formed by intracardiac injection of MDA-MB-231-BR cells in immunocompromised mice is depicted. Scale bar = 200 μm. F) The effect of BARD1 and RAD51 overexpression on multiplicity of brain metastasis clusters of tumor cells was compared with vector control (P = .002 for both comparisons). Mean clusters per section with 95% confidence intervals are shown. P values presented are two-sided and adjusted for multiplicity based on one-way analysis of variance with Dunnett’s correction for multiple comparisons. For all metastasis assays, 10 mice per group were injected.
The effect of BARD1 and RAD51 overexpression was evident in initial brain colonization events. MDA-MB-231-BR metastases developed as clusters of micro- and large metastases in vivo (Figure 4E) that likely resulted from clonal expansion of single initiating cells that eventually coalesced into a single lesion (8). There was a 3.5-fold (P = .002 for both genes) increase in brain metastasis clusters compared with vector controls (Figure 4F), suggesting that both genes increased the number of metastasis-initiating events. To address this possibility, we measured the effects of BARD1 and RAD51 overexpression on single cancer cell multiplicity in the brain—the earliest step of brain colonization. Vector-, BARD1-, and RAD51-transfected MDA-MB-231-BR cells were iron oxide labeled to enable single cell identification in sections by Prussian blue staining (Figure 5A) at three early time points in the experimental brain metastasis assay before the development of micrometastases. No difference was observed between overexpressers and vector controls on day 3 after injection (Figure 5B). A statistical trend toward more single cells was apparent on day 7 after injection for RAD51 overexpressers (P = .08), which was statistically significant for the BARD1-overexpressing group (P = .005). Approximately threefold more BARD1- and RAD51-overexpressing MDA-MB-231-BR cells compared with vector controls were present by day 12 (P = .04 and P = .001, respectively).
Figure 5.

Effect of BARD1 and RAD51 overexpression in MDA-MB-231-BR cells on single-cell populations during early brain colonization. Mice (n = 5 mice per group) were administered an intracardiac injection of 1000000 MDA-MB-231-BR cells transfected with vector, BARD1, or RAD51. On days 3, 7, and 12 after injection, metastatic cells in the brain were visualized by Prussian blue stain of iron oxide–labeled single cells. A) A representative image is shown with a solitary cancer cell indicated by the arrow. Scale bar = 100 μm. B) The number of single Prussian blue cells per section with 95% confidence intervals are presented. Two-sided, multiplicity adjusted P values are presented based on one-way analysis of variance with Dunnett’s correction for multiple comparisons.
RAD51-overexpressing- and short hairpin RNA (shRNA)–mediated RAD51 knockdown polyclonal populations with appropriate vector controls were produced in brain-tropic derivatives of the 4T1 TN mouse mammary carcinoma cell line 4T1-BR (Figure 6A). RAD51 overexpression in 4T1-BR cells resulted in increased multiplicity of brain micrometastases (P = .02) and large brain metastases (P = .006) in an intracardiac injected mouse model (Figure 6, B and C). No effect on lung metastases development was observed with RAD51 overexpression in 4T1-BR cells (Figure 6D). RAD51 knockdown in 4T1-BR cells had the opposite effect, reducing the micrometastasis and large metastasis multiplicity compared with scrambled shRNA controls (2.5-fold; P = .001 for both micro- and large metastases) (Figure 6, E and F). No detectable differences in the multiplicity of lung surface metastases between RAD51 knockdown cells and scrambled shRNA controls were observed in 4T1-BR cells (Figure 6G). Thus, RAD51 expression levels control experimental brain metastatic but not lung metastatic potential in vivo.
Figure 6.
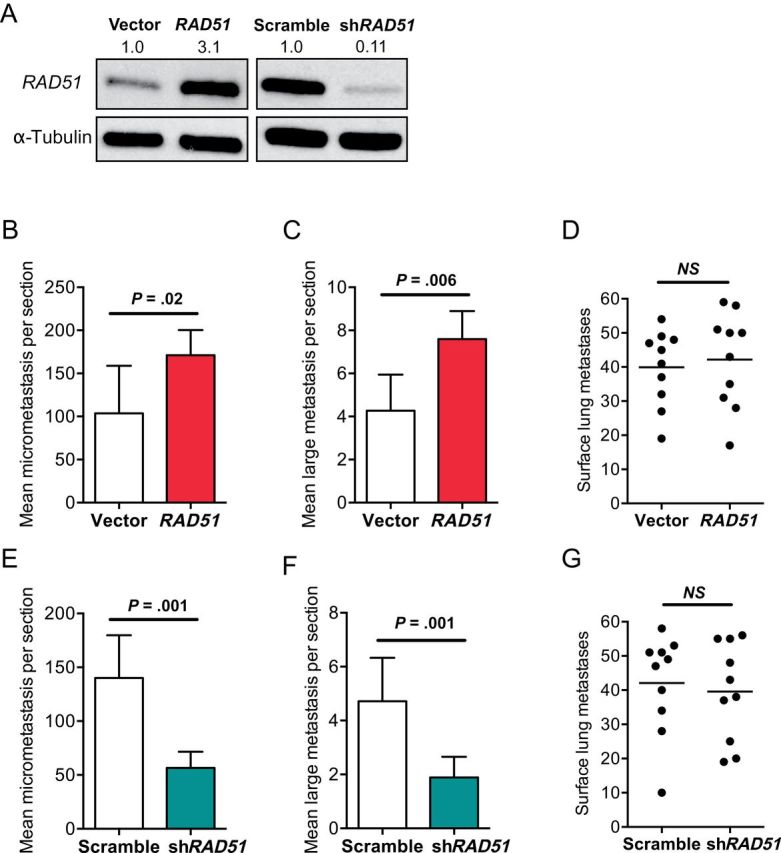
Effect of RAD51 expression level in 4T1-BR cells on brain and lung metastasis development. A) Western blot confirming overexpression and short hairpin RNA (shRNA–mediated knockdown of RAD51 in brain metastatic murine 4T1-BR mammary carcinoma cells is shown. Alpha-Tubulin was used as the loading control. Numbers indicate the expression fold changes relative to Alpha-Tubulin. The effect of RAD51 overexpression on the multiplicity of brain micrometastases (B) and large brain metastases (C) 2 weeks after intracardiac injection was assessed (as described in Figure 4). The mean counts per section with 95% confidence intervals are shown. D) In an experimental lung metastasis assay after tail-vein injection of vector and RAD51 transfectants, the effect of RAD51 overexpression in 4T1-BR cells on multiplicity of lung surface metastases was determined. Individual lung counts with median are shown. NS = not significant. E and F) In the brain metastasis assay, the effects of RAD51 knockdown reduced the multiplicity of brain micrometastases (E) and large brain metastases (F) compared with scrambled shRNA controls. Data are presented as mean counts per section with 95% confidence intervals. G) A similar lung metastasis experiment with 4T1-BR cells was done. P values presented are two-sided and based on unpaired t tests. For all metastasis assays, 10 mice per group were injected.
Effect of BARD1 and RAD51 Overexpression on Resistance to Oxidative Stress in the Metastatic Brain
Given the paradox of brain metastasis promotion by BARD1 and RAD51 overexpression in the absence of any chemotherapeutic DNA DSB induction in vivo, but in vitro evidence that growth and colonization phenotypes were dependent on DNA damage induction, we hypothesized that endogenous genotoxic stresses were present in the brain microenvironment. The brain exhibits one of the highest levels of endogenous ROS compared with other organs, particularly during inflammatory activation of resident microenvironmental cells (52). ROS can induce DNA DSB either directly (53) or indirectly by replication fork failure after single-strand lesions (54). A prominent neuro-inflammatory response forms around brain metastases in both experimental models and in human craniotomy specimens, including activated astrocytes and microglia (8), and is apparent in brains containing vector control– or RAD51-overexpressing metastases (Supplementary Figure 3, A and B, available online). ROS were observed in the normal mouse brain and were more prominent in MDA-MB-231-BR metastases and in the adjacent neuro-inflammatory microenvironment of the brain metastases (Figure 7A). These observations suggested the hypothesis that ROS in the MDA-MB-231-BR cells and the neuro-inflammatory microenvironment provide an endogenous genotoxic stress, which BARD1 or RAD51 overexpression overcomes.
Figure 7.
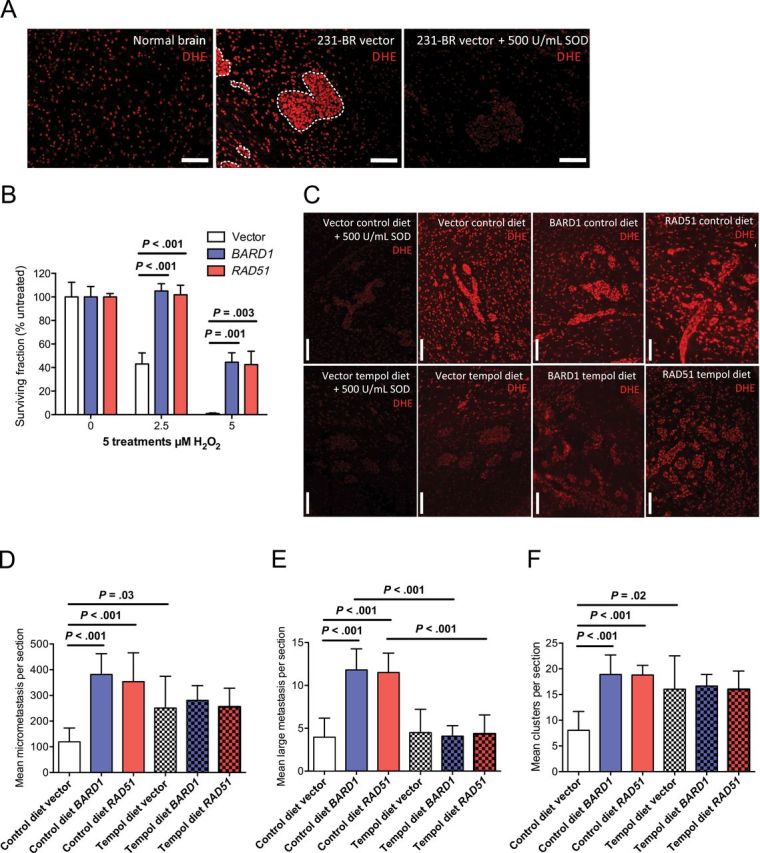
Effect of reactive oxygen species (ROS) in the brain on metastasis-promoting phenotype of BARD1 and RAD51 overexpression. A) Normal uninvolved brain (left) and experimental MDA-MB-213-BR brain metastases with the surrounding neuro-inflammatory microenvironment (center, right) were stained with dihydroethidium (DEH). Superoxide dismutase (SOD) treatment (500U/mL) was done to show staining specificity (right). Scale bar = 100 μm. B) Resistance to hydrogen peroxide (H2O2) in BARD1 and RAD51 transfectants was determined by clonogenic survival assays after five repeated treatments with 0, 2.5, or 5.0 μM of H2O2. Mean percentage surviving colonies relative to untreated ± standard deviation are shown. P values are based on two-sided, unpaired t tests corrected for multiple comparisons by the Holm–Sidak method. C) ROS levels in brains of mice treated with a SOD mimetic, tempol, were measured by DHE staining of brain sections containing experimental MDA-MB-213-BR brain metastases. Scale bar = 100 μm. D−F) An intracardiac-injected MDA-MB-231-BR experimental brain metastasis assay was done. Mice were randomized to control chow or chow containing 10g/kg diet of tempol, both containing flavor additive. Effects of BARD1 and RAD51 overexpression on micrometastasis (D) and large metastasis (E) multiplicity was determined. Mean counts per section with 95% confidence intervals are shown. F) Effects on brain metastasis cluster multiplicity (as described in Figure 4) by BARD1 and RAD51 overexpression in mice maintained on control diet or tempol diet were assessed. Mean clusters per section with 95% confidence intervals are shown. Two-sided, multiplicity adjusted P values are based on one-way analysis of variance with Dunnett’s correction for multiple comparisons. For all metastasis assays, 10 mice per group were injected.
To simulate oxidative stress in vitro, vector-, BARD1-, and RAD51-transfected MDA-MB-231-BR cells were repeatedly treated with hydrogen peroxide in a clonogenic assay. BARD1 and RAD51 overexpressers showed increased resistance to hydrogen peroxide treatments at 2.5 μM and 5.0 μM doses (approximately corresponding to inhibitory concentrations of 50% and 90% for vector controls) in this assay (Figure 7B).
To investigate whether the brain metastasis–promoting phenotype of BARD1 and RAD51 overexpression depends on genotoxic ROS in the brain, we used the brain-permeable radical scavenger tempol, a superoxide dismutase mimic (55). Mice injected with vector-, BARD1-, or RAD51-overexpressing MDA-MB-231-BR cells were randomized to chow containing tempol admixed with a flavor additive or a normal control diet containing the flavor additive. Dihydroethidium (DHE) staining of ROS in brain metastasis sections (Figure 7C) indicated that the tempol diet reduced the amount of ROS in the brain lesions and immediate microenvironment compared with control diet–fed animals; thus, tempol hit its intended target in vivo. In addition to the radical scavenging effects of tempol, the drug had some antiproliferative properties (Supplementary Figure 3C, available online), which were nondifferential with respect to transfection status. In control diet–fed mice, BARD1 and RAD51 overexpression increased the multiplicity of experimental brain micrometastases, large brain metastases, and clusters of metastases (Figure 7, D–F). In contrast, the brain metastasis stimulatory phenotype of the two genes was abrogated in mice fed tempol, when metastases were quantified by any metric. Overall, the data suggest that ROS constitute an endogenous source of DNA damage in the metastatic brain, the effects of which are counteracted by tumor cell BARD1 or RAD51 overexpression to facilitate brain metastatic colonization.
Discussion
We report that human brain metastases of breast cancer frequently overexpress DNA DSB repair genes, including BARD1 and RAD51. Despite a robust literature outlining the contributions of RAD51 to DNA DSB repair and replication fork progression, RAD51 overexpression has only caused phenotypic changes in tumor cells in the context of chemotherapeutic or radiation-induced damage (24–29). To our knowledge, we present the first evidence for a direct biological phenotype of RAD51 or BARD1 overexpression in cancer. RAD51 overexpression in two models of TN breast cancer promoted the formation of brain metastases, but not lung metastases, providing evidence of site specificity in the phenotype. RAD51 knockdown in 4T1 murine mammary cells decreased brain metastases, showing that RAD51 levels are limiting for metastasis. Similar trends were observed for BARD1 in the MDA-MB-231-BR model, and in vitro data suggest that overexpressed BARD1 facilitated RAD51 function.
Given our data that growth and colonization phenotypes in vitro were dependent on a DNA-damaging agent, doxorubicin, we hypothesized that the brain may require increased DNA repair function in cancer progression because of an endogenous source of genotoxic stress. ROS cause a variety of DNA lesions, many of which can lead to DSB (56). The brain has high levels of endogenous ROS, and our data demonstrated that in both MDA-MB-231-BR tumor cells and the metastatic microenvironment, ROS levels are still higher. Tempol, a superoxide dismutase mimic (55), reduced the ROS expression in brain metastases and their microenvironment and abrogated the brain metastasis stimulatory effects of BARD1 and RAD51 overexpression. Quantitatively, the tempol data suggest that nearly the full phenotype of promoting brain metastasis initiation by BARD1 and RAD51 overexpression is explained by resisting the effects of ROS in the brain. The effect of RAD51 or BARD1 overexpression was observed at early points in brain colonization, suggesting that ROS control initial outgrowth of metastases.
A limitation of our study is the relatively small size of our discovery cohort, in which we collected 23 matched sets of brain metastases and primary breast tumors from the same women. We were able to identify the strong signal of DNA repair gene overexpression in brain metastases, but weaker trends in other pathways, as well as subgroup-specific genes, likely elude us because of statistical constrains with small numbers. These matched sets are rare sample sets because brain metastases are infrequently resected and primary tumors precede a brain metastases diagnosis by long periods of time. Other limitations in our investigation were technical in nature. For instance, knockdown of BARD1 and RAD51 was uniformly lethal in MDA-MB-231-BR cells. Even in 4T1-BR cells, which tolerated shRNA-mediated RAD51 knockdown, stable reduction in BARD1 expression could not be achieved. Lastly, tempol treatment was associated with increases in brain metastasis multiplicity, while simultaneously suppressing proliferation of the lesions. It is uncertain whether the suppressed proliferation was on or off target and whether other brain-permeable antioxidants will demonstrate similar effects. From a patient’s perspective, this question warrants further study.
Our study raises a number of clinically relevant questions: Is the DNA repair gene expression status of primary tumors predictive of more frequent or faster brain relapse? Is this measure associated with response to treatment modalities such as focal or whole-brain radiation therapy? And, do differences in inflammatory and/or oxidative brain status between different patient populations affect their risk of brain relapse? Together, these data demonstrate that the brain metastasis–promoting phenotype of BARD1 and RAD51 overexpression is conditional on ROS generated in the brain metastatic microenvironment, identifying a new molecular pathway–promoting specific pattern of breast cancer aggressiveness that is amenable to therapeutic development.
Funding
This work was supported by the intramural research program of the National Cancer Institute and by the US Department of Defense Breast Cancer Research Program grant No. W81 XWH-062-0033 and by intramural grant of the Medical University of Gdańsk, Poland grant No. ST-51.
S. Woditschka and P. S. Steeg designed cell line experiments. S. Badve, R. Duchnowska, and J. Jassem designed DASL analysis in human tissues. R. Duchnowska, E. Szutowicz-Zielińska, T. Mandat, T. Trojanowski, W. Och, and B. Czartoryska-Arlukowicz provided human tissue samples and clinical data for these subjects. J. Jassem and R. Duchnowska coordinated collaborations with different investigators and oversaw and contributed to data generation. S. Badve and G. Sledge performed the DASL analysis. S. Badve and W. Biernat performed central pathological evaluations of human tissues and selected material for molecular analysis. S. Woditschka, L. Evans, L. T. Reed, D. Palmeiri, B. Gril, and Y. Gökmen-Polar performed cell-line experiments. J. B. Mitchell provided Tempol and control chows. M. I. Aladjem and H. Fu designed replication fork progression experiments. S. Woditschka and P. S. Steeg wrote the manuscript, assisted by R. Duchnowska, J. Jassem, and S. Badve. All authors commented and edited the manuscript.
The authors declare no competing financial interests. The study sponsors had no role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication.
References
- 1. Steeg P, Camphausen K, Smith Q. Brain metastases as preventive and therapeutic targets. Nature Rev Cancer. 2011;11(5):352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin N, Bellon J, Winer E. CNS metastases in breast cancer. J Clin Oncol. 2004;22:3608–3617 [DOI] [PubMed] [Google Scholar]
- 3. Brufsky A, Mayer M, Rugo H, et al. Central nervous system metastases in patients with Her2-positive metastatic breast cancer: Incidence, treatment, and survival in patients from registHER. Clin Cancer Res 2011;17(14):4834–4843 [DOI] [PubMed] [Google Scholar]
- 4. Lin N, Claus E, Sohl J, et al. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer. High incidence of central nervous system metastases. Cancer. 2008;113(10):2638–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bendell J, Domchek S, Burstein H, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer. 2003;97(12):2972–2977 [DOI] [PubMed] [Google Scholar]
- 6. Lockman PR, Mittapalli RK, Taskar KS, et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res. 2010;16(23):5664–5678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carbonell W, Ansorge O, Sibson N, et al. The vascular basement membrane as “soil” in brain metastasis. PloS One. 2009;4(6):e5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fitzgerald DP, Palmieri D, Hua E, et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metastasis. 2008;25(7):799–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sierra A, Price J, Garcia-Ramirez M, et al. Astrocyte derived cytokines contribute to the metastatic brain specificity of breast cancer cells. Lab Invest. 1997;77(4):357–368 [PubMed] [Google Scholar]
- 10. Heyn C, Ronald JA, Ramadan SS, et al. In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magn Reson Med. 2006;56(5):1001–1010 [DOI] [PubMed] [Google Scholar]
- 11. Kienast Y.al. E. Real-time imaging reveals the single steps in brain metastasis formation. Nature Med. 2010;16(1):116–122 [DOI] [PubMed] [Google Scholar]
- 12. Bos P, Zhang X, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257 [DOI] [PubMed] [Google Scholar]
- 14. Hashimoto Y, Puddu F, Costanzo V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat Struct Mol Biol. 2012;19(1):17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Long DT, Räschle M, Joukov V, et al. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333(6038):84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feng Z, Zhang J. A dual role of BRCA1 in two distinct homologous recombination mediated repair in response to replication arrest. Nucleic Acids Res. 2012;40(2):726–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Badie S, Escandell JM, Bouwman P, et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat Struct Mol Biol. 2010;17(12):1461–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7(10):739–750 [DOI] [PubMed] [Google Scholar]
- 19. Sage JM, Gildemeister OS, Knight KL. Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J Biol Chem. 2010;285(25):18984–18990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schlacher K, Christ N, Siaud N, et al. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145(4):529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petermann E, Orta ML, Issaeva N, et al. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37(4):492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hashimoto Y, Chaudhuri A, Lopes M, et al. Rad51 protects nascent DNA from MRE11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17(11):1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22(1):106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kiyohara E, Tamai K, Katayama I, et al. The combination of chemotherapy with HVJ-E containing Rad51 siRNA elicited diverse anti-tumor effects and synergistically suppressed melanoma. Gene Ther. 2012;19(7):734–741 [DOI] [PubMed] [Google Scholar]
- 25. Ito M, Yamamoto S, Nimura K, et al. Rad51 siRNA delivered by HVJ envelope vector enhances the anti-cancer effect of cisplatin. J Gene Med. 2005;7(8):1044–1052 [DOI] [PubMed] [Google Scholar]
- 26. Pal J, Fulciniti M, Nanjappa P, et al. Targeting PI3K and RAD51 in Barrett’s adenocarcinoma: impact on DNA damage checkpoints, expression profile and tumor growth. Cancer Genom Proteom. 2012;9(2):55–66 [PMC free article] [PubMed] [Google Scholar]
- 27. Ohnishi T, Taki T, Hiraga S, et al. In vitro and in vivo potentiation of radiosensitivity of malignant gliomas by antisense inhibition of the RAD51 gene. Biochem Biophys Res Commun. 1998;245(2):319–324 [DOI] [PubMed] [Google Scholar]
- 28. Saydam O, Saydam N, Glauser DL, et al. HSV-1 amplicon-mediated post-transcriptional inhibition of Rad51 sensitizes human glioma cells to ionizing radiation. Gene Ther. 2007;14(15):1143–1151 [DOI] [PubMed] [Google Scholar]
- 29. Bertrand P, Lambert S, Joubert C, et al. Overexpression of mammalian Rad51 does not stimulate tumorigenesis while a dominant-negative Rad51 affects centrosome fragmentation, ploidy and stimulates tumorigenesis, in p53-defective CHO cells. Oncogene. 2003;22(48):7587–7592 [DOI] [PubMed] [Google Scholar]
- 30. Brzovic PS, Rajagopal P, Hoyt DW, et al. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001;8(10):833–837 [DOI] [PubMed] [Google Scholar]
- 31. Wu LC, Wang ZW, Tsan JT, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14(4):430–440 [DOI] [PubMed] [Google Scholar]
- 32. Mallery DL, Vandenberg CJ, Hiom K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J. 2002;21(24):6755–6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xia Y, Pao GM, Chen HW, et al. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J Biol Chem. 2003;278(7):5255–5263 [DOI] [PubMed] [Google Scholar]
- 34. Drost R, Bouwman P, Rottenberg S, et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell. 2011;20(6):797–809 [DOI] [PubMed] [Google Scholar]
- 35. Fabbro M, Rodriguez JA, Baer R, et al. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J Biol Chem. 2002;277(24):21315–21324 [DOI] [PubMed] [Google Scholar]
- 36. Simons AM, Horwitz AA, Starita LM, et al. BRCA1 DNA-binding activity is stimulated by BARD1. Cancer Res. 2006;66(4):2012–2018 [DOI] [PubMed] [Google Scholar]
- 37. Westermark UK, Reyngold M, Olshen AB, et al. BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Mol Cell Biol. 2003;23(21):7926–7936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scully R, Chen J, Ochs RL, et al. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90(3):425–435 [DOI] [PubMed] [Google Scholar]
- 39. Chen J, Silver DP, Walpita D, et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998;2(3):317–328 [DOI] [PubMed] [Google Scholar]
- 40. Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 2009;106(17):7155–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Livingston DM. Cancer. Complicated supercomplexes. Science. 2009;324(5927):602–603 [DOI] [PubMed] [Google Scholar]
- 42. Zhang F, Ma J, Wu J, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19(6):524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Irminger-Finger I, Leung WC, Li J, et al. Identification of BARD1 as mediator between proapoptotic stress and p53-dependent apoptosis. Mol Cell. 2001;8(6):1255–1266 [DOI] [PubMed] [Google Scholar]
- 44. Irminger-Finger I, Jefford CE. Is there more to BARD1 than BRCA1? Nat Rev Cancer. 2006;6(5):382–391 [DOI] [PubMed] [Google Scholar]
- 45. Palmieri D, Bronder JL, Herring JM, et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007;67(9):4190–4198 [DOI] [PubMed] [Google Scholar]
- 46. Palmieri D, Lockman PR, Thomas FC, et al. Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer and induces DNA double-strand breaks. Clin Cancer Res. 2009;15(19):6148–6157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu W, Koike A, Takeshita T, et al. The ubiquitin E3 ligase activity of BRCA1 and its biological functions. Cell Div. 2008;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dong Y, Hakimi MA, Chen X, et al. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol Cell. 2003;12(5):1087–99 [DOI] [PubMed] [Google Scholar]
- 49. Duchnowska R, Jassem J, Thorat M, et al. Gene expression analysis for prediction of early brain metastasis (BM) in HER-2-positive (HER2+) breast cancer patients (pts). J Clin Oncol. 2008;26(Suppl.):abstract 1019. [Google Scholar]
- 50. Zhang XH, Wang Q, Gerald W, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16(1):67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin NU, Bellon JR, Winer EP. CNS metastases in breast cancer. J Clin Oncol. 2004;22(17):3608–17 [DOI] [PubMed] [Google Scholar]
- 52. Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev. 2012;2012:428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Karanjawala ZE, Murphy N, Hinton DR, et al. Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr Biol. 2002;12(5):397–402 [DOI] [PubMed] [Google Scholar]
- 54. Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362(6422):709–715 [DOI] [PubMed] [Google Scholar]
- 55. Soule BP, Hyodo F, Matsumoto K, et al. The chemistry and biology of nitroxide compounds. Free Radic Biol Med. 2007;42(11):1632–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dizdaroglu M, Jaruga P. Mechanisms of free radical-induced damage to DNA. Free Radic Res. 2012;46(4):382–419 [DOI] [PubMed] [Google Scholar]