

Abstract
Background
When presenting with advanced stage disease, lung cancer patients have <5% 5-y survival. The overexpression of checkpoint kinase 1 (CHK1) is associated with poorer outcomes and may contribute to therapy resistance. Targeting CHK1 with small-molecule inhibitors in p53 mutant tumors might improve the effectiveness of chemotherapy and radiotherapy in non–small cell lung cancer (NSCLC).
Methods
We evaluatedCHK1 messenger RNA and protein levels in multiple NSCLC cell lines. We assessed cell line sensitization to gemcitabine, pemetrexed, and radiotherapy by CHK1 inhibition with the small molecule AZD7762 using proliferation and clonogenic cell survival assays. We analyzed CHK1 signaling by Western blotting to confirm that AZD7762 inhibits CHK1.
Results
We selected two p53 mutant NSCLC cell lines with either high (H1299) or low (H1993) CHK1 levels for further analysis. We found that AZD7762 sensitized both cell lines to gemcitabine, pemetrexed, and radiotherapy. Chemosensitization levels were greater, however, for the higher CHK1 protein expressing cell line, H1299, when compared with H1993. Furthermore, analysis of the CHK1 signaling pathway showed that H1299 cells have an increased dependence on the CHK1 pathway in response to chemotherapy. There was no increased sensitization to radiation in H1299 versus H1993.
Conclusions
CHK1 inhibition by AZD7762 preferentially sensitizes high CHK1 expressing cells, H1299, to anti-metabolite chemotherapy as compared with low CHK1 expressing H1993 cells. Thus, CHK1 inhibitors may improve the efficacy of standard lung cancer therapies, especially for those subgroups of tumors harboring higher expression levels of CHK1 protein.
Keywords: CHK1, NSCLC cell lines, Combination therapies, AZD7762, Chemosensitivity, Radiosensitivity
1. Introduction
Lung cancer is one of the most commonly diagnosed malignancies in the United States with more than 220,000 new cases each year. Even with advances in chemotherapy and radiotherapy survival rates for patients with advanced stage disease remain largely unchanged [1]. The best chemotherapeutic agents have limited impact with median patient survival only 11–13 mo [2]. Non–small cell lung cancer (NSCLC) comprises 85% of all lung cancers, with three major subtypes: adenocarcinoma, squamous, and large cell carcinoma. These subtypes are extremely heterogeneous with regards to the specific genetic mutations, which drive tumor growth. There are number of factors that contribute to limited chemotherapeutic efficacy, including cellular drug transporters, dose-related toxicities [3] and increased DNA repair mechanisms [4]. Because new oncogenic pathways with novel targets have been identified [5], emerging targeted therapies are an attractive strategy for improving survival in NSCLC. Epidermal growth factor receptor (EGFR) has proven to be an important therapeutic target as EGFR-mutated tumors are an identifiable subgroup of NSCLC’s that may benefit from EGFR-targeted therapeutics. Unfortunately, in the Western population EGFR mutations occur in only 5%–10% of NSCLCs. Other gene mutations identified include anaplastic lymphoma kinase (ALK) fusion genes, p53, and KRAS [6]. With the notable exception of ALK kinase inhibitors in ALK fusion-positive lung adenocarcinomas, most therapies directed at these alterations have not shown favorable responses [7,8].
In recent years, targeting checkpoint kinase 1 (CHK1), an integral component of the DNA-damage response, with small molecule inhibitors has been proposed as one new approach for targeted therapy. A number of CHK1 inhibitors have been developed and have even begun to be used in clinical trials for various cancer types [9,10]. In response to DNA damage induced by cancer treatments, tumor cells activate a complex signaling network to arrest the cell cycle and enable DNA repair [11,12]. Critical molecules in the DNA damage response are p53 and the protein kinases, CHK1 and CHK2. CHK1 has been shown to contribute to therapy resistance and overall cell survival by inducing G2 arrest and activating homologous recombinant repair [13]. It has been shown that inhibition of CHK1 increases chemotherapy and radiotherapy sensitivity in multiple tumor models, including lung [14–16]. Furthermore, sensitization by CHK1 inhibition appears to be tumor cell-selective and preferential in p53 mutant tumor types [17–19]. p53 has been shown to be an important factor in predicting CHK1 inhibitor-mediated sensitization, as p53 wild-type tumors are less sensitized to DNA damage in response to CHK1 inhibition.
We designed a study to assess the effects of the CHK1 inhibitor AZD7762 on NSCLC cell proliferation and clonogenic survival. Prior data from our laboratory [20] have been further analyzed and have suggested that increased CHK1 expression in primary human lung tumors is associated with poor survival. We also found that NSCLC cells have varying levels of CHK1 messenger RNA (mRNA) and protein expression. We hypothesized that the level of CHK1 expression would determine the ability of CHK1 inhibitors to sensitize tumor cells to chemotherapy and ionizing radiation. We sought to determine if AZD7762 in combination with radiation or the anti-metabolite chemotherapies, pemetrexed, or gemcitabine could selectively target lung cancers that overexpress CHK1.
2. Methods
2.1. Cell culture, drugs, and reagents
H1993, H23, H1437 (adenocarcinomas) and H1299, and H460 (large cell carcinomas) cells were obtained from American Type Culture Collection (Manassas, VA) and grown in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS). H460-DNp53 cells were obtained from AstraZeneca [21]. Pemetrexed, and gemcitabine (Eli Lilly Company, Indianapolis, Indiana) were dissolved in phosphate buffered saline as a stock solution at 1 mM. AZD7762 (AstraZeneca) was dissolved in dimethyl sulfoxide as a stock solution at 10 mM. SMARTpool CHK1 or non-targeting–control pool small interfering RNAs were purchased from Dharmacon (Lafayette, CO) and used according to the manufacturer’s protocol.
2.2. Quantitative real-time polymerase chain reaction
RNA was isolated from H1993, H23, H1437, and H1299 cell lines by homogenizing cells in QIAzol reagent (Qiagen, Valencia, CA) and purifying RNA using RNeasy Mini Kits (Qiagen). Two microgram of total RNA was reverse transcribed using a High Capacity complementary DNA Transcription Kit (Applied Biosystems, Foster City, CA). CHK1 transcripts were quantified by quantitative real-time polymerase chain reaction (qRT-PCR) using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen, Grand Island, NJ) in a Rotor-Gene 3000 thermocycler (Corbett Life Science, Valencia, CA). Relative expression levels were normalized to β-actin expression using the 2−ΔΔ computed tomography method [22].
Primer sequences were as follows: ACTB (forward): 5′-\ATGTGGCCGAGGACTTTGATT-3′; ACTB (reverse): 5′-AGTGGGGTGGCTTTTAGGATG-3′ [23]; CHK1 (forward): 5′–CGGTGGAGTCATGGCAGTGCCC-3′; CHK1 (reverse): 5′-TCTGGACAGTCTACGGCACGCTTCA-3′.
2.3. Cell line microarray construction
Formalin-fixed, paraffin-embedded blocks of 48 cell lines were arrayed into a cell line microarray using the methodology of Nocito et al. [24]. Each cell line was represented by two 1 mm diameter cores.
2.4. Immunohistochemistry
Immunohistochemical staining was performed on the Dako Autostainer (Dako, Carpinteria, CA) using Dako EnVision + polymerized horseradish peroxidase and diaminobenzadine as the chromogen. Sections of deparaffinized cell line microarray were labeled overnight with CHK1 (rabbit monoclonal antibody, clone EP691Y, 1:100; Abcam, Cambridge, MA). Microwave treatment in 10 mM Tris buffer pH9/1 mM ethyl-enediaminetetraacetic acid (EDTA) was used for epitope retrieval. Appropriate negative (no primary antibody) and positive controls (breast cancer) were stained in parallel. The immunoreactivity was scored by a three-tier (negative, low-[1+] and highpositive [2+]) modification of the normal grading scheme previously described by Wang et al. [25].
2.5. Chemo- and radiosensitization
Chemosensitization was measured using the cell proliferation reagent WST-1 (Roche Applied Science, Penzberg, Germany) according to the manufacturer’s instructions. In brief, cells were plated into 96-well flat-bottomed microplates in 100 μL of medium containing 10% fetal bovine serum and incubated for 24 h to allow sufficient cell adhesion. This time point was defined as T0 h. Cells were treated with graded concentrations of gemcitabine for 2 h (T = 0–2 h, followed by media T = 2–24 h) or pemetrexed for 24 h (T = 0–24 h) followed by the CHK1 inhibitor, AZD7762, at a 100 nM concentration (T = 24–48 h). After drug exposure cells were washed and cultured in a drug-free medium for an additional 24 h (T = 48–72 h). Ten micro-liter of WST-1 reagent was added to each well and plates were incubated at 37°C for 1–3 h depending on the cell line. Plates were shaken for 1 min and absorbance at 450 nm was measured using a Microplate reader (BioTek, Winooski, VT). A well containing only medium with WST-1 solution was used as a background control. Each experiment was performed using three replicates. Cell viability was expressed as the relative percent absorbance of treated versus nontreated cells. Data were analyzed using Microsoft Excel 2010 (Microsoft, Redmond, WA) and GraphPad Prism version 5.01 software (GraphPad Software, Inc, La Jolla, CA). Chemosensitization was confirmed by clonogenic survival in which cells growing in 100 mm dishes were treated according to the schedule described previously. After the treatment with both drugs, cells were replated at various dilutions, and after 10 d the resulting colonies were fixed with methanol-to-acetic acid (7:1), stained with 0.1% crystal violet and the colonies counted. Cell survival was calculated as the surviving fraction of treated cells as compared with surviving factor of nontreated cells.
Radiosensitization was also evaluated by clonogenic survival. Cells growing in 100 mm dishes were treated with 100 nM AZD7762 (at T = 0 h) and then irradiated with 2, 4, 6, and 8 Gy (T = 1 h) using a Philips RT250 (Kimtron Medical, Oxford, CT) in the University of Michigan Comprehensive Cancer Center Experimental Irradiation Core. At T = 24 h, cells were replated at various dilutions, and after 10 d the resulting colonies were fixed (as mentioned previously) and counted. The cell survival enhancement ratio was calculated as the ratio of the mean inactivation dose under control conditions divided by the mean inactivation dose after drug exposure according to the methods of Fertil et al. [26]. A value significantly >1 indicates radiosensitization.
2.6. Immunoblotting
Cells were washed with phosphate buffered saline and lysed in 150 mM NaCl, 20 mM Tris (pH 7.5), 1 mM ethyl-enediaminetetraacetic acid, 1 mM ethylene glycol tetraacetic acid (EGTA), 2.5 mM Na4P2O4, 1 mM β-glycerol phosphate, 1 mM Na3VO4, 1 μg/mL leupeptin, 1% Triton X-100, and Protease Inhibitor Cocktail (Sigma-Aldrich Corp, St Louis, MO). Protein concentration was determined with DC Protein Assay Reagent (Bio-Rad, Hercules, CA). Samples were diluted in non-reducing lithium dodecyl sulfate sample loading buffer (Thermo Scientific, West Palm Beach, FL) with 2-mercaptoethanol and resolved on 4%–12% gradient Tris-Glycine gels (Life Science Technologies, Grand Island, NY). Separated proteins were transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA) and hybridized overnight at 4°C with antibodies recognizing pS345 CHK1, pS296 CHK1 (Cell Signaling Technology, Danvers, MA), total CHK1 (Abcam), CDC25A (Santa Cruz Biotechnology, Dallas, Texas), or glyceraldehyde-3-phosphate dehydrogenase (Millipore). Membranes were probed with secondary antibodies and incubated with Amersham ECLPlus Western blotting detection system (GE Healthcare, Pittsburgh, PA).
3. Results
To determine if response to CHK1 inhibition is related to the level of CHK1 expression, we identified four p53 mutant NSCLC cell lines with variable CHK1 mRNA expression from publicly available gene expression data (Fig. 1A). H1299 and H23 cells were selected as high CHK1 expressing cells, whereas H1437 and H1993 cells were selected as low CHK1 expressing cells. Using qRT-PCR, we first sought to confirm the relative mRNA expression for the four cell lines. We found that the relative levels of CHK1 mRNA for H23, H1993, and H1299 cells (Fig. 1B) were in general agreement with the publicly available gene expression data. However, H1437 appeared to express relatively high levels of CHK1 when compared with β-actin levels (Fig. 1B), which was inconsistent with the gene expression data (Fig. 1A). Next, we correlated CHK1 mRNA expression to CHK1 protein expression by immunohistochemistry (Fig. 1C) and by Western blot analysis (data are not shown). Protein assays confirmed the results obtained by qRT-PCR, with high levels of CHK1 protein found in H1437, H1299, and H23 cell lines and with almost a complete absence of CHK1 expression in H1993.
Fig. 1.

NSCLC cell lines show variable CHK1 expression. (A) Microarray analysis of CHK1 mRNA levels in 79 lung cancer cell lines. The microarray data were downloaded from Gene Expression Omnibus using the accession number GSE4824 and robust multiarray average (RMA) normalized. P53 mutation information was acquired from http://p53.free.fr/index.html. (B) Relative expression of CHK1 mRNA to β-actin by qRT-PCR in selected four cell lines, H23, H1437, H1993, and H1299. One representative result is shown (C) CHK1 expression by immunohistochemistry of the cell lines showing increased nuclear CHK1 protein expression (brown stain) in the H1299, H23, and H1437 high CHK1 expressing cells as compared with H1993 cells low CHK1 expressing cells. (Color version of figure is available online.)
To begin to determine the impact of CHK1 expression levels on sensitivity to CHK1 inhibitors, we focused on two cell lines, H1299, the most consistent high expressing CHK1 cell line, and H1993, the low expressing CHK1 cell line. First, we evaluated the effect of the CHK1 inhibitor, AZD7762, on gemcitabine or pemetrexed sensitivity using a WST-1 proliferation assay (Fig. 2). We treated cells with graded concentrations of gemcitabine or pemetrexed (followed by treatment with AZD7762; schedule shown in Fig. 2C). This particular treatment schedule was chosen based on previous studies, which showed that chemotherapy with gemcitabine, cisplatin, and paclitaxel results in strong activation and accumulation of CHK1 [27,28]. We observed varying degrees of sensitization to gemcitabine (Fig. 2A) or pemetrexed (Fig. 2B) in both cell lines, but noted more pronounced sensitization by AZD7762 to gemcitabine (13.1-fold) and pemetrexed (15.5-fold) in high CHK1 expresssing-H1299 cells than in low CHK1 expressing-H1993 cells (2.8-and 7.4-fold, respectively). Estimation of inhibitory concentration 50% (IC50) of gemcitabine and pemetrexed showed that AZD7762 caused a greater reduction in the IC50 of gemcitabine and pemetrexed in H1299 cells as compared with H1993 cells (Fig. 2D). In addition, t-test with Welch’s correction showed that at as low as 3 μM treatment, there was statistically significant reduction in proliferation in H1299 (P = 0.0036 with gemcitabine and P = 0.0042 with pemetrexed treatment) as compared with H1993 (P = 0.0393 with gemcitabine and P = 0.0774 with pemetrexed treatment). To determine if sensitization by CHK1 inhibition is selective and preferential in p53 mutant tumor types, we treated p53 wild-type H460 cells and also H460-DNp53 cells with gemcitabine. We found that H460 p53 mutant cells were sensitized to gemcitabine by AZD7762 to much greater extent, 3.5- and 1.5-fold, respectively (data not shown). We have also seen the same selective sensitization to pemetrexed when we treated H1993 and H1299 cells with MK8776, alternate CHK1 inhibitor, or CHK1 small interfering RNA. We found that both treatments produced similar sensitization to pemetrexed, and it was more pronounced in H1299 cells as compared with H1993 (data not shown).
Fig. 2.

Chemosensitization to gemcitabine and pemetrexed in H1299 and H1993 cells as measured by the WST-1 assay. Cells were treated with graded concentrations of gemcitabine (T0–T2 h) or pemetrexed (T0–T24 h) followed by 24 h treatment with AZD7762 at 100 nM (T24–T48 h). H1299, high CHK1 expressing cell line was sensitized to gemcitabine (A) or pemetrexed (B) treatment to a greater extent as compared with H1993, low CHK1 expressing cell line. At 3 μM treatment there was statistically significant reduction in proliferation in H1299 (P = 0.0036 with gemcitabine and P = 0.0042 with pemetrexed treatment) as compared with H1993 (P = 0.0393 with gemcitabine and P = 0.0774 with pemetrexed treatment). (C) The in vitro treatment schedule with gemcitabine, pemetrexed, and AZD7762. (D) Differences in IC50 of gemcitabine and pemetrexed with and without AZD7762 treatment. There is a greater reduction in IC50 in H1299 cell lines with both treatments. (Color version of figure is available online.)
Next, we assessed the effect of AZD7762 on gemcitabine and pemetrexed sensitivity using clonogenic survival assays (Fig. 3A and B). Cells were treated with graded concentrations of gemcitabine or pemetrexed followed by AZD7762 according to the schedule in Figure 2C. We found that AZD7762 in combination with gemcitabine or pemetrexed reduced the surviving fraction only in H1299, high CHK1 expressing cells (2.4- and 2.2-fold changes based on IC50 values for gemcitabine and pemetrexed, respectively), with little to no reduction in the surviving fraction of H1993 cells (no change for gemcitabine, 1.2-fold for pemetrexed). Next, we evaluated the influence of CHK1 expression levels on AZD7762-mediated sensitization to radiation therapy (Fig. 3C). We found a low level of sensitization in both, H1299 and H1993, cells, although there was no significant difference between cell lines. Radiation enhancement ratio was 1.5 in H1993 and 1.4 in H1299 as compared with 1 in controls. Taken together these results suggest that high CHK1 expression may be a useful predictor for the ability of CHK1 inhibitors to sensitize to antimetabolite chemotherapy.
Fig. 3.

Chemo- and radiosensitization by clonogenic survival assay confirmed increased sensitization to both, gemcitabine (A) and pemetrexed (B) in H1299. AZD7762 in combination with gemcitabine or pemetrexed reduced the surviving fraction of H1299 cells by 2.4- and 2.2-fold (based on IC50 values for gemcitabine and pemetrexed, respectively), with little to no reduction in the surviving fraction of H1993 cells (no change for gemcitabine, 1.2-fold for pemetrexed). (C) Both cell lines, H1299 and H1993, showed moderate radiosensitization with AZD7762 treatment (radiation enhancement ratio was 1.4 for H1299 and 1.5 for H1993 cells) with no difference in radiosensitization between the cell lines.
To investigate the mechanism of the sensitization to chemotherapy by CHK1 inhibition in H1299 and H1993 cell lines, we evaluated the effects of pemetrexed and AZD7762 on the levels of phosphorylated CHK1 (S296, an autophosphorylation site and the S345, an ataxia telangiectasia and Rad-3 related/ataxia telangiectasia mutated (ATM/ATR)-mediated phosphorylation site) and CDC25A, which is degraded in response to CHK1 activation and contributes to G2-checkpoint activation in response to DNA damage (Fig. 4). ATR/ATM-mediated phosphorylation of CHK1 at S345 was increased in response to pemetrexed/AZD7762 to a greater extent in H1299 cells than in H1993 cells. This increase in phospho-CHK1 (S345) is likely an indication of increased DNA damage in H1299 cells and suggests an increased dependence of H1299 cells on the CHK1 pathway. Pemetrexed alone resulted in an increased level of pS296 CHK1, which was reduced by treatment with AZD7762. Furthermore, as expected the reduction of CDC25A protein in response to pemetrexed was blocked by AZD7762. This effect of CHK1 inhibition by AZD7762 on S296 CHK1 phosphorylation and CDC25A stabilization was not as easily seen in H1993, but may also reflect the very low basal level of total CHK1 in the H1993 cell line. Taken together, these data indicate that CHK1 inhibition with AZD7762 has a greater effect on CHK1 signaling in high CHK1 expressing H1299 cells as compared with low CHK1 expressing H1993 cells and is consistent with the observed degrees of sensitization to pemetrexed in these cells.
Fig. 4.
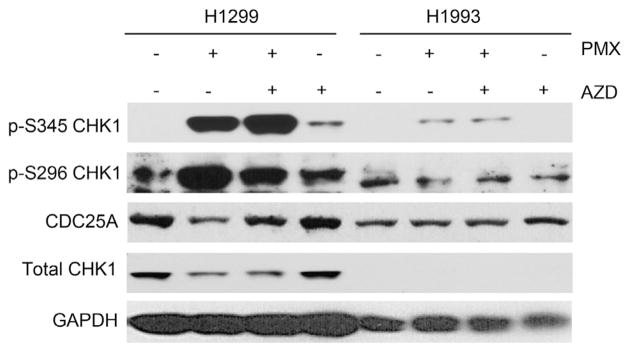
CHK1 signaling in response to pemetrexed and AZD7762 in low (H1993) and high (H1299) CHK1 expressing cell lines.
4. Discussion
Long-term outcomes for patients with advanced lung cancers continue to be poor with 5-y survival rates of <5%. As opposed to finding a single agent or combination to treat all lung cancers, many groups are investigating the targeting of specific subgroups of lung cancers based on specific molecular pathways, that is, EGFR or ALK mutations. Combination therapies that include the suppression of DNA repair mechanisms are being evaluated. Poly (ADP-ribose) polymerase inhibitors have been combined with cisplatin in phosphatase and tensin homolog (PTEN) deficient cells [29], under the premise that phosphatase and tensin homolog deficiency leads to reduced DNA damage signaling and increased sensitivity to the cisplatin therapy. In our study, we focused on p53 mutant cell lines, as these have been shown to potentially be more susceptible to CHK1 inhibition and G2 checkpoint abrogation, as p53 wild-type cells are protected by an intact G1 cell cycle checkpoints that prevents cells from proceeding to cell death without undergoing DNA repair [17,30].
The CHK1 pathway may be one significant mechanism for either primary or acquired chemotherapy resistance and an explanation for the observation that high CHK1 expression correlates with poor prognosis. A recent study demonstrated that increased CHK1 expression in a mesothelioma tumor was associated with acquired resistance to pemetrexed and platin therapy [31]. CHK1 has also been suggested as a possible marker for more aggressive tumors, as higher CHK1 expression is associated with higher grade tumors in gastric cancers [32]. Our prior gene expression data from 442 resected lung adenocarcinoma [20] patient specimens identified CHK1 as one of the top genes that demonstrated highly variable expression and was elevated in patients with poor outcome relative to favorable outcome. CHK1 expression also varies between different cell lines indicating that CHK1 expression is not consistently elevated in all cancers. It has also been shown that CHK1 inhibitors sensitize pancreatic cancer cells to gemcitabine or radiation [32] and that the sensitization mechanism is attributable to G2 checkpoint abrogation and inhibition of homologous recombination repair DNA damage response [14,16]. CHK1 inhibitors have been combined with numerous chemotherapeutics, including cisplatin [33,34] and paclitaxel [35]. Although we did not observe any sensitization when we combined AZD7762 with cisplatin (data not shown), we did see a reduction in both proliferation and clonogenic survival with both gemcitabine and pemetrexed, with a greater reduction in high versus low CHK1 expressing NSCLC cells. Thus, our data suggest that high CHK1 expression may serve as a biomarker for tumors that would benefit most from CHK1 inhibitor treatment, as part of a combination protocol.
When we evaluated the phosphorylation of CHK1 and CDC25A as a downstream molecule, our results appeared consistent with the mechanism described in Parsels et al. [36], 2011. In response to DNA damage from pemetrexed, both cell lines showed an increase in phosphorylation of the S345 site. In the H1299 cells, the AZD7762 blocked the autophosphorylation of the S296 site that is seen with pemetrexed alone, and resulted in a greater stabilization of CDC25A, thought to be by blocking CDC25A degradation. This results in a greater number of cells proceeding to mitotic entry and ultimately cell death. The H1299 data are consistent with a CHK1 pathway dependent response. We do not see the same level of effect in the H1993 cell lines, suggesting that CHK1 does not play a key role in its chemotherapy response.
The combination of radiation therapy and CHK1 inhibition has been well studied, with many showing an improved radiation effect with CHK1 inhibition [16] in lung cancer and pancreatic cancer cells. We saw modest reductions in clonogenic survival in both cell lines when AZD7762 was combined with radiation therapy. However, no differences in radio-sensitization by AZD7762 were observed between the two cell lines suggesting that CHK1 expression levels may not be related to the ability of CHK1 inhibitors to sensitize to radiation.
5. Conclusions
In summary, we have shown that CHK1 levels vary in different cell lines. Furthermore, we found that the ability of CHK1 inhibition to improve the effectiveness of antimetabolite chemotherapy is related to CHK1 expression levels in p53 mutant NSCLC cells, with the greatest sensitization to chemotherapy occurring in high CHK1 expressing cells. It will be important for future studies to evaluate the influence of CHK1 levels on the efficacy of CHK1 inhibitors in combination with chemotherapy in vivo, and to further elucidate the mechanisms behind CHK1 overexpression in certain NSCLCs.
Acknowledgments
Grabauskiene S, Bergeron EJ, Chen G, Thomas DG, Beer DG, and Morgan MA have no financial ties to disclose. Giordano TJ received grant from Asuargen Inc and Reddy RM received grant from GlaxoSmithKline.
Footnotes
The authors have no potential conflict of interest relevant to this article.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Ohe Y, Ohashi Y, Kubota K, et al. Randomized phase III study of cisplatin plus irinotecan versus carboplatin plus paclitaxel, cisplatin plus gemcitabine, and cisplatin plus vinorelbine for advanced non-small-cell lung cancer: four-arm cooperative study in Japan. Ann Oncol. 2007;18:317. doi: 10.1093/annonc/mdl377. [DOI] [PubMed] [Google Scholar]
- 3.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 4.Raymond E, Chaney SG, Taamma A, Cvitkovic E. Oxaliplatin: a review of preclinical and clinical studies. Ann Oncol. 1998;9:1053. doi: 10.1023/a:1008213732429. [DOI] [PubMed] [Google Scholar]
- 5.Bryant CM, Albertus DL, Kim S, et al. Clinically relevant characterization of lung adenocarcinoma subtypes based on cellular pathways: an international validation study. PLoS One. 2010;5:e11712. doi: 10.1371/journal.pone.0011712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dempke WC, Suto T, Reck M. Targeted therapies for non-small cell lung cancer. Lung Cancer. 2010;67:257. doi: 10.1016/j.lungcan.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prudhomme M. Novel checkpoint 1 inhibitors. Recent Pat Anticancer Drug Discov. 2006;1:55. doi: 10.2174/157489206775246520. [DOI] [PubMed] [Google Scholar]
- 10.Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci. 2011;32:308. doi: 10.1016/j.tips.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 11.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 12.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 13.Sorensen CS, Hansen LT, Dziegielewski J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 14.Parsels LA, Morgan MA, Tanska DM, et al. Gemcitabine sensitization by checkpoint kinase 1 inhibition correlates with inhibition of a Rad51 DNA damage response in pancreatic cancer cells. Mol Cancer Ther. 2009;8:45. doi: 10.1158/1535-7163.MCT-08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell JB, Choudhuri R, Fabre K, et al. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin Cancer Res. 2010;16:2076. doi: 10.1158/1078-0432.CCR-09-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan MA, Parsels LA, Zhao L, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70:4972. doi: 10.1158/0008-5472.CAN-09-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levesque AA, Fanous AA, Poh A, Eastman A. Defective p53 signaling in p53 wild-type tumors attenuates p21waf1 induction and cyclin B repression rendering them sensitive to Chk1 inhibitors that abrogate DNA damage-induced S and G2 arrest. Mol Cancer Ther. 2008;7:252. doi: 10.1158/1535-7163.MCT-07-2066. [DOI] [PubMed] [Google Scholar]
- 18.Koniaris LG, Zimmers-Koniaris T, Hsiao EC, Chavin K, Sitzmann JV, Farber JM. Cytokine-responsive gene-2/IFN-inducible protein-10 expression in multiple models of liver and bile duct injury suggests a role in tissue regeneration. J Immunol. 2001;167:399. doi: 10.4049/jimmunol.167.1.399. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, Xiao Z, Gu WZ, et al. Selective Chk1 inhibitors differentially sensitize p53-deficient cancer cells to cancer therapeutics. Int J Cancer. 2006;119:2784. doi: 10.1002/ijc.22198. [DOI] [PubMed] [Google Scholar]
- 20.Shedden K, Taylor JM, Enkemann SA, et al. Gene expression-based survival prediction in lung adenocarcinoma: a multisite, blinded validation study. Nat Med. 2008;14:822. doi: 10.1038/nm.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zabludoff SD, Deng C, Grondine MR, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7:2955. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 2001;25:402. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Lin L, Bass AJ, Lockwood WW, et al. Activation of GATA binding protein 6 (GATA6) sustains oncogenic lineage-survival in esophageal adenocarcinoma. Proc Natl Acad Sci U S A. 2012;109:4251. doi: 10.1073/pnas.1011989109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nocito A, Kononen J, Kallioniemi OP, Sauter G. Tissue microarrays (TMAs) for high-throughput molecular pathology research. Int J Cancer. 2001;94:1. doi: 10.1002/ijc.1385. [DOI] [PubMed] [Google Scholar]
- 25.Wang S, Saboorian MH, Frenkel E, Hynan L, Gokaslan ST, Ashfaq R. Laboratory assessment of the status of Her-2/neu protein and oncogene in breast cancer specimens: comparison of immunohistochemistry assay with fluorescence in situ hybridisation assays. J Clin Pathol. 2000;53:374. doi: 10.1136/jcp.53.5.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fertil B, Dertinger H, Courdi A, Malaise EP. Mean inactivation dose: a useful concept for intercomparison of human cell survival curves. Radiat Res. 1984;99:73. [PubMed] [Google Scholar]
- 27.Morgan MA, Parsels LA, Parsels JD, Mesiwala AK, Mybaum J, Lawrence TS. Role of checkpoint kinase 1 in preventing premature mitosis in response to gemcitabine. Cancer Res. 2005;65:6835. doi: 10.1158/0008-5472.CAN-04-2246. [DOI] [PubMed] [Google Scholar]
- 28.Bartucci M, Svensson S, Romania P, et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death Diff. 2012;19:768. doi: 10.1038/cdd.2011.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minami D, Takigawa N, Takeda H, et al. Synergistic effect of olaparib with combination of cisplatin on PTEN-deficient lung cancer cells. Mol Cancer Res. 2013;11:140. doi: 10.1158/1541-7786.MCR-12-0401. [DOI] [PubMed] [Google Scholar]
- 30.Koniaras K, Cuddihy AR, Christopoulos H, Hogg A, O’Connell MJ. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene. 2001;20:7453. doi: 10.1038/sj.onc.1204942. [DOI] [PubMed] [Google Scholar]
- 31.Roe OD, Szulkin A, Anderssen E, et al. Molecular resistance fingerprint of pemetrexed and platinum in a long-term survivor of mesothelioma. PLoS One. 2012;7:e40521. doi: 10.1371/journal.pone.0040521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao H, Yang Z, Li Y. Expression of checkpoint kinase 1 and polo-like kinase 1 and its clinicopathological significance in benign and malignant lesions of the stomach. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2010;35:1080. doi: 10.3969/j.issn.1672-7347.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 33.Venkatesha VA, Parsels LA, Parsels JD, et al. Sensitization of pancreatic cancer stem cells to gemcitabine by Chk1 inhibition. Neoplasia. 2012;14:519. doi: 10.1593/neo.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson R, Meuth M, Woll P, Zhu Y, Danson S. Treatment with the Chk1 inhibitor Go6976 enhances cisplatin cytotoxicity in SCLC cells. Int J Oncol. 2012;40:194. doi: 10.3892/ijo.2011.1187. [DOI] [PubMed] [Google Scholar]
- 35.Xiao Z, Xue J, Semizarov D, Sowin TJ, Rosenberg SH, Zhang H. Novel indication for cancer therapy: Chk1 inhibition sensitizes tumor cells to antimitotics. Int J Cancer. 2005;115:528. doi: 10.1002/ijc.20770. [DOI] [PubMed] [Google Scholar]
- 36.Parsels LA, Qian Y, Tanska DM, et al. Assessment of chk1 phosphorylation as a pharmacodynamic biomarker of chk1 inhibition. Clin Cancer Res. 2011;17:3706. doi: 10.1158/1078-0432.CCR-10-3082. [DOI] [PMC free article] [PubMed] [Google Scholar]