

Abstract
Purpose
FMS-like tyrosine kinase-3 (FLT3) internal tandem duplication (FLT3-ITD) mutations are common in patients with acute myeloid leukemia (AML). These patients regularly develop resistance to FLT3 inhibitors suggesting that targeted combination drug strategies are needed to enhance AML therapy efficacy.
Experimental Design
Acquired point mutations of FLT3 ITD gene were screened using cDNA-based sequencing approach in vitro sorafenib resistant cells, which were developed by long-term exposure of Ba/F3-ITD to increasing doses of sorafenib, and in FLT3 ITD mutated AML patients, who developed relapse following sorafenib therapy. Drug effects (e.g., proliferation inhibition, apoptosis induction, and changes in signal transduction protein expression) were assessed in AML cells harboring the point mutations in vitro and in FLT3 ITD mutated AML patient samples.
Results
We identified several acquired point mutations in the tyrosine kinase (TK) domains (TKDs) of the FLT3 gene in sorafenib-resistant murine leukemia cell line carrying human FLT3-ITD mutations, which were also detected in two of four sorafenib-resistant patient samples. Engineering these point mutations into Ba/F3-ITD cells generated sub-lines that demonstrated varying degrees of sorafenib (a type II TK inhibitor) resistance. A similar pattern of resistance could be observed by exposing these sub-lines to the other type II TK inhibitors AC220 and MLN518. However, these sub-lines retained sensitivity to the type I TK inhibitors PKC412 or crenolanib. The combination of crenolanib with sorafenib demonstrated marked cytotoxic effects in all of the sorafenib-resistant sub-lines.
Conclusions
These combination strategies could be clinically important in reversing acquired resistance to FLT3 inhibition in AML.
Keywords: FLT3, sorafenib, crenolanib, drug resistance, drug combination strategies
Introduction
One-third of the patients with acute myeloid leukemia (AML) have gain-of-function mutations in the fms-like tyrosine kinase 3 (FLT3) gene. These mutations include FLT3 internal tandem duplication (FLT3-ITD) mutations in the juxtamembrane (JM) domain and point mutations in the tyrosine kinase (TK) domains (TKDs, e.g., mutations at D835/836) (1,2). Leukemias with FLT3 mutations are associated with a higher risk of relapse following therapy and, as a result, shorter overall patient survival (1,3). A number of small molecule inhibitors targeting constitutive FLT3 activation have been developed and are categorized as type I and type II TK inhibitors (4,5). Type I TK inhibitors (e.g., CEP701 and PKC412) bind in and around the region occupied by the adenine ring of ATP and do not require the “DFG-out” conformation (6). Type II TK inhibitors (e.g., sorafenib, AC220, and MLN518) occupy an allosteric site that is directly adjacent to the ATP binding pocket created by the activation-loop that adopts a “DFG-out” inactive conformation (7,8).
We have identified sorafenib as a potent, clinically effective inhibitor of FLT3-ITD mutations and demonstrated an encouraging and effective reduction in leukemia blasts in peripheral blood and bone marrow. However, only 10% of the FLT3-ITD mutated AML patients achieved complete remission (CR), or a complete remission with incomplete platelet recovery (CRp), in our phase I clinical trial (9,10). Furthermore, relapse routinely occurs after prolonged therapy with sorafenib as well as other small-molecule TK inhibitors including type II inhibitors AC220 and MLN518, and the type I inhibitor CEP701 (10-19). These findings would suggest that preventing, or surmounting, the resistance mechanisms associated with FLT3-targeted therapies of AML is of paramount importance for the successful therapy of this disease.
The mechanism (s) of secondary AML resistance to FLT3 inhibitors are poorly understood. Acquired point mutations in the FLT3 gene have been reported in a cell line that exhibited resistance to the type 1 TK inhibitor PKC412 (20). Secondary FLT3 point mutations that conveyed resistance to FLT3 inhibitors were also detected in cultured leukemic cells exposed to the mutagen N-ethyl-N-nitrosourea (21,22), or the type II TK inhibitor MLN518 and AC220 (13,23). Notably, Smith and colleagues recently identified secondary point mutations in either TKD1 or TKD2 region of FLT3 gene in all eight relapsed ITD mutated AML patients who received AC220 monotherapy, which include D835 and F691 mutations. These mutations have also showed cross-resistance to sorafenib (13). Conversely, sorafenib resistance in two patients whose AML harbored FLT3-ITD mutations was reportedly not linked to secondary point mutations in the FLT3 gene (24).Consequently, further investigation is needed to determine the biological significance and potential mechanistic underpinnings of acquired FLT3 point mutations in the development of resistance to FLT3-targeted therapy. This tenet served as the basis for the assessments reported in this study.
Materials and Methods
Chemicals and antibodies
Sorafenib and PKC412 were purchased from American Custom Chemicals Corporation (San Diego, CA) and Sigma-Aldrich (St. Louis, MO), respectively. Crenolanib (CP-868-596) was provided by AROG Pharmaceuticals LLC (Dallas, TX). MLN518, AC220, CI-1040, and CCI-779 were purchased from Skelleck Chemicals (Houston, TX).Rabbit polyclonal antibodies were purchased from the following sources: phospho-FLT3 (Tyr589/591), phospho-ERK (Thr202/Tyr204), phospho-AKT (Ser473), phospho-S6K (Ser240/Ser244), phospho-PI3K, S6K, and cleaved caspase-3 all from Cell Signaling Technology (Beverly, MA); Bim from Calbiochem Co. (San Diego, CA); FLT3 from Santa Cruz Biotechnology (Santa Cruz, CA); and Stat5A/B from R&D Systems (Minneapolis, MN). Mouse monoclonal antibodies were purchased from the following sources: phospho-Stat5 (Tyr694/699) from Upstate, Inc. (Charlottesville, VA) and ERK2 from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell lines
Ba/F3-ITD cells, a murine pro-B lymphocyte line created by transfecting Ba/F3 cells with lentivirus containing human FLT3-ITD mutations were isolated and characterized as described previously (25) and kindly provided by Dr. J. Donald Small (Department of Pediatric Oncology, Johns Hopkins University, Baltimore, MD). The Ba/F3-ITD-Res cells were isolated by exposing Ba/F3-ITD to increasing low doses (i.e., 1 to 5 nmol/L) of sorafenib for up to 3 months, and further exposed for an additional 3 weeks to serial dilutions (5 to 50 nmol/L) of sorafenib. The resistant clones were collected for mutation analysis. The engineered cell lines with point mutations Ba/F3-ITD+651, Ba/F3-ITD+687, Ba/F3-ITD+676, Ba/F3-ITD+842, and Ba/F3-ITD+676/842 were developed by individually introducing D651G, I687F, N676D and/or Y842C mutations, respectively, in the Ba/F3-ITD cells using lentiviral infection. Briefly, the human FLT3-ITD sequence was amplified by RT-PCR. The restriction enzymes were introduced into the cDNA by PCR, and the product was inserted into a CD510B-1 lentiviral vector using a ligation reaction. In each point mutation experiment, pairs of vector primers were prepared and used to amplify all of the vector sequences except for the region containing the sites to be mutated. For PCRs, a QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Inc. Santa Clara, CA) was used following the manufacturer's instructions. The PCR products were transformed into XL10-Gold ultra-competent cells, and the vector clones were analyzed by Sanger-based DNA sequencing. The mutation-containing DNA vectors were produced by transient transfection of 293T cells using FuGENE 6 (Roche, Indianapolis, IN), and supernatants were harvested after 48 hours. Lentiviral infection was performed in Ba/F3-ITD cells, and GFP-positive cell populations were collected using fluorescence-activated cell sorting (FACSAria II, BD Bioscience, San Jose, CA). All cells were maintained in RPMI medium supplemented with 10% fetal bovine serum.
Primary patient samples
Four AML patients who lost response to sorafenib were selected from patients enrolled in phase I/II clinical trials conducted at the University of Texas M. D. Anderson Cancer Center from 2006 to 2009 (10,11,26). Bone marrow or peripheral blood samples were collected when resistance to the therapy was first observed. The patient characteristics are presented in Supplementary Table 1. The mononuclear cells in these samples were purified by Ficoll-Hypaque (Sigma-Aldrich) density-gradient centrifugation, and the cells were cultured as described above prior to treatment.
Screening acquired point mutations of FLT3 gene
Total mRNA from the patient samples was isolated and point mutations were screened for cDNA-based sequencing. Briefly, total mRNA was prepared using TRIzol (Life Technologies, Grand Island, NY) reagent as described by the manufacturer. After reverse transcription with random hexamers (SuperScript III First-Strand Synthesis SuperMix; Life Technologies, Grand Island, NY), a PCR-based mutation screen was performed by screening full length FLT3 gene using the following primer pairs: 5′-GGAGTTGTTTCCATGGTCATT-3′ and 5′-CGTTTCTTGCCACTGATGAT-3′; 5′-GCTTTTGTATCATCAGTGGCA-3′ and 5′-AAGAGGAGACAAACACCAATTG-3′; 5′-GCTTTTGTATCATCAGTGGCA-3′ and 5′-GAAGTTAGCATCAACCGGAAT-3′; and 5′-TGTCGAGCAGTACTCTAAACA-3′ and 5′-CTGAAAGAAAAAGCAGACAGCT-3′.
Cell viability and apoptosis assays
AML cells were plated in 24-well plates at 2 × 105 cells/mL in 2 mL of medium. The indicated drugs, dissolved in dimethyl sulfoxide (DMSO, control, Sigma-Aldrich), were added to the culture medium (i.e., as single agent or multiple agents in combination treatments) for 48- to 72-hour incubations. The Ba/F3-Flt3 cells were treated with the compounds in the presence of IL-3 (2 ng/mL). Cell viability was assessed using the Trypan blue dye exclusion method, and apoptosis was determined via FACS by annexin V positivity as described previously (9). The 50% inhibitory concentration (IC50) for cell growth inhibition and the 50% effective concentration (EC50) for apoptosis were calculated using CalcuSyn software (BioSoft, Cambridge, UK).The Ba/F3 ITD or Ba/F3-ITD+842 cells were treated with indicated compounds in the presence of FL (25 or 50 ng/mL) for 48 hours, and apoptosis induction was determined as previously described above.
Immunoblotting
Phosphorylation and total protein levels were determined using Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE);semi quantitative immunoblotting data was generated using Beta 4.03 imaging software (Scion Corporation, Frederick, MD) by comparing the ratios of phosphoproteins and total proteins, or the loading controls tubulin (or GAPDH) as described previously (27).
In vitro Kinase Assay
Kinase assay was performed in vitro to examine if the inhibitors directly suppress phosphorylation of mutated FLT3 protein. ITD plus Y842C mutated FLT3 protein was isolated from Ba/F3-ITD+842 cells and phosphorylation enzyme reaction was performed in magnesium/ATP-containing reaction buffer in vitro for 30 min at 30°C in the presence/absence of crenolanib (0.5μmol/L) and/or sorafenib (0.5μmol/L) as described previously (9). Phosphorylation level of FLT3 protein was measured using immunoblotting and the ratio of phospho-FLT3 to total FLT3 was determined using Beta 4.03 imaging software as described above.
Statistical analyses
The data are presented as the means ± standard deviation of triplicate samples or assays. The statistical analyses were performed using unpaired Student's t-test. A p≤0.05 was considered statistically significant. Isobologram and combination index analyses were performed using CalcuSyn software (Biosoft) (28,29). A CI value of 1 indicates an additive effect, a value of less than 1 indicates synergy, and a value of greater than 1 indicates antagonism. The average CI values were calculated at different effect levels (50% effective concentration ED50, ED75, and ED90) (30). A two-sided Fisher exact test was used to determine statistical significance between different groups.
Results
Sorafenib resistance reveals distinct mutational profiles in FLT3 TKDs
We screened for acquired mutations of the FLT3 gene in the sorafenib resistant cell line Ba/F3-ITD-Res developed by long-term exposure of Ba/F3-ITD cells to low doses of sorafenib in vitro, and identified N676D and Y842C mutations in TKD1 and TKD2, respectively (Fig. 1A). Assuming that the point mutations in TKDs might be responsible for the resistance, we further studied samples from FLT3 ITD mutated AML patients who relapsed while on sorafenib. Indeed, in addition to the ITD mutations we identified acquired FLT3 point mutations in 2 of 4 patients, which were also located in the TKDs (i.e., G619C, D651G, and I687F) (Supplementary Table 1). We introduced these mutations individually, or tandem mutations in both TKD1 and TKD2, into the Ba/F3-ITD cells and tested their in vitro sensitivity to sorafenib. Varying degrees of resistance were observed in the sub-lines with single point mutations, and the effective concentration in 50% of the treatment population (EC50, mean ± S.D.) for apoptosis induction was 0.69±0.18, 0.61±0.13, and 0.17 ± 0.02 μmol/L for the cells containing mutations D651G, N676D and I687F, respectively, in TKD1, and 2.6±0.81 μmol/L for the cells containing Y842C mutation in TKD2. By comparison, the EC50 for the parental cells Ba/F3-ITD was 0.006±0.002 μmol/L (Fig. 1B). These results suggested that a mutation in either TKD was sufficient to provide resistance to sorafenib-induced apoptosis, and that a mutational alteration in TKD2 was more important to the acquired resistance compared to an analogous alteration in the TKD1. Furthermore, cells with compound mutations in both TKDs (e.g., the Ba/F3-ITD-Res and the engineered Ba/F3-ITD+676/842 cells) in addition to the ITD mutations displayed even greater resistance (EC50 of 4.2 ± 1.50 and 6.6 ± 0.53 μmol/L, respectively, Fig. 1B), indicating a pivotal role for the integrity of both TKDs in maintaining sensitivity of FLT3-ITD AML cells to sorafenib.
Figure 1.
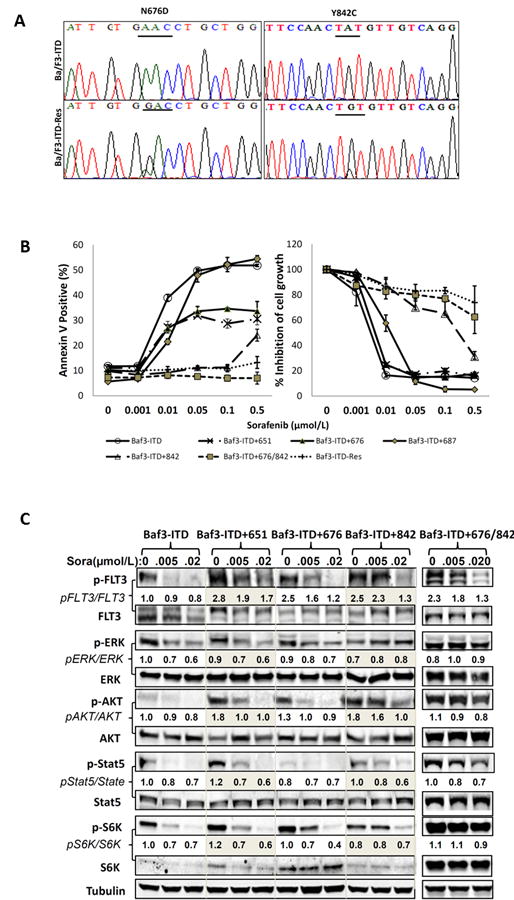
Acquired point mutations of FLT3 TKDs are associated with sorafenib resistance. A, cDNA-based mutation analysis was performed using cDNA sequencing with multiple primers in sorafenib-resistant cells (Ba/F3-ITD-Res) and parental cells Ba/F3-ITD. B, Engineered cells with point mutations were exposed to varying concentrations of sorafenib for 48 hours, and apoptosis induction and cell-growth inhibition were assessed as the percentage of annexin V–positive cells by flow cytometry and by counting the numbers of viable cells using the Trypan blue dye exclusion method, respectively. Growth inhibition was expressed as percentage relative to that in the control group. Data are the mean of three independent determinations. C, Resistant cells and their parental cells Ba/F3-ITD were treated with sorafenib for 2 hours, and phosphorylation levels of FLT3 and its downstream proteins were measured by immunoblot analysis. The ratios are semi-quantitative analyses indicating the expression levels of phosphorylated proteins compared to their total proteins and normalized to the first control lane, which was defined as 1. Sora, sorafenib; p-, phosphorylated.
Relevant biomarkers were investigated in the aforementioned cells to facilitate the understanding of the biological consequences of these mutations. The results illustrated that point mutations in a single TKD augmented the FLT3 activation caused by the ITD mutation. This was accompanied by a modest up-regulation of phosphorylated FLT3 that persisted to varying degrees despite sorafenib treatment (Fig. 1C). The cells harboring TKD2 mutations (e.g., Ba/F3-ITD+842 and Ba/F3-ITD+676/842) were least sensitive to the inhibitory effects of sorafenib, maintaining persistent phosphorylation of FLT3 and its downstream effectors ERK, AKT, Stat5, and S6K after exposure to sorafenib. However, the suppression of phosphorylated FLT3 itself did not parallel those of its downstream effectors, whereby an increased level of phosphorylated ERK was observed in Ba/F3-ITD+842 cells, and persistent phosphorylated-AKT, -Stat5 and -S6K in Ba/F3-ITD+676/842 cells (Fig. 1C). These findings implied that a conformational change of FLT3 protein, resulting from the gene point mutations, may interfere with a binding ability (i.e. accessibility) of sorafenib, and that mutations in TKD2 are more critical for impairing the binding of sorafenib than those occurring in TKD1. Moreover, these results also suggested that a bypass/feedback regulation could be an effective approach in the modulation of FLT3 downstream signaling.
Sorafenib resistant cells retain sensitivity to type I TK inhibitors
We next assessed the sensitivity of either TKD1 and/or TKD2 point mutations to various small molecule FLT3 inhibitors. AC220 (also known as quizartinib), a potent apoptogenic agent in leukemia cells (31), was first tested in our resistant cell lines. AC220 demonstrated a similar pattern of resistance as observed with sorafenib treatment (Fig. 2A). Since both sorafenib and AC220 are type II TK inhibitors, we further tested cell sensitivity by treating these cells with a type I TK inhibitor PKC412 as well as another type II TK inhibitor MLN518. The results demonstrated that all resistant cells retained sensitivity to PKC412 at sub-micromolar concentrations. However, MLN518 was only active in cells with TKD1 mutations, not in the cells with TKD2 or compound TKDs mutations (Fig. 2B). This further suggested that mutations in TKD2 (such as Y842C) might directly interfere with the binding ability of type II TK inhibitors while retaining sensitivity to type I TK inhibitors. A similar mechanism has been suggested in cells with acquired AC220 resistance, but retaining sensitivity to PKC412 (32).
Figure 2.
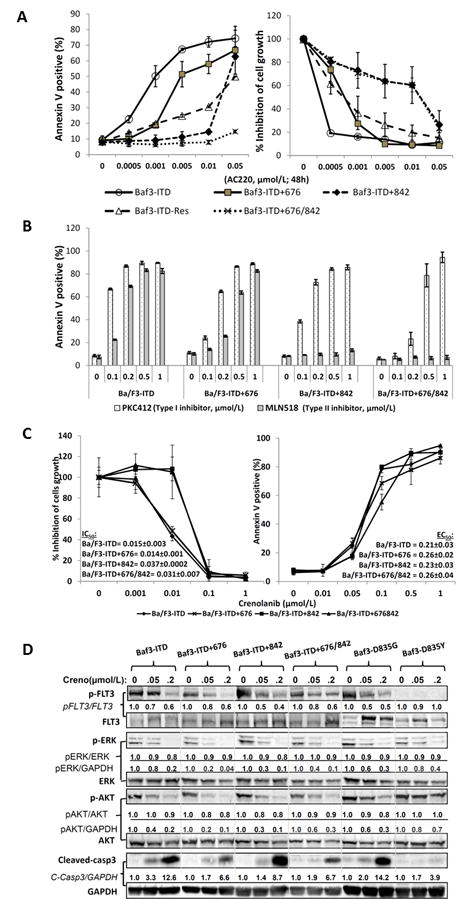
Type I kinase inhibitors retain sensitivity to sorafenib-resistant cells. A, Sorafenib-resistant cells were exposed to the indicated concentrations of AC220 for 48 hours and apoptosis induction and cell-growth inhibition were measured by flow cytometry and Trypan blue dye exclusion method, respectively. B, Sensitivity of resistant cells and their parental Ba/F3-ITD cells to the indicated inhibitor was assessed as described in Fig. 1. C, Sensitivity of novel type I inhibitor crenolanib to sorafenib-resistant cells was assessed by exposing the cells in crenolanib for 72 hours as described in Fig. 1B. D, Phosphorylation levels of FLT3 and its downstream proteins ERK and AKT were determined by immunoblot analysis after 24 hours of treatment with crenolanib. The ratios of semi-quantitative analyses indicated the expression levels of phosphorylated proteins to their respective total proteins or housekeeping protein GAPDH.
The novel type I TK inhibitor crenolanib (CP-868,596) has recently been reported to retain activity against the imatinib-resistant PDGFR-α D842V mutation by targeting the active conformation of a kinase ATP pocket in gastrointestinal stromal tumor (GIST) (33). In the present study, crenolanib demonstrated impressive apoptosis induction in all tested sorafenib-resistant cell lines at sub-micromolar concentrations and, furthermore, the concentrations required for cell growth inhibition were 10-fold lower than that required for apoptosis induction (Fig. 2C). We also tested crenolanib in AML cells harboring FLT3-D835 mutation, which is commonly found in patients with acquired resistance to FLT3 inhibitors. Interestingly, crenolanib induced profound pro-apoptotic effects in Ba/F3-D835G cells (Supplementary Fig. 2).
With the exception of the Ba/F3-D835Y cells, immunoblot analysis revealed that very low concentrations (i.e., 0.05 and 0.2 μmol/L) of crenolanib exerted a marked suppression of FLT3 phosphorylation, as well as that of its downstream effectors ERK and AKT, and induced caspase-3 cleavage in the sorafenib resistant cells (Fig. 2D). Interestingly, Ba/F3-D835Y displayed very low basal levels of phosphorylated FLT3 at the Tyr589/591 sites, which may limit the effectiveness of crenolanib in targeting FLT3. These findings indicated a striking potency of crenolanib in targeting FLT3-ITD cells with acquired point mutations of the TKDs, especially in those with TKD2 mutations.
Combinations of type I and type II TK inhibitors or two type I TK inhibitors exert synergistic anti-leukemic effects in sorafenib-resistant cells
We next assessed the impact of combining the type I TK inhibitor crenolanib with the type II TK inhibitor sorafenib in sorafenib-resistant cells. This combination showed profound synergistic pro-apoptotic effects even at 10-fold lower concentrations than the growth inhibitory concentrations of crenolanib as single agent treatment. The combination indices (CIs) were 0.70±0.01, 0.67±0.03, 0.56±0.12, and 0.82±0.08 in Ba/F3-ITD-RES, Ba/F3-ITD+842, Ba/F3-ITD+676/842, and Ba/F3-ITD cells, respectively (Fig. 3A, Supplementary Fig. 3). Immunoblot analysis demonstrated that the combination enhanced the suppression of FLT3 downstream proteins ERK and AKT, especially in the Ba/F3-ITD-RES cells after a one-hour treatment, but showed modest suppression of phosphorylated FLT3 itself (Fig. 3B). This data imply that the synergistic effects resulted not only from FLT3 suppression but also from blockade of other signaling events involved in the modulation of these pathways, at least in more resistant cell Ba/F3-ITD-RES.In addition, our in vitro kinase assay further demonstrated that these agents were capable of directly targeting the FLT3 protein and suppressing its phosphorylation (Fig. 3C).
Figure 3.

Combination of type I and type II kinase inhibitors or two type I inhibitors shows synergistic pro-apoptotic effects in sorafenib-resistant FLT3-mutant cell lines. A, The sorafenib-resistant and parental cells were treated with a combination of crenolanib and sorafenib at indicated concentrations for 48 hours. Apoptosis induction was determined as described in Fig. 1B. **p< 0.01, ***p< 0.001. B, The modulation of correlative proteins was determined by using immunoblot analysis after 1 hour of single-agent/combination treatments. C, FLT3 with ITD plus Y842C mutant protein was isolated using immunoprecipitation (IP) with anti-FLT3 antibodies and in vitro kinase assay was performed in the presence/absence of crenolanib (0.5 μmol/L) and/or sorafenib (0.5 μmol/L). Phosphorylation levels of FLT3 were determined by anti-phospho-FLT3 antibodies using immunoblotting. D, Sorafenib-resistant cells FLT3-ITD+676/842 were treated with indicated single or multiple agents for 48 hours. Apoptosis induction was determined as described in Fig. 1B. creno: crenolanib; *p< 0.05; **p< 0.01; ***p< 0.001.
To further evaluate if this synergistic effect was exclusive for the combination of sorafenib and crenolanib, the Ba/F3-ITD+676/842 cells were exposed to different combinations of type I and type II TK inhibitors. The results indicated that each type I/type II combination (e.g., crenolanib plus sorafenib, crenolanib plus MLN518, or PKC412 plus sorafenib) exerted identical synergistic pro-apoptotic effects in the resistant cells (Fig. 3D, Supplementary Fig. 4).Interestingly, the combination of two type I TK inhibitors (i.e., crenolanib and PKC412) also triggered a synergistic effect in cells that were resistant to the type II TK inhibitor sorafenib. However, the synergistic effect was not apparent in a combination of two type II TK inhibitors (i.e., sorafenib and MLN518) (Fig. 3D), implying that cells resistant to type II TK inhibitors might still retain sensitivity to a type I TK inhibitor. Of note, these TKI combinations did not show significant anti-leukemic effects in the Ba/F3-FLT3 wide-type cells, even when increasing the drug concentration up to micromolar levels (Supplementary Fig. 5), suggesting that the cytotoxic effect of the combinations was chiefly in the FLT3 mutated AML cells.
Concomitant inhibition of FLT3 and its downstream signaling pathways enhances anti-leukemic effects in sorafenib-resistant cells
Our data demonstrate that sorafenib exhibited impaired ability to suppress phosphorylated ERK or AKT/S6K in the resistant Ba/F3-ITD+842 or Ba/F3-ITD+676/842 cells, but still retained effectiveness, to certain degree, in the suppression of phosphorylated FLT3. This would imply a possible feedback up-modulation of MEK/ERK and/or AKT/S6K signaling pathways in resistant cells. Therefore, we examined the concomitant blockade of FLT3 and ERK/AKT signaling pathways. When we combined sorafenib with the MEK inhibitor CI1040, there was a synergistic pro-apoptotic effect in the sorafenib-resistant cell lines.Less synergistic effect, although statistically significant, could be observed in combination of sorafenib with the mTOR antagonist CCI779 by comparison to the single agent(s) treatments. Triple combination of sorafenib/CI1040/CCI779 triggered marked synergistic pro-apoptotic effect (Fig. 4A and 4B). This was accompanied by a profound suppression of phospho-ERK, -AKT, and -S6K and the upregulation of the pro-apoptotic BH3-only protein Bim (Fig. 4C). These results illustrated that the concomitant inhibition of FLT3, and its downstream targets MEK/ERK, could produce a potentially clinical beneficial result by the killing of sorafenib-resistant cells.
Figure 4.
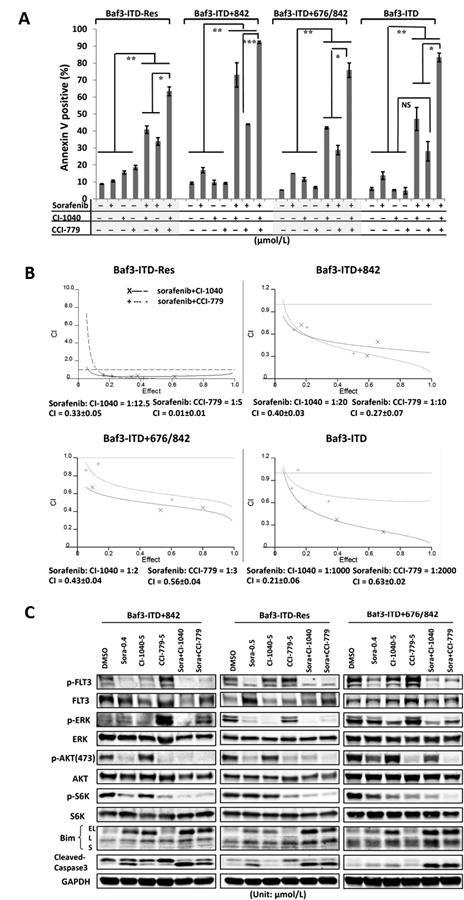
Combination of sorafenib with CI-1040 or CCI-779 induces synergistic pro-apoptotic effects and overcomes sorafenib resistance. A, The sorafenib-resistant and parental cells were treated with combinations of sorafenib with CI-1040 and/or CCI-779 at indicated ratios for 48 hours. DMSO served as a control. Apoptosis induction was determined as described in Fig. 1B. *p< 0.05; **p< 0.01; ***p< 0.001; NS, no significance. B, Isobologram and combination index (CI) analyses were performed using CalcuSyn software (Biosoft). C, The modulation of correlative proteins was determined using immunoblot analysis after 24 hours of single-agent/combination treatments. Bim-EL, extra-long; L, long; and S, short.
Combination of crenolanib with sorafenib demonstrates profound pro-apoptotic effects in FLT3 ITD mutated primary human AML cells ex vivo
Next, we examined the combination effects of sorafenib and crenolanib in FLT3 ITD/D835 mutated primary human AML cells. Two of four the patients received TKIs therapy before current ex vivo study and became to relapsed/refractory (Supplementary Table 1). Synergistic or additive pro-apoptotic effects could be achieved in the AML samples at physiologically-achievable drug concentrations (34,35) (Fig. 5A). Interestingly, this combination had very limited cytotoxicity in normal bone marrow samples or FLT3 wild type AML patient samples (Supplementary Fig. 6, 7), implying specificity for targeting FLT3 ITD mutated AML patients with the combination strategy. Immunoblotting showed the suppression of FLT3 phosphorylation, as well as that of its downstream effectors ERK and AKT, which was observed in either the crenolanib or sorafenib single agent treatments. However, a substantial suppression of these phosphorylated proteins was also observed in the combination treatment (Fig. 5B), suggesting the synergistic suppression of FLT3 and its downstream targets.
Figure 5.

Combination of crenolanib with sorafenib demonstrates synergistic/additive pro-apoptotic effects in primary human AML cells with FLT3 ITD mutations in vitro. A, Primary AML mononuclear cells were exposed to the indicated agents for 48 hours and apoptosis induction was determined as described in Fig. 1B after gating on CD34 or CD33 positive populations. CIs, combination indices; SE, Standard error. B, Relevant proteins were analyzed by immunoblot analysis after treating the primary AML cells with indicated agents for 16 hours. The ratios of semi-quantitative analyses indicated the expression levels of phosphorylated proteins to their respective total proteins or housekeeping protein GAPDH.
FLT3 ligand-mediated resistance to sorafenib-induced apoptosis can be overcome by combination therapies
An increase in FLT3 ligand (FL) as a cellular response to chemotherapy has been reported as a cause of resistance to FLT3-targeted therapy (36). We assumed that FL might activate FLT3 signaling and its downstream cascade signaling MEK/ERK or mTOR/S6K in our AML cell systems. Thus, we first tested the effects of the sorafenib or/and crenolanib in FLT3-ITD mutated cells in the presence or absence of FL. The results showed that apoptosis induction by low doses of either single agent (i.e., sorafenib or crenolanib), or their combination could be abrogated by the presence of 25 ng/mL FL (Fig. 6A). However, apoptosis induction could be partly retained by increasing the drug concentrations to 0.01μmol/L sorafenib or 0.1 μmol/L crenolanib for a 48-hour time period. Impressively, the combination of sorafenib and crenolanib at these concentrations demonstrated marked apoptosis induction even in the presence of FL at high levels (i.e., 50 ng/mL) (Fig. 6B).
Figure 6.
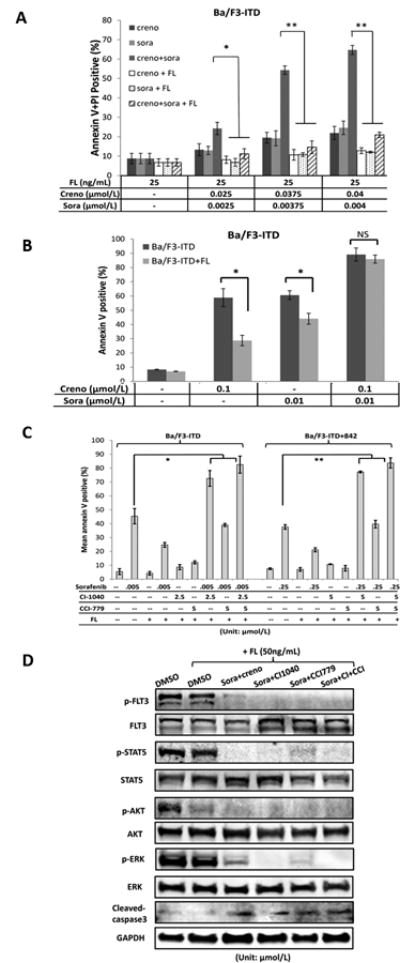
The presence of FLT3 ligand protects FLT3 inhibitors-induced cell apoptosis, which can be overcome by combinatorial treatment of crenolanib and sorafenib in sub-micromolar concentrations. Ba/F3-ITD cells were treated with indicated concentrations of crenolanib or/and sorafenib in the presence/absence of (A) 25 ng/mL or (B) 50 ng/mL FL for 48 hours and apoptosis induction was measured as described in Fig 1B. (C) Ba/F3-ITD and Ba/F3-ITD+842 cells were treated with combinations of sorafenib with CI-1040 and/or CCI-779 in the presence/absence of FL (25 ng/mL) for 48 hours. DMSO served as a negative control. Apoptosis induction was determined by flow cytometry after staining with annexin V. FL, FLT3-ligand; *p<0.05; **p<0.01. (D) Ba/F3-ITD cells were pretreated with FL (50 ng/mL) for 30 min and treated with combination of sorafenib (0.01 μM) and crenolanib (0.1 μM), CI1040 (2.5 μM), or CCI779 (5 μM), respectively, for additional 6 hours. DMSO served as a control. The cells were lysed and phosphorylation levels of FLT3 and its downstream proteins were determined using immunoblot analysis. FL, FLT3-ligand; Sora, sorafenib; creno, crenolanib; CI, CI1040 and CCI, CCI779.
We further tested the effects of combining sorafenib with the MEK inhibitor CI1040 or/and the mTOR inhibitor CCI779. The results showed that co-targeting FLT3-ITD/MEK with sorafenib/CI1040 had a significant synergistic pro-apoptotic effect in the Ba/F3-ITD and Ba/F3-ITD+842 resistant cells despite presence of FL.In turn, co-targeting FLT3-ITD/mTOR with sorafenib/CCI779 showed only limited apoptosis induction in the cells in the presence of FL at 25 ng/mL concentration (Fig. 6C).Immunoblot analysis showed that high phosphorylation levels of FLT3, ERK, and Stat5, but not AKT, could be retained in the presence of FL; in turn, the combination of sorafenib with crenolanib, CI1040 or CI1040/CCI779 profound suppressed phospho-FLT3, its downstream proteins, and induced caspase-3 cleavage after a 6-hour treatment (Fig. 6D). Our findings suggest that these combinations, using the type I/type II inhibitors or co-targeting FLT3 and MEK signaling, could effectively overcome FL-mediated resistance.
Discussion
The efficacy of AML therapy with FLT3 inhibitors is limited by the development of resistance even with next-generation FLT3 inhibitors AC220 (11,12,37). Many point mutations located at either TKD1 (such as N676, F691, and G697) or TKD2 (such as D835 and Y842 in activation loop) have been reported after treatment with various FLT3 inhibitors (excluding sorafenib) in vitro and in vivo and were related to leukemic cell resistance (13,20,22,32). Conversely, no point mutations were detected in two AML patients who reportedly relapsed after an initial response to sorafenib therapy (24), and one study reported that D835Y mutations were detected in 4 of 7 AML patients with FLT3-ITD mutations after sorafenib treatment (38). Although the mutation sites identified in the present study differ from D835Y, they all are located in the FLT3 TKD and several mutations of this type have been observed in patients relapsed during treatment with other FLT3 inhibitors. This would imply instability of these TKDs that are invariably under “selective pressure” by FLT3 inhibition in leukemic cells. Alternatively, these point mutations are too rare to be detected before therapy, and after suppressing the FLT3-ITD allele by FLT3-inhibition, the resistant alleles could conceivably become dominant in the surviving AML clones.
Our engineered cell lines harboring point mutations of TKDs demonstrated varying degrees of resistance to sorafenib, suggesting that mutations of the TKDs are critical for sorafenib resistance. Furthermore, AML cells with TKD2 mutations (i.e., Y842C) showed more resistance to sorafenib compared to those with TKD1 mutations. Cells harboring compound TKD mutations demonstrated even greater resistance to sorafenib implying that conformational changes in the TKD2 might alter sorafenib's binding affinity more than those associated with TKD1, and that mutations of both TKDs might result in further anomalous conformational changes and increased resistance.
Sorafenib-resistant patients with acquired AML point mutations also had a longer length of ITD mutations than those without point mutations (Supplementary Table 1), suggesting instability of the ITD mutated gene may be associated with the length of ITD mutations. Schnittger's group reported that the patients with longer FLT3-ITD mutations had a markedly shorter event-free survival (7.4 versus 12.6 months) (39). This would suggest the possibility that the patients with longer ITD mutations might easily acquire additional point mutations and become resistant to therapy. Since this investigation examined relatively few patient samples, further studies are required to validate this hypothesis.
It was highly likely that sorafenib-resistant cells may display resistance to other type II TK inhibitors. Indeed, our results demonstrate that sorafenib-resistant cells have very similar resistance patterns compared to other type II inhibitors like AC220 and MLN518. However, they retain sensitivity to type I inhibitors like PKC412 and crenolanib. Furthermore, a combination of two type I inhibitors (i.e., crenolanib and PKC412), but not of two type II inhibitors (i.e., sorafenib and MLN518) showed impressive synergistic pro-apoptotic effects in sorafenib-resistant cells Ba/F3-ITD+676/842. Since our in vitro screening for resistant mutations was performed in cells exposed to the type II inhibitor sorafenib, it is reasonable to assume that the mutation-mediated conformational change in FLT3 might impair the accessibility of other type II inhibitors, but retains binding ability with type I inhibitors.Albers and his colleagues also reported an AML patient with acquired point mutation at F691L who developed resistance to Type II kinase inhibitor AC220, but retained sensitivity to type I inhibitor PKC412 (32). This result supports our findings, i.e., the cells resistant to type II kinase inhibitor may remain sensitive to type I kinase inhibitors. On the other hand, Weisberg et al reported that the point mutations of D835Y (or N676K) which developed after therapy with type I inhibitor PKC412, retained sensitivity to highly selective second generation type II inhibitor NVP-AST487 (40), although these mutant cells exhibited resistance to less specific type II inhibitor sorafenib (41). Altogether, these findings strongly suggest that resistance developed by one type kinase inhibitor could be overcome by another type of kinase inhibitor.
The combination of the type I inhibitor crenolanib with a type II inhibitor (i.e., either sorafenib or MLN518) triggered a profound pro-apoptotic effect in the sorafenib-resistant cell line Ba/F3-ITD+676/842, and showed synergistic/additive pro-apoptotic effects in relapsed AML patient samples with FLT3-ITD and/or D835 mutations although some of these patients showed relapsed or refractory to other single TKI therapy (such as PKC412 or AC220). Mechanistically, this seemingly occurred by the suppression of phosphorylation levels of FLT3 and its downstream signaling intermediates. During there vision of this manuscript, Zimmerman and her colleagues also reported a synergistic anti-leukemia effect by combining type I inhibitor crenolanib with type II inhibitor sorafenib in their murine leukemia model which was engrafted with FLT3 ITD mutated cells MV4-11 (42). Taken together, these findings suggest that combination of type I and type II tyrosine kinase may provide a promising strategy for targeting relapsed/refractory AML patient and also be beneficial for preventing resistance of FLT3 targeted therapy by reducing doses and enhancing therapeutic efficacy.
Our data also demonstrate that the suppression of FLT3 activation was not sufficient for inducing apoptosis of leukemia cells, at least in our sorafenib-resistant cells Ba/F3-ITD+842 and Ba/F3-ITD+676/842, which demonstrated a FLT3-independent high activation of ERK and/or AKT (Fig. 1C). This would suggest that these downstream proteins themselves could also be modulated by other signaling pathways and play a pivotal role in maintaining the leukemia cell survival. In fact, aberrant activation of ERK or AKT could be frequently observed in co-culture with bone marrow stromal cells (43) or in hypoxia environment in our resistant cells (Supplementary Fig. 8). Therefore, a strategy for co-targeting FLT3 and MEK/ERK or AKT/mTOR signaling pathways simultaneously could enhance anti-leukemic effects. However, the combination of sorafenib with MEK inhibitor CI1040 showed more impressive anti-leukemic effect than that of sorafenib with mTOR antagonist CCI779 in all tested sorafenib-resistant cells, even in the presence of high concentrations of FL, suggesting that concomitant inhibition of FLT3 and ERK activation might be a potent strategy for overcoming FLT3-inhibition resistance.
In summary, our findings provide an improved understanding of the development of resistance of FLT3-ITD-mutated AML cells. They also extend our knowledge of the role of acquired point mutations in TKDs that are associated with resistance to FLT3 inhibition. The use of combinatorial strategies in this scenario produced effective anti-leukemia effects in most types of sorafenib-resistant cells, which may benefit the design of future clinical trials for the therapy of AML.
Supplementary Material
Translational Relevance.
This study was conducted to determine the effect of acquired FLT3 gene mutations in sorafenib resistance, and to test drug treatment strategies for overcoming this resistance. We here identify acquired point mutations in TKD regions of the FLT3 gene in sorafenib resistant AML cells harboring FLT3-ITDs. The mutations in TKDs were also identified in samples from AML patients who relapsed following sorafenib therapy. We further investigated the effectiveness of combination treatments targeting different TKDs of the FLT3 protein, or different signaling pathways, simultaneously, to overcome sorafenib resistance. Our data demonstrate that combination treatments using type I and type II TK inhibitors, or the simultaneous targeting of FLT3 and its downstream signaling pathways, synergistically induces apoptosis in sorafenib-resistant cell lines. These findings are anticipated to be clinically important with respect to the long-term management of AML as well as enhance our understanding of resistance to FLT3 targeted therapy.
Acknowledgments
The authors would like to thank Wenjing Chen and Sheela V. Mathews who provided follow-up data for this study. Sunita Patterson and Numsen Hail, Jr. provided critical reviews and editorial assistance in preparation of this manuscript.
Grant Support: This work was supported in part by grants from NIH/NCI (CA143805, CA100632, CA055164, and CA049639) and a Cancer Center Support Grant (CA 16672) (to M.A.), and a Leukemia SPORE Career Development Award (CA100632-05) (to W.Z.).
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Author's Contributions: W.Z. contributed to experimental design, performed experiments, analyzed data, and prepared the manuscript; C.G performed drug treatment experiments and plasmid preparation; M.K. contributed to experimental design and data analysis; Y.C. and R.O.J. contributed to lentivial infection; G.B. contributed to patient information analysis and follow-up; J.E.C and F.R. provided patient samples; A.R. contributed to data analysis and provided crenolanib; and M.A. contributed to conceptual design, experimental design, and editing the manuscript.
References
- 1.Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–8. [PubMed] [Google Scholar]
- 2.Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–9. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
- 3.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–42. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 4.Wodicka LM, Ciceri P, Davis MI, Hunt JP, Floyd M, Salerno S, et al. Activation state-dependent binding of small molecule kinase inhibitors: structural insights from biochemistry. Chem Biol. 2010;17:1241–9. doi: 10.1016/j.chembiol.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Traxler P, Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther. 1999;82:195–206. doi: 10.1016/s0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- 7.Kufareva I, Abagyan R. Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J Med Chem. 2008;51:7921–32. doi: 10.1021/jm8010299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–64. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100:184–98. doi: 10.1093/jnci/djm328. [DOI] [PubMed] [Google Scholar]
- 10.Borthakur G, Kantarjian H, Ravandi F, Zhang W, Konopleva M, Wright JJ, et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica. 2011;96:62–8. doi: 10.3324/haematol.2010.030452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, Konopleva MY, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28:1856–62. doi: 10.1200/JCO.2009.25.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma M, Ravandi F, Bayraktar UD, Chiattone A, Bashir Q, Giralt S, et al. Treatment of FLT3-ITD-Positive Acute Myeloid Leukemia Relapsing after Allogeneic Stem Cell Transplantation with Sorafenib. Biol Blood Marrow Transplant. 2011;17:1874–7. doi: 10.1016/j.bbmt.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–3. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeAngelo DJ, Stone RM, Heaney ML, Nimer SD, Paquette RL, Klisovic RB, et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108:3674–81. doi: 10.1182/blood-2006-02-005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management. Am J Hematol. 2013;88:318–27. doi: 10.1002/ajh.23404. [DOI] [PubMed] [Google Scholar]
- 16.Knapper S, Burnett AK, Littlewood T, Kell WJ, Agrawal S, Chopra R, et al. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood. 2006;108:3262–70. doi: 10.1182/blood-2006-04-015560. [DOI] [PubMed] [Google Scholar]
- 17.Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- 18.Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML) Blood. 2009;114:2984–92. doi: 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fathi AT, Chabner BA. FLT3 inhibition as therapy in acute myeloid leukemia: a record of trials and tribulations. Oncologist. 2011;16:1162–74. doi: 10.1634/theoncologist.2011-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- 21.von BN, Engh RA, Aberg E, Sanger J, Peschel C, Duyster J. FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res. 2009;69:3032–41. doi: 10.1158/0008-5472.CAN-08-2923. [DOI] [PubMed] [Google Scholar]
- 22.Williams AB, Nguyen B, Li L, Brown P, Levis M, Leahy D, et al. Mutations of FLT3/ITD confer resistance to multiple tyrosine kinase inhibitors. Leukemia. 2012 doi: 10.1038/leu.2012.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore AS, Faisal A, de Castro DG, Bavetsias V, Sun C, Atrash B, et al. Selective FLT3 inhibition of FLT3-ITD(+) acute myeloid leukaemia resulting in secondary D835Y mutation: a model for emerging clinical resistance patterns. Leukemia. 2012;26:1462–70. doi: 10.1038/leu.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scholl S, Spies-Weisshart B, Klink A, Muegge LO, Fricke HJ, Hochhaus A. Secondary resistance to sorafenib in two patients with acute myeloid leukemia (AML) harboring FLT3-ITD mutations. Ann Hematol. 2011;90:473–5. doi: 10.1007/s00277-010-1027-9. [DOI] [PubMed] [Google Scholar]
- 25.Tse KF, Allebach J, Levis M, Smith BD, Bohmer FD, Small D. Inhibition of the transforming activity of FLT3 internal tandem duplication mutants from AML patients by a tyrosine kinase inhibitor. Leukemia. 2002;16:2027–36. doi: 10.1038/sj.leu.2402674. [DOI] [PubMed] [Google Scholar]
- 26.Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, et al. Phase II study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121:4655–62. doi: 10.1182/blood-2013-01-480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W, McQueen T, Schober W, Rassidakis G, Andreeff M, Konopleva M. Leukotriene B4 receptor inhibitor LY293111 induces cell cycle arrest and apoptosis in human anaplastic large-cell lymphoma cells via JNK phosphorylation. Leukemia. 2005;19:1977–84. doi: 10.1038/sj.leu.2403929. [DOI] [PubMed] [Google Scholar]
- 28.Konopleva M, Milella M, Ruvolo P, Watts JC, Ricciardi MR, Korchin B, et al. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERK-activated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26:778–87. doi: 10.1038/leu.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Zhang W, Konopleva M, Burks JK, Dywer KC, Schober WD, Yang JY, et al. Blockade of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase and murine double minute synergistically induces Apoptosis in acute myeloid leukemia via BH3-only proteins Puma and Bim. Cancer Res. 2010;70:2424–34. doi: 10.1158/0008-5472.CAN-09-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao L, Wientjes MG, Au JL. Evaluation of combination chemotherapy: integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin Cancer Res. 2004;10:7994–8004. doi: 10.1158/1078-0432.CCR-04-1087. [DOI] [PubMed] [Google Scholar]
- 31.Gunawardane RN, Nepomuceno RR, Rooks AM, Hunt JP, Ricono JM, Belli B, et al. Transient exposure to quizartinib mediates sustained inhibition of FLT3 signaling while specifically inducing apoptosis in FLT3-activated leukemia cells. Mol Cancer Ther. 2013;12:438–47. doi: 10.1158/1535-7163.MCT-12-0305. [DOI] [PubMed] [Google Scholar]
- 32.Albers C, Leischner H, Verbeek M, Yu C, Illert AL, Peschel C, et al. The secondary FLT3-ITD F691L mutation induces resistance to AC220 in FLT3-ITD(+) AML but retains in vitro sensitivity to PKC412 and Sunitinib. Leukemia. 2013;27:1416–8. doi: 10.1038/leu.2013.14. [DOI] [PubMed] [Google Scholar]
- 33.Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, et al. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:4375–84. doi: 10.1158/1078-0432.CCR-12-0625. [DOI] [PubMed] [Google Scholar]
- 34.Lewis NL, Lewis LD, Eder JP, Reddy NJ, Guo F, Pierce KJ, et al. Phase I study of the safety, tolerability, and pharmacokinetics of oral CP-868,596, a highly specific platelet-derived growth factor receptor tyrosine kinase inhibitor in patients with advanced cancers. J Clin Oncol. 2009;27:5262–9. doi: 10.1200/JCO.2009.21.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takimoto CH, Awada A. Safety and anti-tumor activity of sorafenib (Nexavar) in combination with other anti-cancer agents: a review of clinical trials. Cancer Chemother Pharmacol. 2008;61:535–48. doi: 10.1007/s00280-007-0639-9. [DOI] [PubMed] [Google Scholar]
- 36.Sato T, Yang X, Knapper S, White P, Smith BD, Galkin S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011;117:3286–93. doi: 10.1182/blood-2010-01-266742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levis MJ. Will newer tyrosine kinase inhibitors have an impact in AML? Best Pract Res Clin Haematol. 2010;23:489–94. doi: 10.1016/j.beha.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Man CH, Fung TK, Ho C, Han HH, Chow HC, Ma AC, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119:5133–43. doi: 10.1182/blood-2011-06-363960. [DOI] [PubMed] [Google Scholar]
- 39.Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- 40.Weisberg E, Roesel J, Bold G, Furet P, Jiang J, Cools J, et al. Antileukemic effects of the novel, mutant FLT3 inhibitor NVP-AST487: effects on PKC412-sensitive and -resistant FLT3-expressing cells. Blood. 2008;112:5161–70. doi: 10.1182/blood-2008-02-138065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams AB, Li L, Nguyen B, Brown P, Levis M, Small D. Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood. 2012;120:3069–79. doi: 10.1182/blood-2012-01-403493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zimmerman EI, Turner DC, Buaboonnam J, Hu S, Orwick S, Roberts MS, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122:3607–15. doi: 10.1182/blood-2013-07-513044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang X, Sexauer A, Levis M. Bone marrow stroma-mediated resistance to FLT3 inhibitors in FLT3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br J Haematol. 2013 doi: 10.1111/bjh.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.