

Abstract
The circadian clock regulates biological processes from gene expression to organism behavior in a precise, sustained rhythm that is generated at the unicellular level by coordinated function of interlocked transcriptional feedback loops and post-translational modifications of core clock proteins. CLOCK phosphorylation regulates transcriptional activity, cellular localization and stability; however little is known about the specific residues and enzymes involved. We have identified a conserved cluster of serines that include, Ser431, which is a prerequisite phosphorylation site for the generation of BMAL dependent phospho-primed CLOCK and for the potential GSK-3 phosphorylation at Ser427. Mutational analysis and protein stability assays indicate that this serine cluster functions as a phospho-degron. Through the use of GSK-3 activators/inhibitors and kinase assays, we demonstrate that GSK-3β regulates the degron-site by increasing CLOCK phosphorylation/degradation, which correlates with an increase in the expression of CLOCK responsive promoters. Stabilization of phospho-deficient CLOCK delays the phase of oscillation in synchronized fibroblasts. This investigation begins the characterization of a complex phospho-regulatory site that controls the degradation of CLOCK, a core transcription factor that is essential for circadian behavior.
Keywords: circadian, CLOCK, GSK-3β, phospho-degron, post-translational modification
Introduction
The intrinsic circadian clock regulates biological processes from gene expression to organism behavior.1 This remarkably precise, sustained, circadian rhythm is generated at the unicellular level allowing for a cell culture approach to the dissection of clock molecular mechanisms. Genetic studies have revealed a network of clock genes that function as transcription-translation feedback loops.2 In mammals, a heterodimer of two basic helix-loop-helix/PAS domain-containing transcription factors, CLOCK and BMAL1, bind to E-box elements and allow for the rhythmic expression of clock genes including Per and Cry. Increasing levels of PER/CRY protein promote translocation to the nucleus where a repressor complex is formed with CLOCK/BMAL1, thereby completing a negative feedback loop. Numerous studies indicate that temporal changes in clock protein post-translational modifications, such as phosphorylation, allow for altered protein-protein interactions, subcellular localization, protein activity and stability, which together provide an important mechanism for fine tuning the 24 rhythmicity of gene expression.3
Similar to other circadian proteins, CLOCK is subject to phosphorylation that displays circadian oscillations in vivo.4 Phosphorylation of CLOCK is triggered by interaction with its transcriptional partner, BMAL1.5, 6 To date, only PKG and PKC have been reported to phosphorylate CLOCK; 7, 8 however their specific phosphorylation sites have not been identified. A recent mass-spectrometry–based study identified Ser38, Ser42 and Ser427 as in vivo phosphorylation sites for CLOCK by yet unknown kinases.9 While the significance of Ser427 has not been reported, Ser38 and Ser42 were shown to regulate nuclear localization and DNA binding. 9
Here we report the identification of a phospho-degron, a phospho-regulatory region that controls CLOCK stability. This region includes a classical GSK-3 consensus site at Ser427 with Ser431 representing the phospho-priming site, which is BMAL –dependent and is a prerequisite for subsequent phosphorylation at Ser427. We provide experimental evidence that CLOCK is a direct substrate for GSK-3-mediated phospho-degradation. Functional assays demonstrated that the CLOCK phospho-degron is an important regulator of transcriptional activity, and consequently of the basal circadian parameters of the molecular clock.
RESULTS
A conserved cluster of phospho-serines mediates BMAL-dependent CLOCK phosphorylation
In order to identify important functional domains of CLOCK, we performed a protein blast search using STAR-DNA proteomic server, and determined CLOCK amino acids 425-461 to be highly conserved across species and within homolog, NPAS2 (Fig. 1A). This stretch of residues contains 52% serines and threonines, which suggests that it controls CLOCK function through phosphorylation. Further use of proteomic servers, NET- PHOSK and NET-PHOS, allowed for a prediction of specific kinases and their specific phospho-residues, respectively. Ser437 is the most probable phosphorylated residue in CLOCK and many other serines/threonines between residues 425-461 exhibited high phosphorylation probability scores suggesting that this is a hot-spot for kinase activity (Supplementary Fig. 1A,B).
Fig. 1. A conserved cluster of serines mediate BMAL-dependent CLOCK phosphorylation.
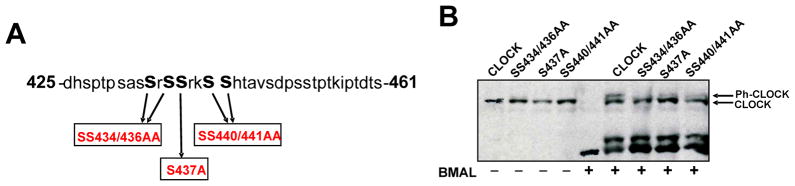
A. The CLOCK amino acid residues 425-461, which includes 14 serines and 4 threonines, are highly conserved across species, as determined by a blast search using STAR-DNA proteomic server. Based on the bioinformatics shown in Supplemental Figures 1A and 1B, bold faced serine (Ser) residues were mutated to alanine. B. Substitution mutants were made by site-directed mutagenesis (Stratagene) using pcDNA plasmid encoding HA-CLOCK as template. The resulting plasmids encoding PD-CLOCK SS434/436AA, S437A, and SS440/441AA were transfected into cells with or without pcDNA-HA-BMAL1 as indicated. Cell lysate loading was adjusted to allow for comparable CLOCK concentrations, which were visualized by anti-HA Western Blot. Arrows depict phosphorylated (Ph-CLOCK) and un-phosphorylated (CLOCK) proteins.
Using site-directed mutagenesis, we generated a set of mutant Clock expression plasmids, in which the conservative-serines were substituted by alanines (Fig. 1A). Western blot analysis of the resulting phospho-deficient (PD) CLOCK proteins SS434/436AA, S437A, and SS440/441AA showed varying degrees of deficiency in phosphorylation (Fig. 1B). At the same time, phosphorylation of BMAL1 was independent of the mutated serine residues since PD-CLOCK mutants are all equally able to induce phosphorylation of BMAL1 (Fig. 1B).
To ensure that the lack of BMAL-mediated CLOCK phosphorylation was not due to loss of interaction of mutant proteins with BMAL1, we preformed co-immunoprecipitation (co-IP) assays using lysates of transfected cells and anti-FLAG (BMAL) antibody. All PD-CLOCK proteins were detected on Western Blot probed with HA (CLOCK)-specific antibody (Fig. 2A, top) A reverse co-IP was performed by using anti-MYC (CLOCK) antibody followed by BMAL detection with anti-HA antibody (Fig. 2A, bottom). Both Co-IP experiments confirmed that BMAL dimerizes with PD-CLOCKs. Western Blot analysis and immunostaining also showed that mutant proteins are translocated to the nucleus upon interaction with BMAL1 (Fig. 2B,C) and therefore have access to nuclear kinases. Together, these data suggest that the identified serine residues are true phosphorylation sites.
Fig. 2. Characterization of phospho-deficient mutant CLOCK proteins.

A. Phospho-deficient CLOCK proteins dimerize with BMAL1. Cells expressing ectopic FLAG-tagged BMAL1 with either HA-tagged PC-CLOCK or individual phospho-deficient mutants were lysed, immunoprecipitated with anti-FLAG antibody and analyzed for CLOCK by Western Blot using HA antibody (top panel). Reverse co-IP was performed with anti-MYC to pull down CLOCK. The HA-tagged BMAL1 associated with this pull down was detected by anti-HA Western Blot (bottom panel).
B. Phospho-deficient CLOCK proteins are translocated to the nucleus in the presence of BMAL1. Cell transfectants expressing various mutant CLOCK proteins individually or in combination with BMAL were fractionated into cytoplasmic and nuclear proteins, resolved by SDS-PAGE and probed with anti-HA antibody. All tested mutant CLOCK proteins are detected in the nucleus, but only phospho-competent CLOCK is phosphorylated in the nuclear fraction.
C. Phospho-competent and phospho-deficient CLOCK proteins showed similar immuno-staining patterns. Cells expressing either wild type PC- or PD-CLOCK proteins showed a predominantly cytoplasmic staining following incubation with primary anti-HA antibody and FITC-tagged secondary antibody; co-expression with Flag-tagged BMAL1 resulted in a predominantly nuclear staining pattern for all CLOCK proteins. A representative image set is shown.
Phosphorylation of serines within the conservative region of CLOCK leads to protein degradation
We and others have previously observed a correlation between CLOCK phosphorylation and degradation.5, 10 Consistent with these observations, our Western Blots showed less protein in cells expressing phospho-competent (PC) CLOCK compared to cells expressing PD mutant proteins, however when phospho-mimic mutants were co-expressed with BMAL1, their abundance was similar to PC-CLOCK (Fig. 3A). The results suggest that a deficiency in phosphorylation within this region leads to protein stabilization. To test whether the loss of CLOCK is the result of protein degradation, 293T cells co-expressing BMAL1 in combination with each of PD-CLOCK mutant proteins were treated at various times with cycloheximide. The results showed that PD-CLOCK proteins degrade slower compared to wild-type CLOCK (Fig. 3B). The data suggest that the identified conserved region between Ser425 and Ser461 represents a phospho-regulatory site, which is involved in phosphorylation-dependent degradation of CLOCK (i.e. “phospho-degron”).
Fig. 3. Identification of a CLOCK phospho-degron.

A. Phosphomimics of CLOCK exhibit stability similar to wild-type CLOCK
293T cells were transfected with Bmal1 cDNA in combination with expression plasmids of either phospho-deficient or phospho-mimic CLOCK mutants as indicated. Resolved CLOCK proteins were detected by anti-HA Western Blot.
B. Phospho-deficient CLOCK mutants are more stable than phospho-competent CLOCK. Cells expressing either PC-CLOCK or PD-CLOCK mutants SS434/436AA and S/T427/429/431/433/434/436/437/440/441A (CLOCKΔ9) were treated with 40ug/ml cycloheximide for the indicated time periods, resolved by SDS-PAGE, and analyzed by Western Blot for CLOCK with anti-HA antibody.
C. Phosphorylation of the degron site is BMAL1-dependent. Wild-type MEFs and MEFs derived from mice with the targeted disruption of Bmal1 gene were transfected with plasmids expressing either PC-CLOCK or PD-CLOCK protein S434/436/437/440/441A and treated with cycloheximide at 3 and 5hrs as indicated. The resulting Western Blot was probed for CLOCK with anti-MYC antibody.
D. Mapping important residues of the phospho-degron site.
Single point mutants of the potential GSK-3 phosphorylation sites Ser427 and Thr429 were generated as well as their putative corresponding phospho-priming sites Ser-431 and Ser433 using myc-Clock cDNA as template. Individual mutants were transfected into 293T cells in combination with BMAL1. The phosphorylation pattern of mutant proteins was visualized by anti-MYC antibody. The amino acid position and phosphorylation pattern of a corresponding mutant are labeled with the same color.
To further confirm the involvement of BMAL1 in phosphorylation-mediated degradation of CLOCK, we compared levels of individual CLOCK proteins ectopically expressed in wild-type MEFs and MEFs derived from mice with a targeted disruption of the Bmal1 gene. In MEF with endogenous BMAL, the level of PC-CLOCK protein was reduced compared to PD-CLOCK proteins, whereas no difference in abundance was observed in Bmal1−/− cells (Fig. 3C). Taken together, these data suggest that BMAL mediates CLOCK degradation through the identified phospho-degron site.
To better define the boundaries of the phospho-degron region, we generated additional mutants of CLOCK, in which serine and threonine residues surrounding Ser434 – Ser441 were substituted with alanine. The subsequent Western Blot showed that the mutants exhibited varying degrees of phosphorylation. Thus, phosphorylation was most significantly impaired in mutant CLOCK proteins SS431/433AA and SS434/436AA whereas CLOCK mutants ST427/429AA and S437A were also phosphorylation deficient, although to a lesser extent (Fig. 3D). Mutations outside of the Ser427 – Ser441 region did not significantly affect the level of CLOCK phosphorylation. These data suggest that individual serine residues within the phospho-degron cluster cooperate and provide differential contribution to the generation of phospho-CLOCK.
The CLOCK phospho-degron is regulated by the activity of RAS/PI3K/AKT/GSK3 pathway
The NetPhosK 1 server predicts that Ser427 and Thr429 are potential sites of GSK-3-mediated phosphorylation (Supplementary Fig. 1B) since they are included in a classical GSK-3 motif ((S/T)XXX(S/T)), which therefore indicate that Ser431 and Ser433 represent potential GSK-3 phospho-priming sites. To test the importance of these residues for CLOCK phosphorylation, we constructed a series of single point mutants (S427A, T429A, S431A, and S433A) and compared their phosphorylation pattern after co-expression with BMAL1 in 293T cells. Western Blot analysis revealed that both Ser427 and Ser431 were required for BMAL-dependent CLOCK phosphorylation. However, while substitution of Ser431 (the potential priming site) with alanine completely blocked CLOCK phosphorylation, a similar mutation of Ser427 (the potential GSK-3 phosphorylation site) only partially blocked phosphorylation (Fig. 3D). These data suggest that phosphorylation of Ser431 is required for subsequent phosphorylation events that generate phospho-primed CLOCK, a substrate for GSK-3 kinase.
To test for a potential physical interaction between GSK-3 and CLOCK, we performed an anti-BMAL co-IP using cell lysates containing Myc-CLOCK and HA-BMAL1 followed by anti-Myc and anti-GSK-3 Western Blots. As shown in Fig. 4A, in addition to CLOCK, anti-BMAL antibody pulled down endogenous GSK-3β. A physical interaction between CLOCK and GSK-3 was further confirmed in complementary experiments, in which cells expressing different combinations of CLOCK, BMAL1 and CRY2 were subject to co-IP with anti-GSK-3 followed by anti-MYC Western Blot for the detection of CLOCK. These results suggest that the dimerization of CLOCK and BMAL allow for an interaction with GSK-3. Greater amount of GSK-3 were pulled-down when CLOCK and BMAL1 were co-expressed with CRY2 (a known substrate of GSK-3) (Fig. 4B).
Fig. 4. CLOCK phospho-degron is regulated by the activity of RAS/PI3K/AKT/GSK3 pathway.

A,B GSK-3β is pulled down in complex with CLOCK and BMAL1
A. 293T cells expressing ectopic CLOCK and BMAL1 were immunoprecipitated with anti-BMAL or normal serum (NS) and analyzed for CLOCK by anti-MYC Western Blot; the same blot was re-probed for the detection of GSK-3. The monoclonal antibody against GSK-3 recognized both alpha and beta forms; GSK-3 α migrated with the IgG heavy chain as denoted by arrows.
B. 293T cells expressing various combinations of clock proteins, as indicated, were immunoprecipitated with anti-GSK-3 antibody followed by CLOCK determination by Western Blot using anti-MYC antibody. Arrows indicate the position of phosphorylated (Ph-CLOCK) and un-phosphorylated CLOCK.
C. GSK-3β phosphorylates CLOCK in vitro. Lysate from cells expressing ectopic CLOCK and BMAL were immunoprecipitated with anti-CLOCK antibody in a stringent RIPA buffer. The immunoprecipitate was incubated in kinase buffer in various combinations of recombinant GSK-3β, BIO, and 32P-ATP followed by incubation at 30 C°. After 1 hr, samples were boiled in SDS buffer, resolved by SDS-PAGE and transferred to nitrocellulose for autoradiogram analysis.
D. Inhibition of GSK-3, via constitutive activation of the RAS/AKT pathway, specifically increases phospho-competent CLOCK levels compared to phospho-deficient CLOCK. 293T cells were transfected with plasmids encoding for BMAL1 and either PC-CLOCK or PD-CLOCK S427/431A, and with either constitutively active RAS or empty vector, as indicated. Cell lysates were analyzed by Western Blot using anti-MYC antibody for the detection of CLOCK.
E. Up-regulation of GSK-3, via inhibition of the RAS pathway, destabilizes CLOCK. 293T cells expressing CLOCK, BMAL and RAS or empty vector, pcDNA3 were treated with either DMSO or PI3K/AKT inhibitor, LY294002 as indicated. A non-specific (n.s.) protein is shown as an indicator of loading.
F. GSK-β3 mediates CLOCK phosphorylation in cell culture. Cells expressing ectopic CLOCK and RAS were co-expressed with either active (CA) or inactive (KD) GSK-3β, as indicated. Cells were treated 3.5 hours with MG132 prior to harvest. CLOCK was visualized by anti-MYC Western Blot.
G. GSK-3β fails to phosphorylate phospho-mutant CLOCK S427A. Cell extract were prepared and analyzed as described in Fig. 4F.
To test whether GSK-3β directly phosphorylates CLOCK, we performed an in vitro kinase assay using recombinant GSK-3β and IP-CLOCK as a substrate. The results showed that recombinant GSK-3β can specifically phosphorylate CLOCK and that this phosphorylation is abrogated in the presence of GSK-3 inhibitor BIO (Fig. 4C).
GSK-3 is constitutively active in resting cells, and is primarily inactivated through direct phosphorylation by AKT, a major player in the RAS/PI3K/AKT survival pathway. To further confirm that the regulation of CLOCK stability is mediated by GSK-3-dependent phosphorylation, we tested the effect of overexpression of constitutively active RAS on the abundance of the CLOCK protein. Signal transduction through the RAS pathway inactivates GSK-3β; therefore if GSK-3β mediates the degradation of CLOCK, then expression of RAS would increase CLOCK protein levels. Indeed, cells co-expressing CLOCK, BMAL1 and RAS showed an increase of PC-CLOCK protein, whereas the protein level of PD-CLOCK was not affected (Fig. 4D). These data suggest that decreased CLOCK stability occurs through phosphorylation of serine residues within the phospho-degron site.
If inhibition of GSK-3 stabilizes CLOCK, then activation of GSK-3 should result in CLOCK degradation. Indeed, treatment with the PI3K/AKT inhibitor, LY294002, resulted in a decrease in CLOCK abundance in cells with ectopically expressed CLOCK, BMAL1 and RAS (Fig. 4E).
To demonstrate the intracellular phosphorylation of CLOCK by GSK-3β, cells were co-transfected with expression plasmids for either CLOCK or phospho-deficient mutant (S427A) and ectopic GSK-3β, either the active (CA) or kinase-dead (KD) form. Ectopic RAS was included to inhibit the constitutively active endogenous GSK-3 activity. A proteasome inhibitor was used to prevent loss of the highly unstable GSK-3β phosphorylated CLOCK. As seen in Fig. 3F, GSK-3β(CA) increased CLOCK phosphorylation while GSK-3β(KD) decreased it. Importantly, S427 is required for this GSK-3β induced phosphorylation (Fig. 3G). Taken together, these data convincingly demonstrate that GSK-3β-mediated phosphorylation of CLOCK targets its degradation through a conservative Ser-rich phospho-degron.
BMAL1-dependent CLOCK phosphorylation increases the transcriptional activity of the complex
In our previous work, we have shown that BMAL1-dependent phosphorylation and degradation of CLOCK are tightly linked to its transactivation function.5 Here, we identified a phospho-degron that requires BMAL1-induction of phospho-primed CLOCK, a target substrate of GSK-3-mediated degradation. This prompted us to test PD-CLOCK mutants for their ability to activate responsive promoters. For this, 293T cells were transiently transfected with plasmids encoding BMAL1 with either PC- or individual PD-CLOCK mutants. To eliminate potential effects of phosphorylation on other characteristics of the CLOCK/BMAL dimer (i.e. DNA-binding activity) and to address its transactivation property in a more direct way, we chose to utilize GAL4 luciferase assay. As shown in Fig. 5A, PD-CLOCK mutants are less transcriptionally active compared to the wild type protein. At the same time, CRY2 represses CLOCK/BMAL1-mediated transactivation independent of the identified phosphorylation sites (Fig. 5A). These results were corroborated in a similar experiment using GAL4-BMAL fusion co-expressed with individual phospho-deficient mutants of CLOCK (data not shown).
Fig. 5. Stabilization of phospho-deficient CLOCK affects CLOCK/BMAL1 functional activity.

A. Phospho-deficient CLOCK proteins are less transcriptionally active compare to phospho-competent CLOCK. 293T were transfected with a GAL4 luciferase reporter and plasmid encoding BMAL1 with either GAL4-PC-CLOCK fusion or GAL4-PD-CLOCK fusion S427/429/431/433/434/436A, followed by luciferase activity measurement. Bars represent relative luciferase signal normalized for efficiency of transfection using β-Gal assay. Experiments were performed at least 3 times in duplicates. Values are mean ± standard error. The transactivation property of all phospho-deficient mutants tested was significantly decreased compared to wild-type CLOCK (p<0.05 as determined by Student’s t-test). The insert shows the abundance and pattern of phosphorylation of CLOCK proteins analyzed by Western Blot.
B. Over-expression of RAS increases transcriptional activity of phospho-competent but not phospho-deficient CLOCK proteins.
A similar GAL4 luciferase assay was executed with co-expression of BMAL1 and either GAL4-CLOCK fusion or GAL4-PD-CLOCK fusion S427/429/431/433/434/436A. Increasing concentrations of the plasmid expressing RAS were used as indicated. Transfected cells were lysed in reporter buffer and luciferase activity was measured and normalized for β-Gal activity. The Western Blot inserts show the abundance and phosphorylation status of CLOCK.
C. Stabilization of phospho-deficient CLOCK protein delays the phase of oscillation in synchronized fibroblasts.
MEFs derived from mice with the targeted disruption of the Clock gene were transfected with mPer1-luciferase reporter gene and BMAL expression construct with either wild type or PD-CLOCK mutant S434/436/437/440/441A. 36 hrs post transfection cells were transferred to 24-well plates and treated with 0.1 uM dexamethasone for 4 hrs. Luciferase activity was measured in cell lysates over a 20-hr period with a 4-hr resolution. Shown is a representative experiment out of three that were each performed in triplicates. Values are normalized by luciferase signal in cells at 0 time point (time of dexamethasone removal) and show mean value ± standard error. Closed circles represent cells transfected with PC-CLOCK, open circles – cells transfected with PD-CLOCK. Black and white arrows indicate peak and trough of Per1-driven luciferase expression by the phospho-competent and phospho-deficient CLOCK/BMAL1 complexes respectively.
The observed decrease in transactivation capacity of PD-CLOCK suggests that GSK-3-dependent control of CLOCK stability may regulate its transcriptional activity. To test this hypothesis, we co-expressed increasing amounts of constitutively active RAS with either PC- or PD-CLOCK in combination with BMAL1 and GAL4-luciferase reporter and monitored CLOCK protein abundance and activity. In line with data presented in Fig. 4E, RAS increases the abundance of both phosphorylated and unphosphorylated forms of PC-CLOCK but had no effect on PD-CLOCK mutants. Importantly, the RAS-induced increase in phospho-CLOCK abundance correlated with increased activity of the responsive promoter (Fig. 5B). Consistently, there was no effect of RAS expression on the transactivation property of mutant CLOCK protein. Thus, RAS-mediated inhibition of GSK-3 reduces CLOCK degradation via the phospho-degron site allowing for increased CLOCK-dependent transcription.
Stabilization of phospho-deficient CLOCK delays the phase of oscillation in synchronized fibroblasts
To address the potential functional significance of GSK-3-mediated CLOCK degradation, we monitored the expression of mPer1-driven luciferase in synchronized cells expressing either wild type PC-CLOCK or the highly phospho-deficient mutant. To eliminate effects of the endogenous CLOCK protein, we utilized MEFs isolated from mice with the targeted disruption of the Clock gene. As shown in Fig. 5C, the peak of mPer1-driven luciferase was significantly delayed in cells expressing PD-CLOCK when compared to PC-CLOCK. Taken together, these data suggest that the newly identified phospho-degron is involved in regulation of circadian molecular mechanism through control of stability of its integral component, CLOCK. A key role of GSK-3β in this regulation is further confirmed by results of a complimentary study demonstrating that GSK-3β can also directly phosphorylate BMAL1 (P. Sassone-Corsi, personal communication).
DISCUSSION
The regulation of the biological clock depends on tightly controlled gene transcription as well as precisely controlled degradation and stability of clock proteins. Post-translational modifications of clock proteins, particularly phosphorylation, regulate the phase and period length of circadian oscillation by controlling the rate of protein accumulation, association and translocation. Phosphorylation is often a signal for the recruitment of an E3 ligase, which ubiquitinates a substrate and allows proteasome-mediated degradation. In mammals, the phosphorylated forms of PER and CRY are targeted for degradation by components of ubiquitin ligase complexes, β-TRCP1 and FBXL3 respectively.11–15 Two kinases have been implicated in mammalian PER/CRY degradation, CK1 and GSK-3β.
GSK-3β is a pro-apoptotic kinase that is constitutively active in resting cells, and is primarily regulated by inactivation involving direct phosphorylation by AKT, a major player in the RAS/PI3K/AKT survival pathway. Unlike most kinases, GSK-3 and CK1 often require that their substrates are phosphorylated by a “priming” kinase, which acts 4 residues C-terminal to the GSK-3 site or 3 residues C-terminal to the CK1 site. GSK-3 is involved in phospho-degrons of many transcription factors and proto-oncogenic proteins including β-catenin, cyclin D1, c-myc, c-jun, and Snail.16–20 More recently, GSK-3 has been implicated in the regulation/degradation of molecular clockwork components.
In Drosophila, GSK-3 homolog phosphorylates TIMELESS, which is required for nuclear translocation of the PER/TIM dimer. 21 Mammalian GSK-3 phosphorylates PER facilitating PER/CRY nuclear translocation, and CRY2. The latter is phosphorylated through a degron-site at Ser553 subsequent to priming phosphorylation at Ser557. 22 Another target of GSK-3 is the nuclear receptor REV-ERBα; an important regulator of Bmal1 gene expression.23 GSK-3 control of circadian regulation is supported by the screening results of a Library of Pharmacologically Active Compounds (LOPAC) for pharmacological modulators of basal circadian parameters in cultured cells. This screening identified several inhibitors of GSK-3β by their ability to shorten the period of circadian oscillation.24 Here we further extend the role of GSK-3 in mammalian circadian clock by showing that this kinase is involved in phosphorylation of a major component of the positive arm of the circadian loop, CLOCK protein.
We have provided several lines of evidence to suggest that CLOCK is a substrate of GSK-3. First, CLOCK contains a classical GSK-3 motif (with Ser427 as a potential GSK-3 site) within a phospho-degron element and mutation of the GSK-3 motif results in loss of CLOCK phosphorylation and increased stability. Second, inhibition of GSK-3 increases CLOCK protein levels while its activation results in the loss of CLOCK. Third, endogenous GSK-3 can be immunoprecipitated with the CLOCK/BMAL1 complex in cultured cells. Fourth, recombinant GSK-3 can specifically phosphorylate CLOCK in vitro, and the over-expression of constitutive active GSK-3β increases an unstable phospho-CLOCK while an inactive GSK-3β inhibits phosphorylation In good agreement with our data, recent report using mass spectrometry approach has confirmed the phosphorylation of Ser427 residue in the mouse liver in vivo. 9
The NET-PHOSK sequence analysis data identified PKC as a high probability candidate to function as a priming kinase for GSK-3-mediated phosphorylation (Supplementary Fig. 1B). This is also supported by previous work showing that CLOCK can be transiently phosphorylated by Ca2+-dependent PKC α and γ within 1 hour of PMA treatment although specific sites for PKC-dependent phosphorylation have not been identified.8 Therefore we were compelled to investigate the potential role of PKC as a priming kinase for the GSK-3-mediated phosphorylation in the phospho-degron site. However, the results of mutational analysis indicated that BMAL1-mediated CLOCK phosphorylation/degradation is independent of classical PKC (Supplementary Fig. 2). Thus, the kinase(s) responsible for BMAL1-dependent “phospho-priming” of CLOCK remain unknown.
It is likely that the identified phospho-degron region of CLOCK represents just one of many regulatory sites that can cooperate in governing the phosphorylation-dependent functions of this core clock component. The phosphorylation of a specific site in CLOCK may trigger a chain of subsequent phosphorylation events performed by either the same or different kinase which will result in generation of differentially phosphorylated forms of CLOCK that may also vary in their functional characteristics. Since the phosphorylation of circadian proteins was originally viewed as a mechanism of targeted degradation that allows for the next circadian cycle, it has been traditionally associated with the reduction in functional activity. However, our data demonstrate that the transactivation function of individual CLOCK mutants is directly correlated with its phosphorylation status and that the more stable PD-CLOCK proteins are less transcriptionally active than the unstable phosphorylated protein. These data are corroborated by a recent study, which showed that regulation of activity and life cycle of the Drosophila homolog of CLOCK is controlled by sequential phosphorylation.25 Detailed investigation of the intracellular distribution of the CLK/CYC heterodimer, the level of its phosphorylation and its functional activity has demonstrated that the most transcriptionally active is an intermediate phosphorylated form of CLOCK whereas PER-dependent hyperphosphorylation of CLK by DOUBLE-TIME kinase results in transcriptional inhibition and degradation of the CLK protein.26 The analogy with the mammalian system would suggest that transactivation properties of CLOCK may depend on the extent and/or specific sites of phosphorylation and that phosphorylation is important both for functional activity and rapid turnover of the protein. The further development of specific tools and techniques that allow for the discrimination between the various CLOCK phosphorylation states will be useful in addressing these issues. In this regards, our work represents an important step in this direction.
The circadian regulatory mechanism described here is based on phosphorylation mediated protein degradation. As we have shown previously, BMAL1-induced CLOCK degradation, at least in part, occurs through the proteasome pathway. 5 A growing body of evidence suggests that the proteasome itself is involved in transcription by promoting the exchange of transcription factors on chromatin and facilitating multiple rounds of transcription initiation.27 According to this model, CLOCK that is bound to the E-box must be removed for the next round of transcriptional activation. The more stable degron-deficient CLOCK would not be efficiently cleared and therefore would exhibit a lower steady-state transactivation property, which is consistent with the luciferase functional assays. Accordingly, the observed delay in phase of circadian oscillation in synchronized cells expressing phospho-deficient CLOCK protein is also likely to result from the deficiency in rapid promoter clearance. Together, our data identifies the regulation of CLOCK turnover as an important mechanism within the molecular clockwork.
Another important aspect of the data presented here is the implication of RAS/AKT pathway in GSK-mediated CLOCK degradation. Previously, we determined that the functional status of the CLOCK/BMAL1 complex correlates with an animal’s response to genotoxic stress induced by DNA damaging agents.28 Since the RAS/AKT pathway plays a central role in growth, proliferation and anti- apoptotic mechanisms to promote cell cycle and survival, our data showing that the RAS/AKT pathway mediate stability of a transcriptionally active phospho-CLOCK may provide clues to the molecular mechanisms involved. This experimental direction would be an important step in the understanding of the molecular basis of a crosstalk between the circadian and stress response pathways. Furthermore, since GSK-3β has been implicated in numerous diseases including diabetes, stroke, Alzheimer and cancer, identification of new targets of this kinase and detailed study of their regulation are important in developing novel therapeutic intervention strategies.
MATERIAL AND METHODS
Animals
The Clock mutant, 29–31 Bmal1−/− knockout 32 and Clock−/− knockout 33 mice are maintained on C57BL/6J background. All animal studies were conducted in accordance with the regulations of the Committee of Animal Care and Use at Roswell Park Cancer Institute.
Cell Culture
HEK-293T cells were obtained from ATCC. Mouse fibroblasts were isolated from 13-day Bmal1+/+, Bmal1−/− and Clock−/− embryos and were immortalized by a retrovirus-mediated expression of GSE56 (a fragment of p53 encoding for a peptide working as dominant-negative inhibitor of p53) as described.34 Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum.
Plasmids
Expression constructs encoding tagged BMAL1 and CLOCK proteins were described previously.5 Substitution mutants were made using QuikChange site-directed mutagenesis kit (Stratagene). Details, including sequence of primers, are provided in Supplementary Information. The mPer1 reporter construct containing 6.4 kb of mPer1 promoter was described in.35 GAL4-fused constructs were generated as described in Supplementary Information. GSK-3β(CA) and GSK-3β(KD) were described in.36
Reagents
BIO, Dexamethasone, DMSO, cycloheximide and LY294002 were purchased through Sigma Inc. Recombinant GSK-3β was purchased through NEN Biolabs. 32P-ATP was purchased from Perkin-Elmer. Luciferin was obtained from Promega.
Transfection, Western Blotting and Antibody
HEK-293T cells were transfected with plasmids encoding clock proteins by using Fugene 6 (Roche Diagnostics) according the manufacturer’s protocol. Whole cell lysates were prepared 24–36 hrs post-transfection by boiling in 2% SDS, and nuclear and cytoplasmic extracts – by using Active Motif Nuclear Extract Kit. Proteins were resolved in 6.5% SDS-PAGE, transferred to nitrocellulose membrane and probed with specific antibody. HA- and FLAG-tagged proteins were detected with mouse anti-HA.11 (Covance Research Products, Richmond, CA) and mouse monoclonal anti-FLAG (Sigma) respectively. Monoclonal anti-GSK-3 and monoclonal anti-Myc antibody 9E10 were purchased from Santa Cruz Biotechnology; Anti-CLOCK and anti-BMAL1 were raised in guinea pig as described in Lee et. al. 4. HRP conjugated anti-rabbit (Santa Cruz Biotechnologies) were used as secondary antibodies. Proteins were visualized with the ECL detection kit (Jackson Research Laboratories).
Immunoprecipitation
HEK-293T cells transfected with plasmids encoding CLOCK and BMAL1 were lysed in a buffer (100mM NaCl, 50mM Tris-HCL at pH 7.5, 2mM EDTA, 1% NP-40, 0.1% SDS), supplemented with proteinase cocktail mixture (Roche Diagnostics), and phenylmethylsulfonyl fluoride (Sigma). The lysates containing equal amounts of total ectopic protein (as determined by Western Blot) were incubated at 4°C with primary antibody for 2 hours and then with protein A/G beads for an additional 2 hours. The beads were washed with TBS washing buffer (50 mM Tris/HCl, pH 7.4 and 150 mM NaCl) supplemented with 0.1% Tween and the precipitates were analyzed for the presence of CLOCK and BMAL1 by Western Blots.
Immunofluorescence
HEK-293T cells were seeded overnight onto coverslips in 35mm 6-well plates. At 30 hours post-transfection, cells were washed in PBS followed by fixation in 3.7% formaldehyde for 15 minutes at room temperature. Cells were then permeabilized in 0.2% Triton-X100 in PBS for 5 minutes and incubated with anti-HA monoclonal antibody (1:500). Coverslips were washed 3 times in PBS-Triton-X100 and incubated at room temperature with the appropriate secondary antibodies: anti-mouse TRITC- and anti-rabbit FITC-conjugated secondary antibodies (Jackson Labs). Cells were mounted by using Vectashield mounting media (Vector Laboratories) and analyzed with a fluorescence microscope (Zeiss) equipped with the appropriate optics and filter modules, and results were recorded with a digital camera (SPOT; Diagnostic Instruments Inc.).
Reporter gene assays
Cells were plated in 24-well plates and transfected by a plasmid mixture containing CLOCK (70ng), BMAL1 (30ng), CRY2 (30ng), reporter plasmids (GAL4-luciferase or mPer1-luciferase) (50ng), and 10ng of pcDNA3-βGAL (to normalize efficiency of transfection). Empty vector pcDNA3 was used to equalize the amount of plasmid DNA for each transfection. Detection of luciferase and β-Galactosidase activities were performed as described previously.37, 38 Results are representative of at least three experiments.
Protein degradation assay
Cells expressing ectopic wild type and mutant CLOCK proteins were treated at various times with 40ug/ml cycloheximide and then harvested in SDS-sample buffer for Western Blot analysis.
GSK-3 in vitro kinase assay
Cell lysates from transfectants of CLOCK were immunoprecipitated with anti-CLOCK antibody using a stringent RIPA buffer (150mM NaCl; 50mM Tris-HCl at pH 8,0; 0.5% Na-DOX, 0.1% SDS and proteinase cocktail). Immunoprecipitates were mixed with recombinant GSK-3β and 5uCi of 32P labeled ATP in 25ul kinase buffer and incubated at 30 C° for 1hr. The reaction was stopped by boiling in SDS sample buffer, and the proteins were resolved by SDS-PAGE.
Circadian synchronization by dexamethasone treatment
MEFs derived from mice with the targeted disruption of the Clock gene were seeded onto 6-well plates and transfected with mPer1-luciferase reporter in combination with either plasmids expressing BMAL and CLOCK or BMAL and CLOCK(S434/436/437/440/441A). The following day cells were collected, evenly transferred to 96-well plates in DMEM supplemented with 2% FBS and allowed to grow until confluent. Cells were synchronized by a 4-hr treatment with 0.1uM dexamethasone as described previously.39 Relative luciferase activity was plotted against time after dexamethasone removal.
Supplementary Material
Acknowledgments
We thank Dr. Kunho Lee for generously providing the plasmid expressing myc-CLOCK; Dr. Helen Piwnica-Worms for plasmids encoding for constitutively active and kinase dead GSK-3β; Dr. Jenny Black for supplying BIM; Dr. Gokul Das for the GAL4-reporter gene, David Weaver for providing Clock knockout animals and Zahra Fayazi for the generation of antibodies. We are grateful to Dr. Kondratov for critical reading of the manuscript and Yan Hu, Marilyn Jackson and Maria Comas Soberats for helpful discussions. We thank Dr. Sassone-Corsi for communicating data prior to publication. This work was supported by the National Institute of Health grant GM550926 to M.P.A.
References
- 1.Bell-Pedersen D, Cassone VM, Earnest DJ, Golden SS, Hardin PE, Thomas TL, Zoran MJ. Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nature reviews. 2005;6:544–56. doi: 10.1038/nrg1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Hum Mol Genet. 2006;15(Spec No 2):R271–7. doi: 10.1093/hmg/ddl207. [DOI] [PubMed] [Google Scholar]
- 3.Virshup DM, Eide EJ, Forger DB, Gallego M, Harnish EV. Reversible protein phosphorylation regulates circadian rhythms. Cold Spring Harbor symposia on quantitative biology. 2007;72:413–20. doi: 10.1101/sqb.2007.72.048. [DOI] [PubMed] [Google Scholar]
- 4.Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–67. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 5.Kondratov RV, Chernov MV, Kondratova AA, Gorbacheva VY, Gudkov AV, Antoch MP. BMAL1-dependent circadian oscillation of nuclear CLOCK: posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes & development. 2003;17:1921–32. doi: 10.1101/gad.1099503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwon I, Lee J, Chang SH, Jung NC, Lee BJ, Son GH, Kim K, Lee KH. BMAL1 shuttling controls transactivation and degradation of the CLOCK/BMAL1 heterodimer. Molecular and cellular biology. 2006;26:7318–30. doi: 10.1128/MCB.00337-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tischkau SA, Mitchell JW, Pace LA, Barnes JW, Barnes JA, Gillette MU. Protein Kinase G Type II Is Required for Night-to-Day Progression of the Mammalian Circadian Clock. Neuron. 2004;43:539–49. doi: 10.1016/j.neuron.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 8.Shim HS, Kim H, Lee J, Son GH, Cho S, Oh TH, Kang SH, Seen DS, Lee KH, Kim K. Rapid activation of CLOCK by Ca2+-dependent protein kinase C mediates resetting of the mammalian circadian clock. EMBO reports. 2007;8:366–71. doi: 10.1038/sj.embor.7400920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshitane H, Takao T, Satomi Y, Du NH, Okano T, Fukada Y. Roles of CLOCK phosphorylation in suppression of E-box-dependent transcription. Molecular and cellular biology. 2009;29:3675–86. doi: 10.1128/MCB.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dardente H, Fortier EE, Martineau V, Cermakian N. Cryptochromes impair phosphorylation of transcriptional activators in the clock: a general mechanism for circadian repression. The Biochemical journal. 2007;402:525–36. doi: 10.1042/BJ20060827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reischl S, Vanselow K, Westermark PO, Thierfelder N, Maier B, Herzel H, Kramer A. Beta-TrCP1-mediated degradation of PERIOD2 is essential for circadian dynamics. Journal of biological rhythms. 2007;22:375–86. doi: 10.1177/0748730407303926. [DOI] [PubMed] [Google Scholar]
- 12.Shirogane T, Jin J, Ang XL, Harper JW. SCFbeta-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. The Journal of biological chemistry. 2005;280:26863–72. doi: 10.1074/jbc.M502862200. [DOI] [PubMed] [Google Scholar]
- 13.Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, Draetta GF, Pagano M. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science (New York, NY. 2007;316:900–4. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- 14.Siepka SM, Yoo SH, Park J, Song W, Kumar V, Hu Y, Lee C, Takahashi JS. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129:1011–23. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Godinho SI, Maywood ES, Shaw L, Tucci V, Barnard AR, Busino L, Pagano M, Kendall R, Quwailid MM, Romero MR, O’Neill J, Chesham JE, Brooker D, Lalanne Z, Hastings MH, Nolan PM. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science (New York, NY. 2007;316:897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- 16.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. The EMBO journal. 1997;16:3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes & development. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9085–90. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nature cell biology. 2004;6:931–40. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 21.Martinek S, Inonog S, Manoukian AS, Young MW. A role for the segment polarity gene shaggy/GSK-3 in the Drosophila circadian clock. Cell. 2001;105:769–79. doi: 10.1016/s0092-8674(01)00383-x. [DOI] [PubMed] [Google Scholar]
- 22.Harada Y, Sakai M, Kurabayashi N, Hirota T, Fukada Y. Ser-557-phosphorylated mCRY2 is degraded upon synergistic phosphorylation by glycogen synthase kinase-3 beta. The Journal of biological chemistry. 2005;280:31714–21. doi: 10.1074/jbc.M506225200. [DOI] [PubMed] [Google Scholar]
- 23.Yin L, Wang J, Klein PS, Lazar MA. Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of the circadian clock. Science (New York, NY. 2006;311:1002–5. doi: 10.1126/science.1121613. [DOI] [PubMed] [Google Scholar]
- 24.Hirota T, Lewis WG, Liu AC, Lee JW, Schultz PG, Kay SA. A chemical biology approach reveals period shortening of the mammalian circadian clock by specific inhibition of GSK-3beta. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20746–51. doi: 10.1073/pnas.0811410106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hung H-C, Maurer C, Zorn D, Chang W-L, Weber F. Sequential and Compartment-specific Phosphorylation Controls the Life Cycle of the Circadian CLOCK Protein. 2009:23734–42. doi: 10.1074/jbc.M109.025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim EY, Edery I. Balance between DBT/CKIepsilon kinase and protein phosphatase activities regulate phosphorylation and stability of Drosophila CLOCK protein. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6178–83. doi: 10.1073/pnas.0511215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins GA, Tansey WP. The proteasome: a utility tool for transcription? Curr Opin Genet Dev. 2006;16:197–202. doi: 10.1016/j.gde.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Gorbacheva VY, Kondratov RV, Zhang R, Cherukuri S, Gudkov AV, Takahashi JS, Antoch MP. Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3407–12. doi: 10.1073/pnas.0409897102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antoch MP, Song EJ, Chang AM, Vitaterna MH, Zhao Y, Wilsbacher LD, Sangoram AM, King DP, Pinto LH, Takahashi JS. Functional identification of the mouse circadian Clock gene by transgenic BAC rescue. Cell. 1997;89:655–67. doi: 10.1016/s0092-8674(00)80246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, Steeves TD, Vitaterna MH, Kornhauser JM, Lowrey PL, Turek FW, Takahashi JS. Positional cloning of the mouse circadian clock gene. Cell. 1997;89:641–53. doi: 10.1016/s0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, Dove WF, Pinto LH, Turek FW, Takahashi JS. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science (New York, NY. 1994;264:719–25. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–17. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debruyne JP, Noton E, Lambert CM, Maywood ES, Weaver DR, Reppert SM. A clock shock: mouse CLOCK is not required for circadian oscillator function. Neuron. 2006;50:465–77. doi: 10.1016/j.neuron.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 34.Ossovskaya VS, Mazo IA, Chernov MV, Chernova OB, Strezoska Z, Kondratov R, Stark GR, Chumakov PM, Gudkov AV. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10309–14. doi: 10.1073/pnas.93.19.10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilsbacher LD, Sangoram AM, Antoch MP, Takahashi JS. The mouse Clock locus: sequence and comparative analysis of 204 kb from mouse chromosome 5. Genome research. 2000;10:1928–40. doi: 10.1101/gr.10.12.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang T, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung MC, Piwnica-Worms H. GSK-3 beta targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3 beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kondratov RV, Kondratova AA, Lee C, Gorbacheva VY, Chernov MV, Antoch MP. Post-translational regulation of circadian transcriptional CLOCK(NPAS2)/BMAL1 complex by CRYPTOCHROMES. Cell Cycle. 2006;5:890–5. doi: 10.4161/cc.5.8.2684. [DOI] [PubMed] [Google Scholar]
- 38.Kondratov RV, Shamanna RK, Kondratova AA, Gorbacheva VY, Antoch MP. Dual role of the CLOCK/BMAL1 circadian complex in transcriptional regulation. FASEB J. 2006;20:530–2. doi: 10.1096/fj.05-5321fje. [DOI] [PubMed] [Google Scholar]
- 39.Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, Schutz G, Schibler U. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science (New York, NY. 2000;289:2344–7. doi: 10.1126/science.289.5488.2344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.