

Abstract
Rapid growth of sequencing technologies has greatly contributed to increasing our understanding of human genetics. Yet, in spite of this growth, mainstream technologies have been largely unsuccessful in resolving the diploid nature of the human genome. Here we describe statistically aided long read haplotyping (SLRH), a rapid, accurate method based on a simple experimental protocol that requires potentially as little as 30 Gbp of sequencing in addition to a standard (50x coverage) whole-genome analysis for human samples. Using this technology, we phase 99% of single-nucleotide variants in three human genomes into long haplotype blocks of 200 kbp to 1 Mbp in length. As a demonstration of the potential applications of our method, we determine allele-specific methylation patterns in a human genome and identify hundreds of differentially methylated regions that were previously unknown. Such information may offer insight into the mechanisms behind differential gene expression.
In spite of rapid advances throughout genomics and a plethora of genomes that have been sequenced, most genomics studies to date have given little consideration to a crucial aspect of human genetics1. Humans are diploid organisms and typically possess two copies of each chromosome: one inherited from the mother, and one from the father. To date, mainstream technologies have been largely unsuccessful in resolving this key facet of the human genome2.
[Also, why focus exclusively on human? There would seem to be many other important areas where SLRH can be applied (Polyploid plants? Other diploid organisms?).]
[We have not tested the method or non-humans, and so we feel like we should not make too many claims about that.]
In previous studies, the assignment of variants to alleles was carried out by sequencing the parents of an individual3 or by using specialized molecular approaches that involved physically separating the chromosomes during cell division45, analyzing long-range chromosomal interactions using a proximity ligation-based method6, or recovering haplotypes from long DNA fragments, such as fosmid clones7,8 (a technique also known as dilution haplotyping910). Each of these methods has shortcomings: separating chromosomes requires complex specialized devices and careful manipulation of cells; proximity ligation-based methods leave unphased many variants6; cloning fragments in fosmids typically involves at least a week of library preparation7. Recently, haplotyping methods based on long fragments were modified to use multiple displacement amplification (MDA) instead of fosmid-based cloning1112. Using MDA reduces library preparation time to a day, but the method suffers from a high amplification bias12 and therefore requires very deep sequencing of the samples (often exceeding 500 Gbp11).
Even at a high coverage, MDA-based methods may leave up to 5% of variants unphased81112. Such considerable sequencing requirements have prevented these haplotyping methods from being widely deployed.
Here we describe statistically aided long read haplotyping, which involves as little as 30 Gbp of sequencing in addition to a standard (50X coverage)whole-genome analysis to haplotype a human genome. It recovers haplotypes from long DNA fragments which are obtained using techniques recently developed for the study of the genome of Botryllus schlosseri13. Unlike earlier technologies based on fosmid cloning or MDA, SLRH uses PCR to amplify fragments. PCR exhibits less amplification bias than MDA and therefore does not require as much coverage. However, the amplified fragments rarely exceed 10 Kbp, whereas some MDA-based protocols generate 80 Kbp fragments. This relatively short length required the development of a phasing algorithm, Prism, that augments long-fragment haplotyping with statistical techniques. Starting with shorter fragments, Prism produces haplotype blocks that are of equal or greater quality to ones obtained from existing haplotyping technologies. Furthermore, the resulting protocol can phase 99% of single-nucleotide variants (SNVs); current technologies typically phase about 95–97%11,12. See Supplementary Table 1 for a more detailed comparison of SLRH with current haplotyping technologies.
As a demonstration of an application that is made possible by readily available haplotype information, we use SLRH to determine allele-specific methylation patterns in a human genome. Differentially methylated regions affect the expression of many genes; yet, little is known about the details of this process14, largely owing to the difficulty in obtaining accurate haplotypes. Our analysis yields a base-resolution map of DNA methylation across a human genome, which is a valuable resource for understanding mechanisms involving allele-specific DNA methylation. Our map contains several hundred differentially methylated genomic regions, most of which to our knowledge have not been described previously.
Results
Statistically aided long read haplotyping
Essentially, SLRH is a form of dilution haplotyping similar to previous approaches based on fosmid clones7 or long fragment reads (LFR)11. It involves placing a small number of large ~7–10 kbp DNA fragments into separate pools. Each pool has a unique barcode that identifies its fragments, which are then recovered from short-read sequences and assembled into long haplotype blocks using a phasing algorithm (Fig. 1).
Figure 1.
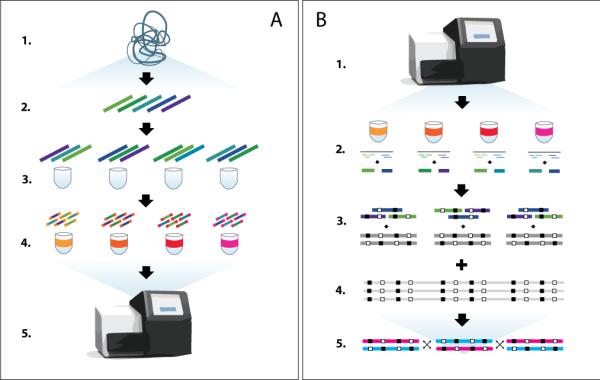
Statistically aided long read haplotyping (a) Overview of the library preparation protocol. The subject's DNA (1) is sheared into fragments of about 10 kbp (2), which are then diluted and placed into 384 wells, at about 3,000 fragments per well (3). Within each well, fragments are amplified through long-range PCR, cut into short fragments and barcoded (4), before being finally pooled together and sequenced (5). (b) Overview of the bioinformatics pipeline. Sequenced short reads are aligned and mapped back to their original well using the barcode adapters (1). Within each well, reads are grouped into fragments (2), which are assembled at their overlapping heterozygous SNVs into haplotype blocks (3). These blocks are assigned a phase statistically based on a phased reference panel (4), which produces very long haplotype contigs (5).
The preparation of each phasing library starts by shearing DNA into fragments of approximately 10 kbp in size; these fragments are gel-purified and ligated with amplification adaptors at both ends. Fragments are then diluted into a 384-well plate containing 3,000–6,000 molecules per well and PCR-amplified using adapter-specific primers (Supplementary Fig. 1). The number of fragments amplified (3,000–6,000) is chosen to minimize the probability of two fragments overlapping, which later simplifies fragment calling in the informatics pipeline. Each resulting pool of amplified molecules is prepared into a sequencing library using the Nextera DNA transposase, and sequencing adapters with barcodes unique to each well are incorporated through limited-cycle PCR. The resulting sub-libraries are pooled and sequenced on the Illumina platform (Fig. 1a). A single phasing library typically corresponds to about 30 Gbp of 101-base, paired-end Illumina reads.
The sequenced reads are then aligned to the reference genome and mapped back to their original wells as specified by the barcode adapters. Mapped reads within each well are clustered into groups that are believed to come from the same fragment. Variants in each fragment are called based on the subject's whole-genome genotyping; in our experiments, the subject's genotypes were determined from a 50X read coverage on the Illumina platform (Online Methods). Fragments called at this stage have N50 lengths of about 7–9 kbp (i.e. at least half of all sequenced bases are within fragments of at least 7-9kbp) and cover the genome to a depth of about 4–8X (Supplementary Tables 3 and 4). The relatively short length of these reads compared to older technologies1115 required the development of a haplotyping algorithm, Prism (Fig. 1b and Supplementary Data), which augments dilution haplotyping with statistical techniques2.
In brief, Prism proceeds in two stages. First, in a local_stage, it assembles fragments into haplotype blocks by connecting them together at their overlapping heterozygous SNVs8. This step is similar to existing algorithms for dilution haplotyping technologies. Then, in a global stage, Prism exploits linkage disequilibrium patterns2 to assemble local blocks into long and accurate global haplotype contigs. Such contigs can phase up to 99% of heterozygous SNVs and up to 95% of heterozygous variants. Between each local block, Prism produces confidence scores that indicate the likelihood of introducing a switch error owing to statistical phasing.
In applications where a high level of accuracy is desired, the user may introduce breaks in statistically assembled haplotype contigs whenever a confidence score falls below a certain threshold. This reduces the length of the haplotype blocks, but increases their accuracy. The ability to trade-off between accuracy and completeness is a feature of Prism that to our knowledge is not provided by other phasing algorithms that we expect to be useful in applications that demand a high a level of precision.
The global phasing stage is an essential component of the SLRH pipeline. Simply connecting long DNA fragments at heterozygous SNVs results in local blocks of about 60 kbp in length (Supplementary material); existing technologies produce haplotype blocks of more than 600 kbp1115. The global phasing performed by Prism increases the lengths of these blocks by more than ten-fold; it also increases SNV phasing rates from 94% to more than 99%.
The key algorithmic tool used by Prism is a hidden Markov model (HMM; Supplementary material). At a high level, the HMM tries to represent the partially phased haplotypes of the sample as an “imperfect mosaic” of haplotypes from a pre-phased reference panel from the 1000 Genomes Project16. It then assigns to each block the phase that best matches this representation. Confidence scores between adjacent blocks are derived from the forwards-backwards variables of the hidden Markov model.
The Prism HMM extends the work of Li and Stephens17, on which modern statistical phasing packages such as IMPUTE2 (ref.18) or SHAPE-IT19 are based. Unlike existing packages, our method is able to use partially phased information obtained from long reads, which allows us to achieve greater accuracy than traditional statistical methods. The only statistical package able to leverage partial phase information is the recently introduced SHAPEIT220; its underlying algorithm follows the same basic approach as Prism.
Phasing three genomes using SLRH
As a demonstration of the ability of SLRH to accurately phase human genomes, we prepared libraries for three HapMap samples: NA12878, NA12891 and NA12892 (Supplementary Table 2). These genomes were previously phased at a high quality using familial information3. Two phasing libraries were prepared for each member of the trio. A summary of the results appears in Table 1.
Table 1.
Summary of haplotyping performance. We used SLRH to phase three human genomes from the HapMap project. Two libraries were prepared for each subject, and each was evaluated at a fixed accuracy threshold.
| Haplotype block N50 length (bp) | Phasing rate over SNVs | Switches per Mbp | |
|---|---|---|---|
| NA12878 (two libraries) | 563,801 | 99.00% | 0.47 |
| NA12891 (two libraries) | 647,599 | 99.25% | 0.68 |
| NA12892 (two libraries) | 531,804 | 98.84% | 0.75 |
| NA12878 (library #1) | 401,342 | 98.49% | 0.51 |
| NA12878 (library #2) | 405,472 | 98.44% | 0.49 |
We evaluated the performance of SLRH at different accuracy thresholds by introducing breaks in the global haplotype contigs whenever the confidence score of a local block was below a threshold. Depending on the threshold, the N50 lengths of haplotype blocks varied between 130 and 750 kbp; the percentage of SNVs phased varied between 96.0% and 99.4% (Fig. 2 and Supplementary Tables 5, 6).
Figure 2.
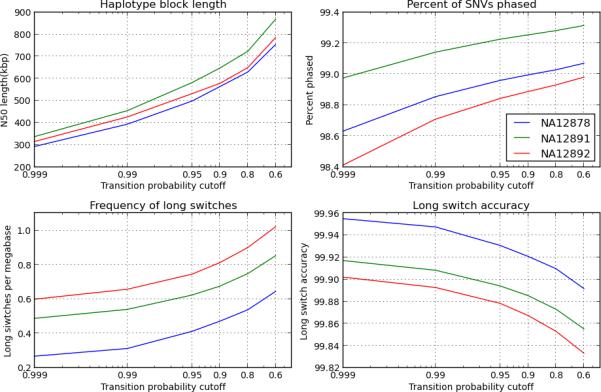
Haplotyping results at several accuracy thresholds. Long statistically constructed haplotype contigs are cut at positions where confidence scores are below a certain threshold (x axes), forming shorter but more accurate haplotype blocks. We evaluate the completeness (top panels) and the switch accuracy (bottom panels) of the smaller blocks at a series of thresholds. The blocks are evaluated only over SNVs.
In particular, at the 0.9 confidence score threshold, about 99% of all SNVs were phased in blocks with N50 lengths of 560–650 kbp (Supplementary Tables 7, 8). About 89% of 23,645 genes derived from the UCSC dataset21 were fully contained in a single haplotype block (Table 2 and Supplementary Table 9). The haplotype blocks also contained more than 73% of novel SNVs and indels (Supplementary Table 9), although the accuracy over these variants was lower than average, partly because of the increased difficulty in genotyping such rare variants.
Table 2.
Overview of heterozygocity patterns. Within each subject, SLRH phases about 90% of all genes. Of the genes containing variants, the majority (85%) contains heterozygous variants and a very large fraction (74%) contains compound heterozygous variants. Moreover, the phased genes harbor about 2,500 SNVs that were found to be damaging by the SIFT software package. About 1,500 genes are affected by such variants, and about 500 have both of their copies damaged.
| NA12878 | NA12891 | NA12892 | |
|---|---|---|---|
| Genes considered | 23,410 | 23,410 | 23,410 |
| Genes phased | 21,018 | 20,804 | 20,711 |
| Genes containing variants | 14,799 | 14,630 | 14,571 |
| Genes containing heterozygous variants | 12,634 | 12,573 | 12,339 |
| Genes containing compound heterozygous variants | 11,076 | 10,970 | 10,790 |
| Number of damaging SNVs | 2,460 | 2,422 | 2,323 |
| Number of damaging heterozygous SNVs | 1,597 | 1,667 | 1,583 |
| Genes with a damaging SNV on one strand | 1,573 | 1,579 | 1,507 |
| Genes with a damaging SNV both strands | 518 | 481 | 466 |
The majority of phased genes contained compound heterozygous SNVs (about 75% of all genes with SNVs); HLA-C, a gene whose haplotypes are used to predict immune response during organ transplantation4 is an example of a compound heterozygous gene (Supplementary Fig. 2). Phased genes also contained about 2,500 SNVs that were found to be damaging by the SIFT software package22. About 1,500 genes were affected by these mutations, and about 500 were found to have at least one damaging SNV on both the maternal and the paternal copy (Table 2).
To assess the accuracy of SLRH, we compared the above haplotypes to ones derived from applying Mendelian inheritance rules to our three samples. Even the long statistically assembled haplotypes obtained from a 0.5 probability threshold were highly accurate: for sample NA12878, SLRH produced long blocks with N50 lengths of 1.1 Mbp that contained long switching events at an average rate of 0.85 per Mbp (Supplementary Tables 5, 6).
Breaking these haplotypes at low-confidence positions further improved their precision: in every sample, long switch errors occurred at an average rate between 0.2 and 0.9 per Mbp, depending on the chosen accuracy threshold (Fig. 2 and Supplementary Tables 5, 6). At the 0.9 threshold, long switch events occurred at a rate of 0.47 per Mbp for sample NA12878 (Supplementary Tables 7, 8). At a small number of positions, we observed short (one-base) discordances with parental haplotyping. This affected about 0.15% of heterozygous positions (Supplementary Tables 5, 6), which were often associated with centromeres and copy number variations. Finally, the absolute accuracy of the haplotype blocks (i.e. without correcting for haplotype switching) varied between 93% and 96%, depending on the sample.
Whole-genome phasing from 30 Gbp of sequencing
Next, to demonstrate the low sequencing requirements of SLRH, we ran the Prism algorithm separately on each individual phasing library. We observed only a small loss of haplotyping performance, highlighting the robustness of our statistically aided approach.
Overall, we obtained haplotype blocks that were almost as accurate and only 100 kbp shorter than ones derived from 60 Gbp of sequencing (Fig. 3).
Figure 3.
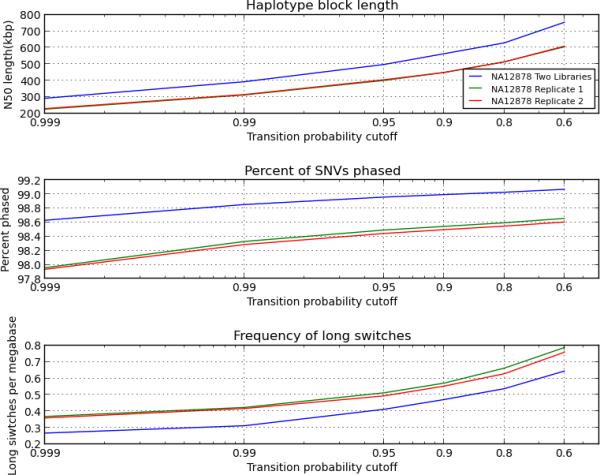
Haplotyping performance from 30 Gbp of sequencing. We ran the bioinformatics pipeline independently on two 30 Gbp replicate libraries of the sample NA12878. The resulting haplotype blocks are almost as accurate and only 100 kbp shorter than ones derived from two phasing libraries. Moreover, results from the two replicates are highly concordant.
In particular, at the 0.9 accuracy threshold, 98–99% of all SNVs were phased in blocks with N50 lengths of 400–500 kbp, depending on the sample (Supplementary Fig. 3 and Supplementary Tables 10, 11). Long switching events always occurred at rates below one switch per Mbp, and at the 0.9 threshold, we measured 0.5–0.8 switches per Mbp. This corresponds to accuracies of 99.87–99.90%, a drop of only about 0.01% with respect to two phasing libraries per sample.
Finally, results for the two replicate libraries of the HapMap sample NA12878 were highly concordant (Fig. 3 and Supplementary Table 12). The two libraries were prepared with exactly the same input parameters (e.g. fragment length, number of fragments per well); their performance metrics differed by less than 1% and the two replicates assigned the same phase to most SNVs (Online Methods).
Determining allele-specific DNA methylation
Next, as a demonstration of scientific applications based on the phased genome made possible by SLRH, we performed an analysis of differential DNA methylation across the genome of the HapMap sample NA12878 (lymphoblastoid cell line GM12878) based on its haplotype information. We thus obtained a detailed map of allele-specific methylation patterns within a human genome; such maps are useful for understanding biological mechanisms such as genomic imprinting.
In brief, we performed MethylC-seq experiment on GM12878 cell line and assigned methylated short reads to their closest haplotype. With the reads coverage and bisulfite conversion rates on both alleles, we then quantified allele-specific DNA methylation (ASM) using Fisher's exact test (Online methods). We found 216,034 statistically significant ASM events that clustered in 992 differentially methylated regions (DMRs) ranging in size from 6 to 3,181 bp (median size 190 bp, Online methods). Ten of the DMRs were located at previously studied areas of the genome, such as in the upstream region of the H19 gene23 (Fig. 4). The full list of DMRs and their associated genes is available in the Supplementary Material.
Figure 4.
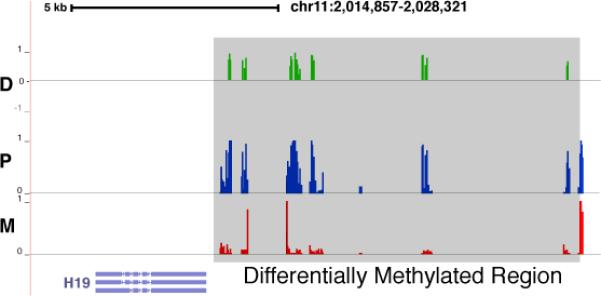
Genome browser view of differentially methylated regions at the promoter of the H19 gene. Differences in DNA methylation levels (green tracks, D) and the absolute DNA methylation level at the two parental alleles (blue tracks for paternal methylation (P) and red tracks for maternal methylation (M)) are shown around the H19 locus. The shaded regions show significant (P<0.05; Fisher's exact test) difference in DNA methylation levels between the two parental alleles and are identified as a DMR.
To gather more insight into how differential methylation may affect gene expression, we determined the overlap between the DMRs and transcription start sites (TSSs), transcription end sites (TESs), exons and intergenic regions defined by Genecode v14. Consistently with previous findings, the DMRs were significantly enriched at gene promoters (P < 2.2E-16, binomial test). About 20% of the DMRs were located at gene TSSs, and an additional 42% were located within annotated genes (which include TESs, introns and exons); the remaining 38% were found at distal intergenic regions (Supplementary Fig. 4). We further explored the regulatory role of the majority of DMRs that are not in gene promoters by assessing the overlap between the DMRs and DNase I hypersensitive sites and TF binding sites identified by ENCODE. We found that about 55% of the DMRs overlapped with TF binding sites and 82% overlapped with DNaseI hypersensitive sites (Supplementary Figs. 4 and 5). Overall, the above findings support the fact that differential methylation plays a role in gene regulation, particularly in the differential expression of genes.
We compared the ASM events we found with a previous study24 that studied methylation patterns within the HapMap sample NA12878 using reduced-representation bisulfite sequencing (RRBS). We discovered substantially more ASM events (216,034, compared to 2,998) than were previously found using RRBS, a method that targets only GC-enriched regions. Since MethylC-seq can detect DNA methylation in the whole genome while RRBS only detect DNA methylation in GC- enriched regions, our results suggest the prevalence of ASM events outside of CpG islands captured by RRBS technology. To our surprise, although 326 cytosines that were identified as ASM in the RRBS study also passed the criteria for testing in our study, only 96 were significantly (P<0.05, Fisher's exact test) differentially methylated between the two alleles. We suspect the RRBS technology may introduce high bias from the amplification that leads to high false positive rates.
Effects of PCR and Nextera on haplotyping performance
Both PCR and the Nextera transposase introduce errors in the haplotyping process; we assessed the significance of these errors by running Prism on a high-quality synthetic data set obtained by sampling 7 kbp reads uniformly at random from the trio-phased genome of NA12878 (Online Methods). Analysis of the synthetic data resulted in more complete haplotypes with a 0.4% higher SNV phasing rate. A further analysis of PCR amplification bias (Online methods) suggested that some areas of the genome exhibit a systematically lower amplification rate, and are covered by fewer long fragments.
The long-range switch accuracy on both datasets was similar, but the short switch accuracy was much higher on the synthetic dataset. This suggests that PCR and Nextera mainly introduce gaps in the phased haplotypes as well as point errors at individual variants; however, their impact on long-rage phase information appears to be small.
Discussion
The wealth of information one can obtain from a haplotype-resolved genome promises new advances in both biology and medicine. SLRH represents a step towards making such haplotype information easily obtainable.
Compared with existing dilution haplotyping methods71112, SLRH produces haplotypes of equal or greater quality using substantially less sequencing effort (Supplementary Table 1). Whereas existing methods require from 110 Gbp7 to 496 Gbp11 of sequencing, SLRH requires as little as 30 Gbp. Moreover, our method phases up to 99% of all SNVs, whereas others exhibit phasing rates of at most 97%12, and typically less than 95%7811. SLRH haplotypes also retain long-range phase information, with N50 lengths of 450–560 Kbp; alternative methods have N50 lengths from 350 Kbp712 to 600Kbp11.
Notably, SLRH achieves this performance without sacrificing accuracy: long-range switching events occur less than once per Mbp on average (99.90–99.92% long switch accuracy). For applications demanding an even higher level of precision, SLRH provides confidence scores that may be used to trade-off haplotype completeness for increased accuracy. At the most stringent thresholds, the method yields short and highly accurate regions that may be valuable in clinical applications.
The two components of SLRH that enable these advances are a low-bias PCR-based amplification step, and the Prism statistical phasing algorithm, which compensates for having relatively little input data. The two components naturally complement each other: although long fragments cannot span across long regions of low heterozygosity, such regions typically exhibit high linkage disequilibrium, and are more amenable to statistical phasing. The limitations of SLRH include the need to use a compute cluster for statistical phasing, and a lower phasing accuracy in statistically-assembled regions (Supplementary Table 13). The statistical component of SLRH also cannot be applied to species other than human due to the lack of a suitable reference panel. However, we expect that the molecular component can be applied to species with genomes of at least 100 Mbp, which are large enough for long fragments to be sufficiently diluted.
Finally, compared to a recently introduced proximity ligation-based method (HaploSeq6), our approach produces shorter, but more complete haplotype blocks. While HaploSeq phases 81% of SNVs in a human genome, SLRH phases 99%. The errors of HaploSeq mostly affect individual positions without altering the global haplotype structure; SLRH produces much fewer errors, but some of them may introduce long-range switching events. Overall, the two methods appear to have complementary strengths and weaknesses.
As an example of the scientific studies that are made possible by SLRH, we determined the allele-specific methylation patterns across a phased human genome. We observed many methylation events, and found that the DRMs are often associated with cis-regulatory regions. In previous studies, differential methylation patterns were determined either by purely statistical methods2526 or from Mendelian inheritance rules24. Such methods may be inaccurate and may not scale to large studies owing to the need to sequence the parents of every subject. Here, we were able to reproduce the work of previous studies? without relying on parental information or large amounts of sequencing.
Besides differential methylation studies, haplotype information has applications in many areas of genomics, including: (i) the analysis of disorders affected by compound heterozygocity, such as blistering skin27, cerebral palsy28, deafness29 and others;1 (ii) population genetics, where population-specific haplotype blocks are currently resolved using lower-accuracy statistical methods2; (iii) the detection of structural variations, which has been shown to benefit from phase information7; (iv) the matching of hosts and donors in organ transplantation based on the HLA region of the genome4; (v) the evolution of genomes across species30. The wide range of these fields highlights the importance of phase information in human genetics. Tools that facilitate access to this information such as ones we presented here will lay the foundation for further advances throughout genomics.
Supplementary Material
Acknowledgments.
We thank Dr. Cuiping Pan for assistance in coordinating contacts and discussions. This work is funded by NIH grants HL107393-02, HG004558-05, and the Genetics Department of Stanford University.
Footnotes
Author contributions
D.P. and M.K. developed the laboratory preparation protocol. V.K. developed the Prism phasing algorithm. Z.M. and R.C. performed the Methyl-seq experiments. T.B. prepared the phasing libraries. V.K., D.X. and D.P. performed computational analysis. V.K., D.X., and M.S. wrote the manuscript. R.C. and M.K reviewed and revised the manuscript. M.K. and M.S. supervised the research.
Competing financial interests
V.K., D.P., T.B. and M.K. performed the research at Moleculo Inc. (acquired by Illumina Inc.). D.P., T.B., and M.K. are employed by Illumina Inc. and V.K. is a consultant to Illumina Inc. M.K., D.P., T.B., and V.K. are listed as inventors on a patent filed for the SLRH technology. The library preparation protocol is covered by U.S. and international patents with numbers 61/532,882 and 13/608,778 on which D.P and M.K. are listed as inventors. The SLRH technology is offered commercially by Illumina Inc.
References
- 1.Tewhey R, Bansal V, Torkamani A, Topol EJ, Schork NJ. The importance of phase information for human genomics. Nat Rev Genet. 2011;12:215–223. doi: 10.1038/nrg2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Browning SR, Browning BL. Haplotype phasing: existing methods and new developments. Nat Rev Genet. 2011;12:703–714. doi: 10.1038/nrg3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roach JC, et al. Chromosomal haplotypes by genetic phasing of human families. Am. J. Hum. Genet. 2011;89:382–397. doi: 10.1016/j.ajhg.2011.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan HC, Wang J, Potanina A, Quake SR. Whole-genome molecular haplotyping of single cells. Nat Biotechnol. 2010;29:51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang H, Chen X, Wong WH. Completely phased genome sequencing through chromosome sorting. Proceedings of the National Academy of Sciences. 2011;108:12–17. doi: 10.1073/pnas.1016725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selvaraj S, R Dixon J, Bansal V, Ren B. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat Biotechnol. 2013;31:1111–1118. doi: 10.1038/nbt.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitzman JO, et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat Biotechnol. 2010;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duitama J, et al. Fosmid-based whole genome haplotyping of a HapMap trio child: evaluation of Single Individual Haplotyping techniques. Nucleic Acids Res. 2012;40:2041–2053. doi: 10.1093/nar/gkr1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruano G, Kidd KK, Stephens JC. Haplotype of multiple polymorphisms resolved by enzymatic amplification of single DNA molecules. Proc. Natl. Acad. Sci. U.S.A. 1990;87:6296–6300. doi: 10.1073/pnas.87.16.6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeffreys AJ, Neumann R, Wilson V. Repeat unit sequence variation in minisatellites: a novel source of DNA polymorphism for studying variation and mutation by single molecule analysis. Cell. 1990;60:473–485. doi: 10.1016/0092-8674(90)90598-9. [DOI] [PubMed] [Google Scholar]
- 11.Peters BA, et al. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature. 2012;487:190–195. doi: 10.1038/nature11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaper F, et al. Whole-genome haplotyping by dilution, amplification, and sequencing. Proceedings of the National Academy of Sciences. 2013;110:5552–5557. doi: 10.1073/pnas.1218696110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voskoboynik A, et al. The genome sequence of the colonial chordate, Botryllus schlosseri. Elife. 2013;2:e00569. doi: 10.7554/eLife.00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daelemans C, et al. High-throughput analysis of candidate imprinted genes and allele-specific gene expression in the human term placenta. BMC genetics. 2010;11:25. doi: 10.1186/1471-2156-11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suk E, et al. A comprehensively molecular haplotype-resolved genome of a European individual. Genome Res. 2011;21:1672–1685. doi: 10.1101/gr.125047.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McVean GA, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li N, Stephens M. Modeling Linkage Disequilibrium and Identifying Recombination Hotspots Using Single-Nucleotide Polymorphism Data. Genetics. 2003;165:2213–2233. doi: 10.1093/genetics/165.4.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howie BN, Donnelly P, Marchini J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delaneau O, Zagury J, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Meth. 2012;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 20.Delaneau O, Howie B, Cox AJ, Zagury J, Marchini J. Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 2013;93:687–696. doi: 10.1016/j.ajhg.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu F, et al. The UCSC Known Genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 22.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 23.Edwards CA, Ferguson-Smith AC. Mechanisms regulating imprinted genes in clusters. Current Opinion in Cell Biology. 2007;19:281–289. doi: 10.1016/j.ceb.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 24.Gertz J, et al. Analysis of DNA Methylation in a Three-Generation Family Reveals Widespread Genetic Influence on Epigenetic Regulation. PLoS Genet. 2011;7:e1002228. doi: 10.1371/journal.pgen.1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, et al. The DNA Methylome of Human Peripheral Blood Mononuclear Cells. PLoS Biol. 2010;8:e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welch KO, Marin RS, Pandya A, Arnos KS. Compound heterozygosity for dominant and recessive GJB2 mutations: effect on phenotype and review of the literature. Am. J. Med. Genet. A. 2007;143A:1567–1573. doi: 10.1002/ajmg.a.31701. [DOI] [PubMed] [Google Scholar]
- 28.Fong CYI, Mumford AD, Likeman MJ, Jardine PE. Cerebral palsy in siblings caused by compound heterozygous mutations in the gene encoding protein C. Dev Med Child Neurol. 2010;52:489–493. doi: 10.1111/j.1469-8749.2010.03618.x. [DOI] [PubMed] [Google Scholar]
- 29.Shimizu H, et al. Epidermolysis bullosa simplex associated with muscular dystrophy: phenotype-genotype correlations and review of the literature. J. Am. Acad. Dermatol. 1999;41:950–956. doi: 10.1016/s0190-9622(99)70252-5. [DOI] [PubMed] [Google Scholar]
- 30.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.