

Abstract
Antigen derived from viral infections with influenza and Vesicular Stomatitis Virus (VSV) can persist after resolution of infection. Here we show that antigen can similarly persist for weeks following viral challenge and vaccination. Antigen is captured by Lymphatic Endothelial Cells (LECs), under conditions that induce LEC proliferation. Consistent with published data showing that viral antigen persistence impacts on the function of circulating memory T cells, we find that vaccine elicited antigen persistence, found on LECs, positively influences the degree of protective immunity elicited by circulating memory CD8+ T cells. The coupling of LEC proliferation and antigen capture identifies a mechanism by which the LECs store, or “archive”, antigens for extended periods of time after antigen challenge, thereby increasing IFNγ/IL-2 production and enhancing protection against infection. These findings therefore have the potential to impact future vaccination strategies and our understanding of the role for persisting antigen in both vaccine and infectious settings.
Introduction
In conjunction with generating robust, enduring cellular immunity, antigen derived from infectious agents often persists for an extended period of time after the infection is cleared from the host 1–5. Flu antigens persist in the lung-draining lymph nodes 5–7 and within the respiratory tract 1,3 well after the clearance of the primary infection. While it is clear that the maintenance of CD8+ T cell memory does not require the presence of antigen over time 8,9, it is also clear that antigen persistence after viral infections can have an overall positive influence on the maintenance of protective T cell immunity. Flu-related antigens within the lung draining nodes or inducible Bronchus Associated Lymphoid Tissue (iBALT) periodically stimulate circulating memory cells to proliferate and differentiate into more effector-like cells 2,3,5,10. The proliferation that accompanies this process serves to increase the concentration of viral specific effector T cells within the local tissue, enhancing protective immunity by initiating a rapid response in situ to rechallenge 1,5. This type of antigen persistence is in sharp contrast to the high antigen loads resulting from chronic viral infections, which typically produce a hypo-responsive memory T cell pool 11. Thus, the functional significance of both persisting and chronic viral antigen are well established.
Considerably less clear are the precise cellular locations of persisting antigen that can stimulate antigen-specific CD8+ T cells. Follicular Dendritic Cells (FDCs) are a non-hematopoietic cell (CD45-) characterized by gp38 and CD21/35 expression, the latter (complement receptors 1 and 2) being important for the function of FDCs in holding antigen/antibody complexes for extended periods of time 12. Historically, FDCs have been documented as a source of persisting antigen 13 associated with antigen presentation to B cells and helper T cells. Specifically, persisting antigen was thought to be part of the process of follicle maturation and B cell memory formation where antigen is hidden within the tendrils of the FDC for later presentation to memory B cells 14. More recently, an elegant imaging study showed the mechanism by which antigen/antibody complexes are maintained for long periods of time, being continually internalized and resurfaced on the FDC 12.
While FDCs have been long thought to be the CD45- cell associated with all reservoirs of persisting antigen, there are other CD45- stromal subsets that line the subcasular sinus (lymphatic endothelial cells -LECs), blood vessels (Blood Endothelial Cells –BECs), or act as conduits for the migration and trafficking of T cells and DCs (Fibroblastic Reticular Cells-FRCs) within the lymph node or white pulp. Besides lining the lymph vessels, LECs project into the interfollicular ridges and T cell zones and are important for lymph node structure as well as some antigen presentation and dendritic cell trafficking 15–19. Most stromal cells express toll like receptors (TLRs) and may play a role during inflammatory responses as well 17. While unique mechanisms behind how FDCs hold onto whole antigen and cooperate with B cells were recently identified 12, no reports to date have established whether stromal cell types other than FDCs can facilitate antigen persistence following infection or vaccination.
Here, we demonstrate that both viral challenge and subunit vaccination induce antigen to persist in the host for times extending well beyond the peak of the T cell response to the immunologic challenge. Surprisingly, we found that the persisting antigen resides on LECs, a stromal cell subset not previously characterized for antigen capture and persistence. Perhaps most significant is our observation that antigen persistence is dependent not specifically on T or B cells, but instead on LEC proliferation in the context of a productive immune response. The coupling of LEC proliferation and antigen capture identifies a previously unappreciated mechanism by which the stroma in secondary lymphoid tissue maintains antigens against which a robust response has been initiated, in effect “archiving” these antigens for periods of time lasting well after the peak of normal immune response. Consequently, the presence of archived antigen on LECs results in a memory CD8 T cell pool with increased effector function and protective capacity. Thus, given the functional consequences of antigen persistence on T cells after viral infection 1–5 and vaccination, as we show here, these results are significant as the first report to identify LEC involvement in this process.
Results
Antigen persists following viral infection
Previous studies have shown that viral antigens can be detected in mice well after the peak of the CD8 T cell response and clearance of the infection. We obtained similar results using vaccinia virus as a model viral challenge. Persistence of viral antigen was observed by using the transfer of CFSE labeled transgenic T cells (OT1 transgenic cells, specific for the SIINFEKL peptide derived from ovalbumin) at various time points after infectious challenge with an ovalbumin-expressing strain of vaccinia virus (VV, VV-ova). The proliferation of the transferred cells, as gauged by the loss of CSFE fluorescence, was used as a functional readout for the presence or absence of persisting antigen upon transfer. Compared to infection with an ovalbumin-expressing strain of Listeria monocytogenes (LM, LM-ova) which resulted only in a brief period of antigen persistence 20, VV infection resulted in antigen persistence (Fig 1) long after (>30 days) the endogenous T cell response had peaked and declined to <1% of the total CD8+ T cells. This time frame of vaccinia virus antigen persistence is similar to that observed for flu antigens 1,3,5. We conclude from these data that virus-associated antigen persists for periods of time well after clearance of infection, and the magnitude of antigen persistence is sufficient to result in substantial proliferation of naïve antigen specific CD8+ T cells in the host.
Figure 1.
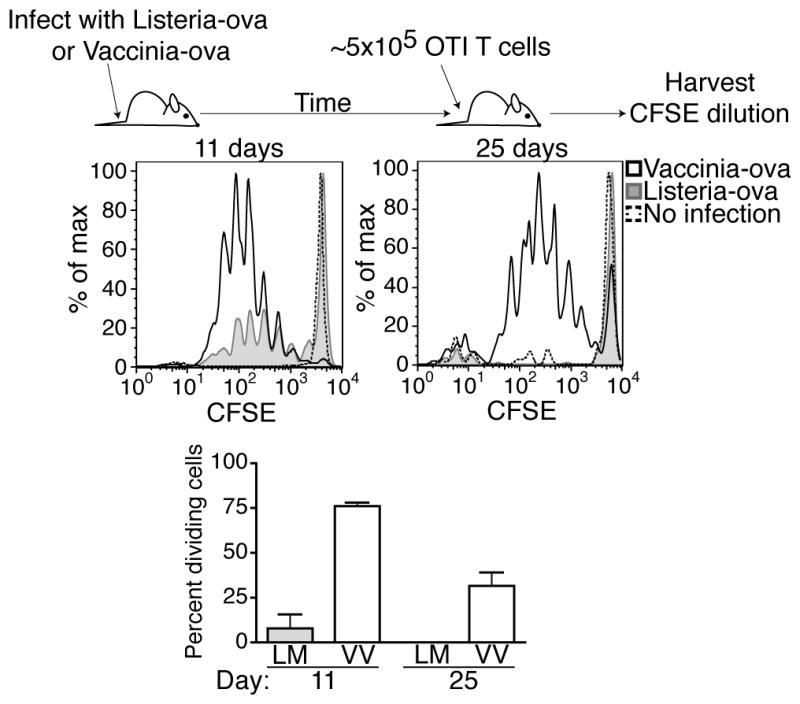
Antigen persists following infection with Vaccinia Virus. Mice were challenged with either VVova or LMova. CFSE labeled congenically different (CD45.1+) OT1 T cells were transferred on day eight or day twenty-two after challenge. Three days post T cell transfer, mice were sacrificed and single cell suspensions were generated from each spleen. Cells were positively gated for CD8 and CD45.1 and T cell proliferation was assessed by CFSE dilution by flow cytometery. The percent of OT1 cells that are dividing was calculated as described in the Methods. Data representative of 2 experiments with at least three mice per group with similar results. Error bars represent the standard error.
Antigen persists following subunit vaccination
To date, all model systems in which antigen persistence has been described utilize a viral challenge. However, vaccinia stimulates many different inflammatory pathways and expresses many different antigens. To simplify, we used one antigen and one TLR agonist along with a DC activator, anti-CD40, to be able to understand the mechanism by which antigen persists. We 21–28 and others 29,30 have published extensively on this TLR/αCD40 vaccination method. The reasoning behind examining antigen persistence to this particular vaccine was two fold. First, its capacity to elicit protective cellular immunity and its underpinning molecular mechanisms, are well-established 21–28. Second, the magnitude of the T cell response (CD4 and CD8) following combined TLR/CD40 vaccination is comparable to that observed following model infections such as Vaccinia (10–25% of the total T cell population end up as antigen-specific CD4 or CD8 T cells 21–31).
Mice were immunized with ovalbumin in the context of combined αCD40/polyI:C and at indicated time-points after immunization, OT1 T cells were transferred and their proliferation was assessed as an indicator of antigen persistence. As with viral challenge, antigen was not cleared rapidly post immunization, but instead was retained in the lymph node for at least 3–4 weeks (Fig 2A). Antigen persistence was not specific to ovalbumin because a similar amount of T cell proliferation was observed after immunization with HSV glycoprotein B (HSVgB) and transfer of T cells from the gBT-1 TCR transgenic (specific for the SSIEFARL epitope from HSVgB) (Fig 2B). Persistence was also not specific to the use of a particular TLR agonist or the use of anti-CD40 in the adjuvant as it was observed following TLR1/2 agonist (Pam3cys)/anti-CD40 (Fig 2C) or TLR7 agonist-antigen conjugate (3M019) 32,33 vaccination (Fig 2D). Thus, similar to viral challenge, immunization with a spectrum of vaccine adjuvants and antigens results in antigen persistence within the secondary lymphoid tissue.
Figure 2.

Antigen persists following subunit vaccination. A. Mice were immunized with 100μg of ovalbumin+50μg of polyI:C+50μg of αCD40 or with ovalbumin antigen alone (Ag only). Ten or twenty days post immunization CFSE labeled congenically labeled OT1 T cells were transferred into immunized mice and their division in the spleen assessed 3 days later as in Fig 1. B. As in (A) except using 50μg of whole HSVgB protein as the antigen and Violet Proliferation Dye (VPD) labeled HSVgB-specific TCR transgenic, gBT, as the transferred T cells. C. As in (A) except using 25μg pam3cys as the adjuvant. D. Mice were immunized subcutaneously with 50μg of a TLR7 agonist conjugated to ovalbumin. Antigen persistence was measured by proliferation of transferred OT1 cells in the draining lymph node as in A. The average percent of dividing cells and standard error of all mice in each group, calculated as described in the Methods are shown within each plot. Data representative of 2 experiments with at least three mice per group with similar results. Error bars represent the standard error.
Persistence is influenced by antigen dose and pro-inflammatory stimuli
With this reductionist model in hand, we next examined what aspects of the combined adjuvant were required to result in persistence of the antigen past the peak of the endogenous T cell response. Consistent with the results from the TLR7 conjugate vaccine, we observed that antigen persistence was independent of the anti-CD40 stimulation but dependent on the pro-inflammatory (TLR agonist) portion of the vaccine adjuvant (Fig 3A). This was also true of agonists for TLR2 (Supplementary Fig. 1). Additionally, antigen dose correlated with the length of time antigen persists (Fig 3B) and surprisingly small amounts of polyI:C (0.5μg) (Fig 3C) were sufficient to induce persistence of minimal amounts of antigen (10μg). PolyI:C is also an agonist for Mda5 34,35 and induces T cell responses independent of TLR3. This suggests that inducing antigen persistence may be more broadly applied to innate receptors in general and is not unique to the TLRs.
Figure 3.

Antigen persistence is dependent on innate receptor stimulation and is influenced by antigen dose. A. Mice were immunized IP with 10μg of ovalbumin in conjunction with either anti-CD40, polyI:C, or both. Six days post immunization OT1 T cells were CFSE labeled, transferred, and their division was assessed 3 days later as in figure 2A. B. Mice were immunized with the indicated amount of ovalbumin alone or with 50μg of polyI:C and 50μg of anti-CD40. OT1 division was visualized as in part A. C. Mice were immunized with 10μg of ovalbumin and the indicated amount of polyI:C and OT1 division was assessed as in part A. Data representative of 2 experiments with at least three mice per group with similar results.
Antigen persists independent of FDCs or B cells or CD4 help
FDCs are a radio-resistant stromal cell subset that can capture and harbor antigen over extended periods of time 1,12–14,36. The mechanism by which FDCs hold antigen is largely through antigen/antibody complexes and their interaction with CR2 (CD21)12,37–40. As such, we expected that if antigen persistence following viral infection or combined TLR/CD40 vaccination was dependent on this mechanism, then antigen persistence would be compromised in a CR2 deficient host. However, antigen persistence was largely intact in a CR2 deficient mouse following vaccination (Fig 4A). Furthermore, in hosts with either limited (HEL-specific BCR transgenic, MD4) or absent (μMT) B cell repertoires we found no defect in the ability to retain antigen (Fig 4B and C). As FDCs are absent from B cell deficient mice 41,42 and complement receptors are necessary for FDC antigen capture 12,37–40, this further diminishes a potential role for FDCs in antigen persistence. Using a mouse deficient for MHC class II we also confirmed that CD4 T cell help was also dispensable for antigen persistence (Fig 4D). Collectively these data are consistent with the conclusion that antigen persistence can occur independent of CD4+ T cells, B cells, FDCs, antibody production or antigen-antibody complexes.
Figure 4.

Lymphatic Endothelial Cells capture persisting antigen. A. Experiments performed as in Fig 2A, where Cr2−/− mice, B. μMT mice, C. MD4 mice, or D. MHC class II−/− mice were assessed for their ability to retain antigen. The average percent dividing cells and standard error of all mice in each group were calculated as described in the Methods. E. Mice were injected subcutaneously with 20μg of ovalbumin conjugated to AlexaFluor488, 2μg polyI:C and 2μg anti-CD40. Post immunization, mice were sacrificed and draining lymph nodes harvested and prepared and stained as described in the Methods. Cells shown are CD45-. Arrows indicate which numbered quadrant in the left most dot plot is being analyzed in the same numbered column of plots on the right to visualize fluorescent antigen on I. Fibroblastic Reticular Cells (FRC), Follicular Dendritic Cells (FDC), or Marginal Reticular Cells (MRC); II. Lymphatic Endothelial Cells (LEC); or III. Blood Endothelial Cells (BEC) one day (acute) or six days (persistent) post immunization. Experiments in E were repeated (10+ times) with at least 3 mice per group with similar results. Numbers not in parentheses show the percent of cells with the region in that plot. Numbers in parentheses show the average percent and standard error of the percent of cells within the region calculated from all mice in each group. F. Mice were immunized with 10μg of fluorescently labeled ovalbumin, 2μg of polyI:C, and 2μg of αCD40 subcutaneously as in E and draining LN removed, prepared and stained as in A on days 7, 14, or 21 post immunization. G. Mice were injected in the footpad with fluorescent antigen as in F. 13 days later, T cells and B cells labeled with VPD or CMTMR, respectively, were transferred intravenously into immunized mice and anti-Lyve-1 antibody (Alexa Fluor 647) was injected the footpad. 12–18 hours after transfer mice were sacrificed and popliteal lymph nodes were imaged by multi-photon microscopy as described in Methods. The bar represents 70μm in naïve, 100μm in Day14 and 50μm in the Day14 zoomed in box. Imaging experiments in G were repeated twice with similar results.
Antigen persists on Lymphatic Endothelial Cells
An important role for radio resistant cells in holding antigen following viral infection was shown in a previous study 1, though the precise radio resistant stromal cell subset was never specifically identified. Using established markers (Fig 4E and 19), we sought to determine if any of the known stromal cell subsets could be observed capturing and/or maintaining fluorescently labeled antigen at late time points after subunit vaccination. As we predicted from Fig 4A, FDCs possessed little to no detectable antigen 6 days after vaccination (Fig 4E). However, fluorescent antigen associated with LECs persisted for more than three weeks after vaccination (Fig 4F and G). This apparent antigen capture by LECs was not related to the use of ovalbumin as a model antigen since similar results were obtained with fluorescently labeled influenza nucleo-protein (NP) (Supplementary Fig. 2).
We further examined LECs and their capture of fluorescent antigen through the use of multi-photon microscopy 2 weeks after immunization with fluorescent antigen. To examine the localization of fluorescent antigen within the 3 dimensional architecture of the lymph node, fluorescently labeled polyclonal T and B cells were transferred intravenously, along with subcutaneous injection of fluorophore conjugated αLyve-1 antibody, into the immunized mice the day before lymph node excision and microscopy. This method effectively identified T cell and B cell regions as well as the LECs lining the subcapsular sinus and the interfollicular ridges, respectively. Consistent with the flow cytometric data, fluorescently labeled antigen could be seen encapsulating the lymph node and branching into the interfollicular ridges where LECs reside, and indeed specifically co-localized with a portion of the αLyve-1 staining (Fig 4G and Supplementary Movie 1).
Persisting antigen is not presented by LECs
Given that LECs can present antigens to the naïve T cell pool 16, we next examined whether or not the LECs were presenting the persisting vaccine-derived antigen directly to the circulating T cells. We initially used a pH sensitive fluorophore-coupled antigen (ova-DQ) to investigate whether LEC-acquired antigen was endocytosed into endocytic compartments involved in antigen processing and presentation. Mice were vaccinated with either ova-AF488 or ova-DQ in combination with polyIC/CD40 and one week later the stromal cells were examined by flow cytometry for the amount of fluorescence derived from either antigen. As before, greater than 50% of LECs were marked with the ova-AF488 antigen, indicating robust antigen capture. In contrast, ova-DQ fluorescence on LECs was minimal (Fig 5A), suggesting that the majority of injected ova-DQ failed to access a late endosomal compartment. These data indicate one of two possibilities. First, the majority of persisting antigen is maintained either on the cell surface or within a non-lysosomal endocytic compartment. If correct, then LECs would not be expected to directly present antigen to circulating T cells. Second, persisting antigen may be so slowly taken up for processing that it provides only a minimal fluorescent signal with the DQ reagent. If correct, then LECs would be expected to directly present antigen to T cells.
Figure 5.

Hematopoietically derived APCs are required for presenting persisting antigen to circulating T cells. A. B6 mice were vaccinated with either ova-AF488 or ova-DQ + adjuvant as in Fig 4E. Seven days later, mice were sacrificed and LEC fluorescent antigen capture (AF488) or LEC fluorescence (Ova-DQ) was determined by flow cytometry as described in Fig 4E and the Methods. Numbers not in parentheses show the percent of cells with the region in that plot. Numbers in parentheses show the average percent and standard error of the percent of cells within the region calculated from all mice in each group. B. WT or BM8 recipient mice were lethally irradiated and reconstituted with either WT or BM8 donor bone marrow. Reconstituted mice were immunized with ovalbumin, polyI:C and αCD40 and two weeks post immunization, CFSE labeled OT1 T cells were transferred monitored for CFSE dilution as in Fig 1. The average percent dividing cells and standard error of all mice in each group, calculated as described in the Methods, are shown within each plot. C. WT or BM8 mice were immunized and analyzed for fluorescently labeled ova on LECs as in B. Numbers in plot are as in A. D. Schematic representation of experimental method for E. Irradiated B6 mice reconstituted with CD11c-DTR bone marrow were injected with ova/polyIC/antiCD40 and at the indicated times post vaccination were injected with DT as described in the Methods. Fourteen days after vaccination, CFSE labeled OT1s were transferred and monitored for division as in Fig 1. E. Percent OT1s divided from each treatment group was calculated as described in Methods. Significance was calculated using a student’s t test. Asterisk represents a p-value <0.05. Experiments were repeated at least twice with three mice per group with similar results.
To distinguish between these two possibilities and address whether LECs could present captured antigen to circulating T cells, we utilizing mice with a mutant form of MHC (Kbm8) which cannot stimulate OT1 transgenic T cells with the SIINFEKL peptide 43,44. Using bone marrow chimeras, we determined whether hematopoietic and/or non-hematopoietic cells could present persisting antigen. Following reconstitution of WT or Kbm8 recipients with WT or K bm8 bone marrow, we observed that stimulation of T cells by persisting antigen required the presence of hematopoietic cells that can present the antigenic peptide (Fig 5B). The persistence of fluorescent antigen could be detected on WT or Kbm8 LECs (Fig 5C). Thus, antigen is able to persist on LECs despite the inability of antigen to be presented. However, no proliferation of the transferred OT1 cells occurred in the WT mice reconstituted with K bm8 bone marrow (Fig 5B), suggesting that even though antigen persists on LECs, it is not presented by LECs.
These data indicated that while persisting antigen was harbored by the LECs, antigen presentation to T cells required the participation of cells of hematopoietic origin. Dendritic cells (DCs) are critical to the response of naïve T cells in the majority of experimental systems, and as such were the main hematopoietic cell type we expected to be involved in presenting persisting antigen. Indeed, previous studies documented a central role for DCs in presenting antigen persisting after viral infection 1,20. We therefore utilized the CD11c-DTR mice which express the diphtheria toxin receptor on DCs, allowing the deletion of DCs by administration of Diphtheria Toxin (DT). Due to toxicity issues when using the intact CD11c-DTR host, we made chimeric mice by transferring bone marrow from CD11c-DTR mice into irradiated WT recipients. After reconstitution, mice were immunized with antigen and combined αCD40/polyI:C. DT was administered 5 or 13 days post immunization with a dose of DT determined to transiently deplete (for 24–48 hours) >98% of all CD11c+ cells from the secondary lymphoid tissue 23. Fourteen days after immunization (9 or 1 day after DC depletion) TCR transgenic T cells were transferred into DT treated and control hosts and the extent of their proliferation was again used to identify the persistence of antigen (Fig 5D). DC depletion 5 days after immunization (9 days before T cell transfer) failed to reduce antigen persistence as measured by the percent of transferred T cell proliferation (Fig 5E), confirming that DCs do not harbor the persisting antigen. In contrast, administration of DT one day before T cell transfer resulted in a significant (~60–70%) reduction in the proliferation of the transferred T cells (Fig 5E). Thus, consistent with previous data in viral antigen persistence, DCs play a central, though not exclusive, role in presenting the persisting antigen to circulating T cells. Collectively, we conclude that persistence of antigen on the LECs occurs independent of hematopoetically derived antigen presenting cells. However, LECs do not directly stimulate T cells specific for the persisting antigen. Rather, hematopoetic cells in general, DCs in specific, are required for the presentation of persisting antigen to circulating antigen specific CD8 T cells. Interestingly, these data also indicate that transfer of antigen between LECs and DCs or other antigen presenting cells (APCs) must occur for persisting antigen to be seen by circulating antigen specific T cells.
Acquisition of antigen by LECs as a consequence of LEC proliferation
Consistent with our data using the OT1 transfer system (Fig 4), we observed capture of fluorescent antigen by LECs in both μMT and Cr2−/− mice (Fig 6A). We next examined whether antigen specific T cell responses were necessary for antigen persistence. We initially used a Rag−/−×gBT-1 TCR transgenic to address this question as this host possesses T cells but cannot mount an ovalbumin specific response. Using both the adoptive transfer and fluorescently labeled antigen methods it was clear that there was a dramatic defect in the Rag−/−×gBT-1 mice to support persisting antigen (Fig 6A). However, closer examination of the LECs in this host revealed two interesting observations. First, in contrast to the WT hosts, both μMT and Rag−/−× gBT-1 mice had significantly fewer LECs than their WT counterparts (Fig 6B). Second, LECs in WT and μMT mice responded to vaccination by expanding between 3–6 fold (Fig 6B). This increase in LECs appeared to be the result of proliferation as noted by Ki67 staining (Fig 6C). In contrast, LECs in the Rag−/−× gBT-1 host failed to expand after immunization (Fig 6B).
Figure 6.

Antigen persists only under conditions of LEC proliferation. A. WT, μMT, gBT rag−/− mice were immunized with ova-488 + polyI:C + anti-CD40 and 7 days later, persisting antigen was measured by examining fluorescent antigen capture by LECs as in Fig 4E (for WT, Cr2−/− and gBT Rag−/−) and by CFSE dilution of labeled and transferred OT1 T cells as in Fig 1 (for WT and gBT Rag−/−). Numbers not in parentheses show the percent of cells with the region in that plot. Numbers in parentheses show the average percent and standard error of the percent of cells within the region calculated from all mice in each group. For CFSE dilution histograms, the numbers shown in each plot are calculated as in Figure 3. B. Total numbers of LECs were calculated from draining lymph nodes (axillary, brachial, inguinal, and popliteal). P-values were calculated using student’s T-test where one asterisk represents a p-value less than 0.05, two asterisks represent a p-value less than 0.01 and three asterisks represent a p-value less than 0.001. C. Mice were immunized as in A and sacrificed on the indicated days. Draining LNs were prepared and stained for identification of Ki67 staining in LECs. Experiments we repeated at least twice with 3–4 mice per group with similar results. Average and standard error from the experiment are shown in parentheses on the respective plot.
T cell expansion allows for antigen archiving during an immune response
There are two possible explanations for the lack of antigen persistence in the Rag−/−× gBT-1 host: i) an antigen specific T cell response is required for antigen acquisition/persistence by LECs, or ii) LEC expansion/cell division, independent of the specificity of the T cell response, is a necessary prerequisite to antigen acquisition/persistence. In this second possibility, vaccine-induced inflammation and the subsequent expansion of T cells within the draining LN would simply provide the necessary stimuli to the stroma to induce LEC proliferation via inflammation and/or baroreceptors. To distinguish between these possible mechanisms, we established a model that could track antigen persistence during a T cell response in which the persisting antigen and responding T cells were unrelated. We transferred CD4 TCR transgenic T cells specific for the Eα-derived peptide 3K 45 into WT or Rag−/−× gBT-1 hosts. One day after CD4 T cell transfer, mice were immunized with fluorescent ovalbumin with or without the 3K peptide. If the first model described above is correct (an antigen specific T cell response is required for the persistence of that antigen), then none of the Rag−/−× gBT-1 mice will demonstrate persistence of the ovalbumin antigen as this host cannot mount any response to ovalbumin. However, if the second possibility is correct (any T cell proliferation promotes antigen persistence), then the Rag−/−× gBT-1 hosts which are co-immunized with the 3K peptide will result in persistence of ovalbumin while the Rag−/−× gBT-1 hosts not immunized with 3K peptide will not.
Upon analysis of the LECs 12 days after immunization we observed that both WT and Rag−/−× gBT-1 hosts with expanded 3K specific CD4 T cells captured fluorescent antigen (Fig 7A and B) while the Rag−/−× gBT-1 mice not immunized with the 3K peptide failed to capture antigen. Additionally, the number of LECs in the CD4-expanded Rag−/−× gBT-1 host proliferated to a similar degree as the LECs in the WT host (Fig 7C–3K peptide).
Figure 7.
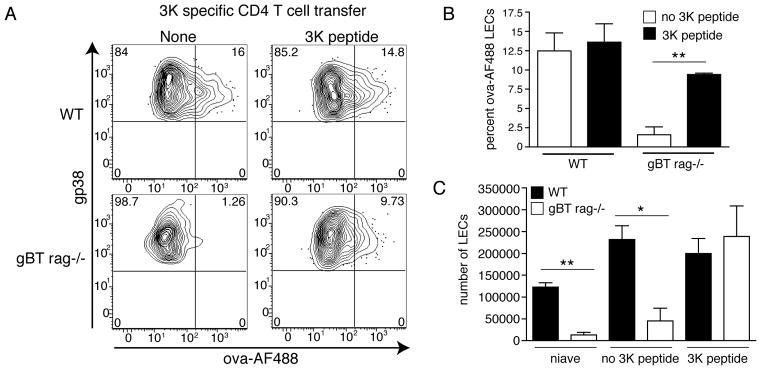
Non-antigen specific T cell expansion facilitates LEC proliferation and antigen archiving. A. One thousand CD4 T cells from a 3K specific CD4 T cell transgenic mouse were transferred into WT or gBT rag−/− mice 12–18 hours prior to immunization. Mice were left unimmunized or immunized subcutaneously with 10ug ova-488+2μg polyI:C+2μg αCD40 (as above) +/− 3K peptide. Mice were sacrificed 12 days post immunization and draining lymph nodes prepared as in Fig 4. Shown are CD45−, CD31+, gp38+ LECs. B. Quantitation of A. C. Total number of LECs expanded post immunization from A.
Collectively, we conclude form these data that a T cell response is a necessary component for inducing LEC expansion and subsequent antigen acquisition, not because of the specificity of the response, but because the expanding T cells promote the necessary stimuli to the stroma to induce LEC proliferation. These data support the model that LECs capture antigens involved in robust immune responses, in effect, “archiving” them based on whether or not the ensuing immune response stimulated LECs to proliferate.
CD8+ T cell memory formed under conditions of persisting antigen is beneficial to the maintenance of protective memory
Antigen persistence in viral model systems affects the functional status of circulating memory cells, increasing their effector functions and peripheral trafficking patterns 2,3,5,10. We therefore established a model system in which we could interrogate the functional response of memory CD8+ T cells formed in the presence or absence of persisting antigen. As a source of effector/memory T cells, we isolated T cells from an ovalbumin-immunized, congenically marked (CD45.2) Vβ5 TCR transgenic host. These mice express only the TCRβ chain of the OT1 TCR (Vβ5), requiring the recombination of, and pairing with, endogenous TCRα chains. As a result, the T cell repertoire has an elevated frequency of ova-specific T cells in the midst of a diverse repertoire of other specificities 46. Besides the advantage of having a polyclonal response to ovalbumin, the Vβ5 transgenic host does not show any of the functional abnormalities sometimes associated with traditional TCR transgenic cells 47.
Utilizing a modified version of the model described above, Rag−/−× gBT-1 (CD45.1) mice were transferred with 3K CD4 T cells and immunized with 3K peptide +/− whole ovalbumin (Fig 8A). Two weeks after immunization, Vβ5 effector/memory T cells were transferred IV into the different recipients. Effectively, these T cells then circulated in animals with (3K+ova) and without (3K only) LEC archived ovalbumin. The T cells were allowed 2 more weeks to complete memory formation in the presence or absence of persisting antigen (4 weeks after initial immunization) at which point all recipient mice were challenged with LM-ova. The phenotype and function of the transferred T cells were examined and bacterial loads in the liver determined in each host.
Figure 8.

Archived antigen enhances protective immunity. A. Schematic of the experimental design for parts B and C. 1000 3K specific CD4 T cells were transferred 1 day prior into gBTxrag−/− mice. gBTxrag−/− (with 3K cells) were immunized subcutaneously with the 100μg of the 3K peptide,10μg polyI:C, and 10μg α CD40 +/− 50μg ovalbumin (as in Fig 6). Simultaneously, Vβ5 mice were immunized with 100μg of ovalbumin, 50μg aCD40, and 50μg polyI:C. 2 weeks later, CD8+ T cells were isolated form the Vβ5 mice and 3×104 SIINFEKL-specific CD8 cells/recipient were transferred into the previously immunized gBTxrag−/− mice with (Ag+) or without (Ag−) ovalbumin. Two weeks later mice were infected with 1×105 LM-ova and 5 days after challenge, mice were sacrificed and analyzed for bacterial load and T cell phenotype/function. B. Spleen T cells from recipients described in A, stimulated directly ex-vivo with SIINFEKL peptide in brefeldin A and stained for intracellular IFNγ and IL-2. Cells are gated on live, CD8+, B220−, and CD45.1/2. Representative flow plots of the expression of IL-2 and IFNγ are shown with or without archived antigen. The number of IFNγ+ cells are quantified in the right panel of B. C. The number of antigen specific T cells from each mouse with archived or acute antigen exposure was calculated and plotted. D. The number of colony forming units was calculated per spleen on day 5 post infection. The limit of detection is displayed as a dotted line between 102 and 103 CFU. These experiments were repeated three times with 3–4 mice per group with similar results.
The transferred memory cells in the hosts with archived antigen showed increased IFNγ and IL-2 production after challenge with LM-ova as compared to memory T cells circulating in the hosts without archived antigen (Fig 8B). Consistent with the increased IL-2 production of these cells, there was a greater expansion of the transferred T cells in the host with archived antigen compared to the host without (Fig 8C), despite similar frequencies of antigen specific T cells before LM challenge (Supplementary Fig. 3). Further, archived antigen had a dramatic impact on protection against LM challenge, promoting a 2–3 logs reduction in bacterial load (Fig 8D). We conclude from these studies that LEC-archived antigen has a positive impact on the maintenance of protective CD8 T cell memory by influencing the capacity of the circulating memory cells to expand in response to secondary challenge.
Antigen archiving on LECs following Vaccinia infection
The mechanism by which antigen is captured and archived on LECs in our vaccine model system suggested that the same process should likely be involved during a viral infection. As mentioned above, stromal cell involvement in viral antigen persistence has been previously reported but has never ruled in or out any possible involvement of LECs. Indeed, challenge of both WT and CR2−/− hosts showed that Vaccinia-derived antigen persisted equally well in both of these hosts (Fig 9A), indicating that that FDC capture of antigen/antibody complexes is not responsible for antigen persistence following viral challenge. We therefore challenged mice with a strain of Vaccinia 48 expressing the Influenza NP protein conjugated to the fluorescent protein mCherry (NP-S-mCherry). Mice were infected in the footpad with NP-S-mCherry48 or control VV-ova. To identify sites of viral replication/antigen, lymph node sections from infected mice were stained to visualize the LECs, subcapsular macrophages, and B cell follicles by confocal microscopy (Fig 9B). At time points toward the end of viral replication within the node (three days post infection 49), virus fluorescence was easily detected in close proximity to the subcapsular region and interfollicular ridges (Fig 9C, E), consistent with previous reports 49,50. Well after the clearance of actively replicating virus 49 we observed a substantial reduction in the viral antigen fluorescence within the lymph node sections, as expected (Fig 9D). However, the association of viral antigen fluorescence with LECs was dramatically enriched over time as determined by proximity of viral fluorescence to αLyve-1 staining (Fig 9D, E). Thus, LECs appear to archive viral antigen for extended periods after viral challenge, indicating that LEC antigen capture and archiving is an active process for both infectious and vaccination settings.
Figure 9.

Vaccinia-related antigen persists on LECs. A. As in Fig 1, WT and Cr2−/− mice were challenged with VV-ova and antigen persistence was determined by transfer of CFSE labeled OT1 cells at 18 days post infection. As shown and calculated in Fig 1, the data is expressed as percent OT1 cells that divided 3 days after T cell transfer. The percent of OT1 cells that are dividing was calculated as described in the Methods. B. Schematic and staining of a lymph node from a mouse infected with Vaccina expressing ova (VV-ova) without the mCherry fluorophore. B cells are depicted in white, MOMA-1 subcapsular macrophages are depicted in green, and Lyve-1+ LECs are depicted in blue. Bar represents 15μm. C. Lymph node sections from mice 3 days after S.C. challenge with Vaccinia-NP-S-mCherry. VV-mCherry fluorescence is depicted in red. Bar represents 50μm. The white box indicates the area of the image analyzed under higher magnification on the right. The higher magnification image shows only the Lyve-1 and VV-mCherry fluorescence. Bar represents 10μm. D. Lymph node sections from mice 14 days after S.C. challenge with Vaccinia-NP-S-mCherry, analyzed as in B. In the left image the bar represents 50μm and at higher magnification the bar represents 10μm. E. Distance between Lyve-1 and mCherry fluorescence was calculated using the surfaces function in IMARIS. The image shown on the left demonstrates how the surfaces were made and the calculation on the right is the average of multiple images from at least 3 separate LN. The bar in the surfaces image represents 50μm. This experiment was repeated three times and images from multiple different lymph nodes were taken. The data shown are representative images from each time point.
Discussion
Our data demonstrate an unexpected role for LECs in harboring antigen following the induction of cellular immunity in response to vaccination or viral challenge. The capture/archiving of persisting antigen by this stromal cell requires a productive T cell response but is independent of the responding T cell specificity, instead coinciding with LEC proliferation within the local lymphoid tissue. These results present a novel mechanism by which the local secondary lymphoid tissue stroma can sense the degree of local inflammation and archive antigens against which a strong immune response has been generated. Though antigen persistence after infectious challenge is well-documented 1–5, our data show for the first time that vaccination can also lead to persistence of antigen for periods of time well beyond the rise and fall of the T cell response. Our data is also the first to demonstrate that LECs can serve as a reservoir for antigen that can be subsequently presented to antigen specific CD8 T cells by hematopoietically derived APCs, in turn leading to increased effector function and immune protection by circulating memory CD8 T cells.
It is necessary to contrast the antigen persistence observed in our data, as well as in the infectious models discussed above, from the high levels of antigen observed following some chronic viral infections and vaccine-related antigen depots. Though both result in the maintenance of antigen within the host for extended periods of time after the peak adaptive response, differences in the location of the antigen seem to correlate with the vastly different effects each type of persisting antigen has upon the development of protective immune memory. Chronic viral infections (such as LCMV clone13) produce high levels of antigen which persists within the peripheral tissues as well as within lymphoid tissues. In these cases, the T cell response ultimately suffers from tolerance mediated by clonal exhaustion 51. In contrast, antigen persistence in response to acute viral infections (influenza, vaccinia) or subunit vaccination as we show here, is largely, if not exclusively, localized within the secondary lymphoid organs (SLO). Data from the viral models show that this SLO-associated antigen periodically stimulates circulating memory cells to proliferate and/or differentiate into more effector-like cells 2,3,5,10, enhancing the trafficking of antigen-specific cells into the peripheral tissues and ultimately promoting more immediate protection against potential re-infection 1–5. Consistent with this, we found an increase in the number of IL-2+IFNγ+ cells and an increased protection against bacterial challenge in mice with archived antigen. Collectively the data suggest that chronic peripheral (non-lymphoid tissue) sites of antigen deposition are detrimental to the development and/or maintenance of immune memory while SLO sites of antigen promote protective memory by modulating the differentiation status of the circulating memory pool.
An important confirmation of this prediction is seen in the detrimental effect that vaccine emulsions can have on the magnitude/maintenance of immune memory. We 52 and others 53,54 have shown that the immune memory derived from subunit vaccination can be all but eliminated by simply formulating the vaccine into an emulsion 52,53. A recent publication by Hailemichael et al. elegantly shows how the emulsion forms a peripheral antigen depot which sequesters effector/memory cells, inducing their dysfunction and deletion 54. This is consistent with older data on antigen specific CD4+ T cells and their tendency to localize in the injection site 55. Thus, chronic viral infections and vaccine emulsion antigen depots have a similar negative impact on productive immune memory, likely through a common mechanism of tolerance/deletion enforced by long-lived antigen deposition within a peripheral tissue.
Ultimately, these data indicate that a better delineation between the various forms of antigen persistence within the vaccinated/challenged host is necessary. We propose the term “antigen archive” to describe residual antigen that persists within the SLO following vaccination or acute infection. An archive can be loosely defined as an active process by which material or data is preserved, a definition that matches well with the various observations of antigen capture and maintenance by SLO stromal cells. In this regard, our documentation of a central role for proliferating LECs in antigen archiving is both highly unexpected and significant. Previous work investigating how antigen persists showed necessary functions for the B cell follicle, follicular dendritic cells, and/or antigen antibody complexes in long term antigen persistence 1,36,40,56. A broad diversity of model systems, from infections to pregnancy, have documented various mechanisms by which FDCs capture and archive antigen for future B and T cell stimulation 36,57–60. Indeed we anticipated FDCs as the repository of archived antigen in our model system as well, making our demonstration of LECs as the antigen archive following subunit vaccination all the more significant.
Perhaps even more surprising is the role for LEC proliferation in the process of archiving antigen. It is as of yet unclear what specific signals are required to induce LEC proliferation in the context of vaccination and viral infection. Our data indicate that inflammation (Fig 2), and T cell expansion (Fig 7) are both necessary to induce LEC proliferation. The simplest model would suggest that these signals are perhaps necessary only to promote lymph node swelling which signals through baroreceptors to stimulate LEC proliferation in order to keep up with the growing size of the SLO. However, the data resulting from vaccination using αCD40 alone, or with LM infection, suggests a more complex model. Anti-CD40 vaccination of the host typically results in expansion of B cells in the SLO not to mention a degree of T cell expansion that is typically greater than that seen in response to other single adjuvants 21,22,27. Further, LM challenge clearly elicits robust CD4 and CD8+ T cell immunity in the presence of numerous inflammatory signals. However, neither promotes antigen archiving nearly to the degree that viral challenge or polyI:C do (Fig 1,2,3), indicating more specific signals are required for inducing antigen archiving than lymph node swelling alone. Given the propensity for antigen to persist following viral challenge and not bacterial, there may be additional requirements for direct activation of the stromal cells by the adjuvant/virus via specific innate receptors in order to induce antigen capture by LECs 17,61,62.
LECs have been shown to tolerize tumor/self-specific CD8 T cells by presenting self antigen 15,16,18,19,63. We were therefore somewhat surprised at the inability of the LECs to present archived antigen to circulating T cells (Fig 5). However, given the fact that only limited subsets of professional APCs are capable of MHC class I presentation of exogenous antigens (cross presentation)64–67, perhaps it is not surprising that the LECs can only present endogenous/self antigens via class I MHC but not the exogenously provided vaccine/viral antigens. The requirement for hematopoietic cells in the presentation of persisting antigen indicates that some form of antigen transfer between the LECs and the APC must occur, perhaps similar to that observed between different DC subsets 68, or FDCs and DCs 36. How and in what form the antigen might be captured and maintained is as yet undetermined. Our use of DQ-coupled antigen indicates that little of the retained antigen finds it way into late endosomal compartments (Fig 5A), suggesting that the retained antigen may be surface bound or perhaps involved in a form of recycling of surface bound antigen as has been recently demonstrated for FDCs 12. Antigen transfer between B cells and FDCs has been previously shown to involve CR1/2 which are not expressed on LECs 19,40. Whatever the mechanisms of antigen transfer, DCs are clearly responsible for the majority, though not all, of the presentation of the persisting antigen that is archived by the LECs (Fig 5). A reasonable prediction is that DCs from the peripheral tissues acquire antigen from the LECs as they migrate through the inter-follicular ridges and into the T cell zones of the local node. The molecular means by which the proliferating LECs capture and hold antigen and the long term impact on vaccine elicited cellular immunity is the focus of future work.
Methods
Mice
In general, 6–8 week old female WT, BM8, gBT-1 rag−/−, μMT, MHC classII−/−, Cr2−/−, MD4, 3K, OT1, and OT1 Vβ5 mice were all bred on a C57bl/6 background. Mice were purchased from either the National Cancer Institute or The Jackson laboratory unless otherwise stated and bred in house. Cr2−/− mice were a kind gift from Michael Holers laboratory at the University of Colorado, Denver, CO. OT1 mice are a TCR transgenic strain specific to the SIINFEKL peptide of ovalbumin (OVA257-264) in the context of H-2Kb. OT1 Vb5 TCR mice only express the transgene for the beta chain of the OT1 TCR. Normal thymic selection at the alpha chain leads to a diverse repertoire that has 1–3% ova specific CD8+ T cells. gBT-1 (gBT) mice (a kind gift from Bill Heath, University of Melbourne) are a TCR transgenic strain specific to the SSIEFARL peptide of Herpes Simplex Virus Glycoprotein B, HSV-1 gB 498–505 in the context of H-2Kb. 3K mice are a TCR transgenic specific to the 3K peptide which is a variant of the Ea peptide in the context of IAb 45. These mice were a kind gift from the Kappler/Marrack lab at National Jewish Health, Denver, CO. B6.C-H-2Kbm8 (BM8) H-2Kb mutant which cannot present the SIINFEKL peptide to OT1 T cells. BM8 mice were a kind gift from Larry Pease, Mayo Foundation, Rochester, MN. MD4 mice are an IgMa BCR transgenic strain specific to hen egg lysozyme (HEL). MD4 mice were a kind gift from John Cambier, University of Colorado, Denver, CO. All animal procedures were approved by the Institutional Animal Care and Use Committee at National Jewish Health.
Stromal and dendritic cell harvesting and staining
Mice were immunized with 0–20μg of antigen (ova-AF488 or ova-DQ (Life Technologies)) with 2μg polyI:C and 2μg αCD40 into 5 sites subcutaneously (footpads, flanks, scruff). Mice were sacrificed 1–21 days and popliteal, inguinal, axillary, and brachial lymph nodes were harvested into EHAA media without L-glutamine (GIBCO, Grand Island, NY). Lymph nodes were dissected using 22 guage needles to separate the tissue. Tissue was digested using 0.25mg of Liberase DL (Roche, Indianapolis, IN)/ml of EHAA media and DNAse (Worthington, Lakewood, NJ) for 1 hour at 37 degrees Celsius. An equal volume of 0.1M EDTA in Hank’s Buffered Saline Solution without calcium or magnesium (HBSS) was added and incubated for 5 minutes at 37 degrees. Digested LNs were then pushed through a screen and washed with 5mM EDTA in EHAA. All flow cytometry antibodies were purchase from Biolegend (San Diego, CA) unless otherwise stated. Stromal cells were stained with CD45 Pacific Blue clone 30-F11 (1:300), Gp38/podoplanin APC clone 8.1.1 (1:200), CD31 PerCp/Cy5.5 clone 390 (1:200), and CD21/35 PE clone 7E9 (1:200). As published 19 stromal cell subsets were identified as follows: BECs, CD45− gp38− CD31+ ; LECs, CD45− gp38+ CD31+ CD21/35−; FDCs, CD45− gp38+ CD31− CD21/35+ ; FRCs, and MRCs CD45− gp38+ CD31− CD21/35−. Dendritic cells were stained with CD11c APC/Cy7 clone N418 (1:400), B220 PE clone RA3-6B2 (1:300), CD8 PerCP/Cy5.5 clone 53–67 (1:200), and CD11b Pacific Blue clone M1/70 (1:750) (eBioscience, San Diego, CA). Dendritic cells shown are gated on live, CD11c+, B220−, and CD8+ CD11b− or CD8− CD11b+ cells. Cells were run on the DakoCytomation CyAn ADP flow cytometer (Fort Collins, CO) and acquired using Summit acquisition software. FlowJo software (Tree Star, Ashland, OR) was used to analyze flow cytometry data and cells were counted using a ViCell (Beckman Coulter, Brea, CA).
Protection Assay
Approximately 1000 3K CD4 T cells were transferred intravenously into gBT rag−/− mice (CD45.1). Cells were allowed to “park” overnight before immunizing with 100μg 3K peptide (ASFEAQKAKANKAVDKA)+ 10μg polyI:C+10μg anti-CD40 +/−50μg ovalbumin protein per mouse divided into the flanks, scruff, and footpads. The same day Vβ5 (CD45.2) mice were immunized with 100μg ovalbumin+50μg polyI:C+50μg anti-CD40. Immunization of Vβ5 with ovalbumin produces a substantial pool of memory cells within a diverse background of other specificities. Two weeks later the Vβ5 mouse was sacrificed and CD8 T cells were isolated from the spleen and lymph nodes using the STEMCELL CD8 T cell isolation kit. Isolated CD8s were stained with antibodies against, CD8 APC-Cy7 (Biolegend clon 53–6.7 1:600), CD45.1 PerCP Cy5.5 (Biolegend clone A20 1:200), CD45.2 APC (Biolegend clone 104 1:200), CD44 Pacific Blue (Biolegend clone IM7 1:400), and SIINFEKL specific tetramer (PE in house 1:200). 3×104 SIINFEKL specific T cells were transferred into each mouse intravenously. Two weeks post CD8 T cell transfer mice were challenged with 1×105 listeria monocytogenes expressing the ovalbumin protein. 4.5–5 days after LM challenge mice were sacrificed and spleens were cut in half. Half of the spleen was placed into 10 ml of 0.2% Igepal CA 630 (Sigma) and dissociated using a tissue grinder. Ground spleens were diluted in PBS. All dilutions were plated onto BHI (Bacto-Brain Heart Infusion-BD) media with 5μg/ml erythromycin (sigma) for LM-ova selection. Plates were incubated at 37 degrees Celsius for 2 days and colonies were counted. The other half of the spleen was placed into complete media and macerated with glass slides, red blood cells were lysed, and the single cell suspension was stimulated in Brefeldin A with or without the SIINFEKL peptide for 4 hours at 37 degrees Celsius. After stimulation, cells were stained with Biolegend antibodies: CD8 APC-Cy7 (clone53-6.7 1:600), B220 AF488 (clone RA3-6B2 1:300), CD45.1 PerCP Cy5.5 (clone A20 1:200), and CD45.2 Brilliant Violet (clone 104 1:200). Cells were fixed with 1% PFA in 4% sucrose for 10 minutes and washed twice with FACs buffer. Cell membranes were then permeabilized with perm wash (BD) and intracellular cytokines IL2 PE (clone JE56-5H4 1:100) and IFNγ APC (clone XMG1.2 1:100) were used to stain cells overnight in perm wash. Cells were washed and run on the flow cytometer as described above.
OT1 transfer assays
Mice were immunized 1–30 days prior to OT1 T cell transfer. CD45.1 congenically marked OT1 T cells were isolated by negative selection using the Milltenyi (Milltenyi, Auburn, CA) or Stem Cell Technologies (Vancouver, BC) CD8 negative selection kits. Greater than 90% CD8 T cell purity was obtained by either method. Following negative selection OT1s were labeled with CFSE or Violet Proliferation Dye (VPD) (Invitrogen) and 2×105–1×106 labeled T cells were transferred into immunized C57bl/6 mice (CD45.2). 3 days post transfer mice were sacrificed and spleens and draining lymph nodes were harvested. Tissues were macerated with glass slides, RBC lysed with ACK buffer, and stained with CD45.1, CD45.2, B220, CD8, CD44 and SIINFEKL specific tetramer (as above) to assess transferred OT1 T cell proliferation. Determination of the fraction of cells that have divided at least once (percent dividing cells) was calculated as previously described 69 using the equation fraction diluted= Σi1 Ni/2i/Σi0 Ni/2i where i is the generation number (0 is the undivided population), and Ni is the number of events in generation i.
Vaccines and Pathogen Challenge
Draining lymph nodes and spleen were used to gather activated T cells. Ovalbumin (Sigma-Aldrich, St. Louis, MO) or HSVgB (produced in the lab using baculovirus expression) were decontaminated of lipopolysaccharide (LPS) using a Triton X-114 LPS detoxification method 70 were used in combination with polyI:C (GE Healthcare), pam3cys [N-palmitoyl-S-2,3-bis(palmitoyloxy)-(2R,S)-propyl]-(R)-cysteine-(S)serine-(S)lysine 4] (InvivoGen, San Diego, CA), anti-CD40 (clone FGK4.5, BioXCell, West Lebanon, NH) or the TLR7a agonist, 3M012, conjugated to ovalbumin 32,33. For pathogen challenge, female B6 mice were injected with either 1×107 pfu/mouse of Vaccinia Virus Western Reserve Strain expressing the ovalbumin protein (VV-ova) or 1×105 Listeria Monocytogenes (Lm) expressing whole Ovalbumin protein (Lm-ova).
Bone Marrow chimeras
WT or BM8 mice were irradiated with 900 Rads and rested for 4 hours before reconstitution with either WT, BM8, or CD11c-DTR bone marrow. Bone marrow was isolated and red blood cells were lysed prior to intravenous transfer. At 12 weeks post reconstitution, mice were immunized as described above. For CD11c DTR experiments, mice were vaccinated and 100ng of diptheria toxin (DT) (Sigma Aldrich) was injected intraperitoneally at the indicated time points as previously described 23.
Multi-photon Microscopy
Multiphoton imaging was done using an Olympus FV1000MPE microscope with a XLPLN25XWMP Super 25x 1.05-N.A. water immersion objective and a Spectra Physics 10-W Mai-Tai DeepSee-OL laser. Bulk T cells were purified as described above with a miltenyi column and labeled with Violet proliferation dye (VPD) (BD biosciences) for 25 minutes at 37 degrees C and washed. Bulk B cells were purified using negative selection with PE conjugated antibodies (0.014μl/106 cells) to CD3 (Biolegend clone 145-2C11), CD4 (BD Pharmingen clone GK1.5), CD8 (ebioscience clone 53-6.7), CD11c (Biolegend clone N418), CD11b (BD Pharmingen M1/70), NK1.1 (Biolegend clone DK136), IA/IE (Biolegend clone M5/114), and CD49d (Biolegend clone DX5). Cells were bound to anti-PE magnetic beads and run over a miltenyl LS column and the flow through was taken. The purified B cells were labeled with CMTMR (Invitrogen) for 25 minutes at 37 degrees C and washed. 5×10^6 total B and T cells were injected into mice intravenously 12–18 hours prior to imaging. 2–5 μg of αLyve-1 conjugated to Alexa fluor 647 was injected subcutaneously into the footpad of immunized and transferred mice 12–18 hours prior to imaging. Popliteal lymph nodes were excised and fat was removed. Explanted lymph nodes were immobilized to coverslips with efferent lymphatics adhered to coverslips. A 3×3 grid of Z-stack images with each XY plane spanning 509μm × 509μm at a resolution of 0.994μm/pixel was acquired to encompass the whole lymph node. Each Z-stack covered a total depth of 300μm in 5μm Z-step increments. T cell VPD fluorescence was captured by the 450–490 nm filter, ovalbumin conjugated to Alexa fluor 488 was captured by the 500–550nm filter, B cell CMTMR fluorescence was captured by the 575–640nm filter and Lyve-1 alexa fluor 647 was captured by the 645–685 filter for channel acquisition.
Analysis of lymph node Z-stack images was done using Xuv stitch GUI 1.8.099×64.Ink software to reassemble together the 9 images taken from the explanted lymph node in order to visualize the entire node. The channel arithmetics function in the Imaris software (Bitplane) was used to compensate between colors on the lymph node images with no antigen. The same settings were used for lymph node images from the antigen-immunized mice that were calculated using the control naïve mouse. Using the colocalization function in Imaris, Lyve-1 and antigen co-staining were made into a new channel and visualized as yellow.
Confocal Microscopy
Mice were infected with 1×105 VV-ova or VV-NP-S-mcherrry (Yewdell) subcutaneously in the foot pad 3 or 14 days prior to sacrifice. Popliteal lymph nodes were harvested from infected mice and fixed for 45 minutes in 1% paraformaldehyde before imbedding the node into OCT. 10um sections were cut using a cryostat and stained with Lyve-1 (R&D systems) either directly conjugated to AF647 (Invitrogen) (1:50) or αLyve-1 biotin (R&D systems 1:100) followed by strepavidin pacific blue (R&D systems 1:500), MOMA-1 FITC (AbC Serotec clone MCA947F 1:50), and B220 biotin (BD Pharmingen RA3-6B2 1:100) followed by strepavidin pacific blue or IgM AF650 (A kind gift from Raul Torres-clone R33.24.12 1:100). Sections were imaged using a Zeiss LSM 700 confocal microscope at 40x magnification. For VVmCherry infected nodes, 3×3 scans were acquired with approximately 20 0.5um Z-sections. Images were analyzed using Imaris software as described above. Following channel arithmetics surface masks were created for LECs, B cells and VV-associated mcherry fluorescence. The number of mcherry surfaces less than two microns from other created surfaces was calculated using a script created in Matlab (Mathworks). Multiple lymph node sections were analyzed from 5 lymph nodes of infected mice.
Statistical analysis
Statistical analysis was done using the Student’s t test in prism4 (GraphPad, San Diego, CA). One asterisk represents a p-value of less than 0.05 and two asterisks a p-value of less than 0.01. Each analysis was done with at least 3 mice per group and each experiment was done at least twice with the same results.
Supplementary Material
Acknowledgments
We would like to thank Larry Pease for BM8 mice, Pippa Marrack for μMT mice, 3K peptide and 3K TCR transgenic mice, Jon Yewdell for VV-NP-S-mcherry, and Michael Holers for the Cr2−/− mice. We would also like to thank Jordan Jacobelli and Rachel Friedman for their guidance with multi-photon microscopy as well as Peter Beemiller and Bonnie Levitt for programming of image analysis scripts. This work was funded by an American Cancer Society fellowship (BAT), and Cancer Research Institute Irvington Post Doctoral Fellowship (MAB), and NIH grants AI099863 and AI06612.
Footnotes
Author contributions: BAT, MAB, and RMK designed and conceived experiments, BAT and MAB performed experiments and BAT and RMK wrote the paper.
Competing financial interests: There are no competing financial interests from any of the authors.
References
- 1.Kim TS, Hufford MM, Sun J, Fu YX, Braciale TJ. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J Exp Med. 2010;207:1161–1172. doi: 10.1084/jem.20092017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim TS, Sun J, Braciale TJ. T cell responses during influenza infection: getting and keeping control. Trends Immunol. 2011;32:225–231. doi: 10.1016/j.it.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takamura S, et al. The route of priming influences the ability of respiratory virus-specific memory CD8+ T cells to be activated by residual antigen. J Exp Med. 2010;207:1153–1160. doi: 10.1084/jem.20090283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zammit DJ, Cauley LS, Pham QM, Lefrancois L. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity. 2005;22:561–570. doi: 10.1016/j.immuni.2005.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zammit DJ, Turner DL, Klonowski KD, Lefrancois L, Cauley LS. Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity. 2006;24:439–449. doi: 10.1016/j.immuni.2006.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jelley-Gibbs DM, et al. Unexpected prolonged presentation of influenza antigens promotes CD4 T cell memory generation. J Exp Med. 2005;202:697–706. doi: 10.1084/jem.20050227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jelley-Gibbs DM, et al. Persistent depots of influenza antigen fail to induce a cytotoxic CD8 T cell response. J Immunol. 2007;178:7563–7570. doi: 10.4049/jimmunol.178.12.7563. [DOI] [PubMed] [Google Scholar]
- 8.Murali-Krishna K, et al. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 1999;286:1377–1381. doi: 10.1126/science.286.5443.1377. [DOI] [PubMed] [Google Scholar]
- 9.Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol. 2003;4:835–842. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- 10.Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol. 2009;9:153–161. doi: 10.1038/nri2496. [DOI] [PubMed] [Google Scholar]
- 11.Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol. 2007;19:408–415. doi: 10.1016/j.coi.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Heesters BA, et al. Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity. 2013;38:1164–1175. doi: 10.1016/j.immuni.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tew JG, Phipps RP, Mandel TE. The maintenance and regulation of the humoral immune response: persisting antigen and the role of follicular antigen-binding dendritic cells as accessory cells. Immunol Rev. 1980;53:175–201. doi: 10.1111/j.1600-065x.1980.tb01044.x. [DOI] [PubMed] [Google Scholar]
- 14.Szakal AK, Tew JG. Follicular dendritic cells: B-cell proliferation and maturation. Cancer Res. 1992;52:5554s–5556s. [PubMed] [Google Scholar]
- 15.Cohen JN, et al. Lymph node-resident lymphatic endothelial cells mediate peripheral tolerance via Aire-independent direct antigen presentation. J Exp Med. 2010;207:681–688. doi: 10.1084/jem.20092465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lund AW, et al. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012;1:191–199. doi: 10.1016/j.celrep.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol. 2009;9:618–629. doi: 10.1038/nri2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nichols LA, et al. Deletional self-tolerance to a melanocyte/melanoma antigen derived from tyrosinase is mediated by a radio-resistant cell in peripheral and mesenteric lymph nodes. J Immunol. 2007;179:993–1003. doi: 10.4049/jimmunol.179.2.993. [DOI] [PubMed] [Google Scholar]
- 19.Roozendaal R, Mebius RE. Stromal cell-immune cell interactions. Annu Rev Immunol. 2011;29:23–43. doi: 10.1146/annurev-immunol-031210-101357. [DOI] [PubMed] [Google Scholar]
- 20.Turner DL, Cauley LS, Khanna KM, Lefrancois L. Persistent antigen presentation after acute vesicular stomatitis virus infection. J Virol. 2007;81:2039–2046. doi: 10.1128/JVI.02167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahonen CL, et al. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J Exp Med. 2004;199:775–784. doi: 10.1084/jem.20031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahonen CL, et al. Enhanced efficacy and reduced toxicity of multifactorial adjuvants compared with unitary adjuvants as cancer vaccines. Blood. 2008;111:3116–3125. doi: 10.1182/blood-2007-09-114371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kurche JS, Burchill MA, Sanchez PJ, Haluszczak C, Kedl RM. Comparison of OX40 ligand and CD70 in the promotion of CD4+ T cell responses. J Immunol. 2010;185:2106–2115. doi: 10.4049/jimmunol.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurche JS, Haluszczak C, McWilliams JA, Sanchez PJ, Kedl RM. Type I IFN-dependent T cell activation is mediated by IFN-dependent dendritic cell OX40 ligand expression and is independent of T cell IFNR expression. J Immunol. 2012;188:585–593. doi: 10.4049/jimmunol.1102550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McWilliams JA, Sanchez PJ, Haluszczak C, Gapin L, Kedl RM. Multiple innate signaling pathways cooperate with CD40 to induce potent, CD70-dependent cellular immunity. Vaccine. 2010;28:1468–1476. doi: 10.1016/j.vaccine.2009.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez PJ, Kedl RM. An alternative signal 3: CD8(+) T cell memory independent of IL-12 and type I IFN is dependent on CD27/OX40 signaling. Vaccine. 2011;30:1154–1161. doi: 10.1016/j.vaccine.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez PJ, McWilliams JA, Haluszczak C, Yagita H, Kedl RM. Combined TLR/CD40 stimulation mediates potent cellular immunity by regulating dendritic cell expression of CD70 in vivo. J Immunol. 2007;178:1564–1572. doi: 10.4049/jimmunol.178.3.1564. [DOI] [PubMed] [Google Scholar]
- 28.Tamburini BA, Kedl RM, Bellgrau D. IL-6-inducing whole yeast-based immunotherapy directly controls IL-12-dependent CD8 T-cell responses. J Immunother. 2011;35:14–22. doi: 10.1097/CJI.0b013e3182356888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bullock TN, Yagita H. Induction of CD70 on dendritic cells through CD40 or TLR stimulation contributes to the development of CD8+ T cell responses in the absence of CD4+ T cells. J Immunol. 2005;174:710–717. doi: 10.4049/jimmunol.174.2.710. [DOI] [PubMed] [Google Scholar]
- 30.Van Deusen KE, Rajapakse R, Bullock TN. CD70 expression by dendritic cells plays a critical role in the immunogenicity of CD40-independent, CD4+ T cell-dependent, licensed CD8+ T cell responses. J Leukoc Biol. 2010;87:477–485. doi: 10.1189/jlb.0809535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lind EF, et al. Dendritic cells require the NF-kappaB2 pathway for cross-presentation of soluble antigens. J Immunol. 2008;181:354–363. doi: 10.4049/jimmunol.181.1.354. [DOI] [PubMed] [Google Scholar]
- 32.Oh JZ, Kedl RM. The capacity to induce cross-presentation dictates the success of a TLR7 agonist-conjugate vaccine for eliciting cellular immunity. J Immunol. 2010;185:4602–4608. doi: 10.4049/jimmunol.1001892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh JZ, Kurche JS, Burchill MA, Kedl RM. TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood. 2012;118:3028–3038. doi: 10.1182/blood-2011-04-348839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCartney S, et al. Distinct and complementary functions of MDA5 and TLR3 in poly(I:C)-mediated activation of mouse NK cells. J Exp Med. 2009;206:2967–2976. doi: 10.1084/jem.20091181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Cella M, Gilfillan S, Colonna M. Cutting edge: polyinosinic:polycytidylic acid boosts the generation of memory CD8 T cells through melanoma differentiation-associated protein 5 expressed in stromal cells. J Immunol. 184:2751–2755. doi: 10.4049/jimmunol.0903201. [DOI] [PubMed] [Google Scholar]
- 36.McCloskey ML, Curotto de Lafaille MA, Carroll MC, Erlebacher A. Acquisition and presentation of follicular dendritic cell-bound antigen by lymph node-resident dendritic cells. J Exp Med. 2010;208:135–148. doi: 10.1084/jem.20100354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klaus GG, Humphrey JH, Kunkl A, Dongworth DW. The follicular dendritic cell: its role in antigen presentation in the generation of immunological memory. Immunol Rev. 1980;53:3–28. doi: 10.1111/j.1600-065x.1980.tb01038.x. [DOI] [PubMed] [Google Scholar]
- 38.Qin D, et al. Evidence for an important interaction between a complement-derived CD21 ligand on follicular dendritic cells and CD21 on B cells in the initiation of IgG responses. J Immunol. 1998;161:4549–4554. [PubMed] [Google Scholar]
- 39.Roozendaal R, Carroll MC. Complement receptors CD21 and CD35 in humoral immunity. Immunol Rev. 2007;219:157–166. doi: 10.1111/j.1600-065X.2007.00556.x. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J Exp Med. 2009;206:1485–1493. doi: 10.1084/jem.20090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cerny A, Zinkernagel RM, Groscurth P. Development of follicular dendritic cells in lymph nodes of B-cell-depleted mice. Cell Tissue Res. 1988;254:449–454. doi: 10.1007/BF00225818. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez M, Mackay F, Browning JL, Kosco-Vilbois MH, Noelle RJ. The sequential role of lymphotoxin and B cells in the development of splenic follicles. J Exp Med. 1998;187:997–1007. doi: 10.1084/jem.187.7.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nikolic-Zugic J, Bevan MJ. Role of self-peptides in positively selecting the T-cell repertoire. Nature. 1990;344:65–67. doi: 10.1038/344065a0. [DOI] [PubMed] [Google Scholar]
- 44.Nikolic-Zugic J, Carbone FR. The effect of mutations in the MHC class I peptide binding groove on the cytotoxic T lymphocyte recognition of the Kb-restricted ovalbumin determinant. Eur J Immunol. 1990;20:2431–2437. doi: 10.1002/eji.1830201111. [DOI] [PubMed] [Google Scholar]
- 45.Dai S, et al. Crossreactive T Cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity. 2008;28:324–334. doi: 10.1016/j.immuni.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25:261–270. doi: 10.1016/j.immuni.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Badovinac VP, Haring JS, Harty JT. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity. 2007;26:827–841. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hickman HD, et al. Chemokines control naive CD8+ T cell selection of optimal lymph node antigen presenting cells. J Exp Med. 2011;208:2511–2524. doi: 10.1084/jem.20102545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol. 2002;3:265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- 50.Hickman HD, et al. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol. 2008;9:155–165. doi: 10.1038/ni1557. [DOI] [PubMed] [Google Scholar]
- 51.Ciurea A, et al. Persistence of lymphocytic choriomeningitis virus at very low levels in immune mice. Proc Natl Acad Sci U S A. 1999;96:11964–11969. doi: 10.1073/pnas.96.21.11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burchill MA, et al. T cell vaccinology: exploring the known unknowns. Vaccine. 2013;31:297–305. doi: 10.1016/j.vaccine.2012.10.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Celis E. Toll-like receptor ligands energize peptide vaccines through multiple paths. Cancer Res. 2007;67:7945–7947. doi: 10.1158/0008-5472.CAN-07-1652. [DOI] [PubMed] [Google Scholar]
- 54.Hailemichael Y, et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat Med. 2013;19:465–472. doi: 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reinhardt RL, Bullard DC, Weaver CT, Jenkins MK. Preferential accumulation of antigen-specific effector CD4 T cells at an antigen injection site involves CD62E-dependent migration but not local proliferation. J Exp Med. 2003;197:751–762. doi: 10.1084/jem.20021690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol. 2008;20:14–25. doi: 10.1016/j.smim.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carrasco YR, Batista FD. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity. 2007;27:160–171. doi: 10.1016/j.immuni.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 58.Junt T, et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450:110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 59.Martinez-Pomares L, Gordon S. Antigen presentation the macrophage way. Cell. 2007;131:641–643. doi: 10.1016/j.cell.2007.10.046. [DOI] [PubMed] [Google Scholar]
- 60.Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol. 2007;8:992–1000. doi: 10.1038/ni1494. [DOI] [PubMed] [Google Scholar]
- 61.Kataru RP, et al. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity. 2011;34:96–107. doi: 10.1016/j.immuni.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 62.Pegu A, et al. Human lymphatic endothelial cells express multiple functional TLRs. J Immunol. 2008;180:3399–3405. doi: 10.4049/jimmunol.180.5.3399. [DOI] [PubMed] [Google Scholar]
- 63.Yip L, et al. Deaf1 isoforms control the expression of genes encoding peripheral tissue antigens in the pancreatic lymph nodes during type 1 diabetes. Nat Immunol. 2009;10:1026–1033. doi: 10.1038/ni.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bedoui S, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol. 2009;10:488–495. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- 65.den Haan JMM, Lehar SM, Bevan MJ. CD8+ but Not CD8− Dendritic Cells Cross-prime Cytotoxic T Cells In Vivo. J Exp Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heath WR, Carbone FR. Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol. 2001;19:47–64. doi: 10.1146/annurev.immunol.19.1.47. [DOI] [PubMed] [Google Scholar]
- 67.Hildner K, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Allan RS, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 69.Roederer M. Interpretation of cellular proliferation data: avoid the panglossian. Cytometry A. 2013;79:95–101. doi: 10.1002/cyto.a.21010. [DOI] [PubMed] [Google Scholar]
- 70.Anis MM, Fulton SA, Reba SM, Harding CV, Boom WH. Modulation of naive CD4+ T-cell responses to an airway antigen during pulmonary mycobacterial infection. Infect Immun. 2007;75:2260–2268. doi: 10.1128/IAI.01709-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.