

Abstract
Defining the role of epigenetic regulators in hematopoiesis has become critically important, as recurrent mutations or aberrant expression of these genes has been identified in both myeloid and lymphoid hematological malignancies. We found that PRMT4, a type I arginine methyltransferase, whose function in normal and malignant hematopoiesis is unknown, is overexpressed in AML patient samples. Overexpression of PRMT4 blocks the myeloid differentiation of human stem/progenitor cells (HSPCs) while its knockdown is sufficient to induce myeloid differentiation of HSPCs. We demonstrated that PRMT4 represses the expression of miR-223 in HSPCs via the methylation of RUNX1, which triggers the assembly of a multi-protein repressor complex that includes DPF2. As part of a feedback loop, PRMT4 expression is repressed post-transcriptionally by miR-223. Depletion of PRMT4 results in differentiation of myeloid leukemia cells in vitro and their decrease proliferation in vivo. Thus, targeting PRMT4 holds potential as a novel therapy for acute myelogenous leukemia.
Introduction
Arginine methylation is a common post-translational modification that regulates the function of a wide range of proteins. There are ten members of the protein arginine methyltransferase (PRMT) family, eight of which catalyze the formation of either asymmetric di-methylarginine (the type I enzymes) or symmetric di-methylarginine (the type II enzymes) (Bedford and Clarke, 2009). The type I protein arginine methyltransferase 4 (PRMT4), also called co-activator-associated arginine methyltransferase 1 (CARM1), functions as a co-activator of nuclear hormone receptors as well as other transcription factors including p53 (An et al., 2004), NF-kappa B (Covic et al., 2005), β-catenin (Koh et al., 2002) and Mef2c (Chen et al., 2002). PRMT4 can methylate the transcriptional co-activator, p300 (Xu et al., 2001) (Lee et al., 2011), and several histone substrates, in particular H3R17 and H3R26 (Daujat et al., 2002); (Schurter et al., 2001). PRMT4 plays an important role in a number of biological processes including muscle differentiation (Chen et al., 2002), T cell development (Kim et al., 2004) and adipocyte differentiation (Yadav et al., 2008). PRMT4 maintains embryonic stem cell (ESC) pluripotency and inhibits ESC differentiation (Torres-Padilla et al., 2007a); (Wu et al., 2009). Although other members of the PRMT family have been implicated in hematopoiesis and acute leukemia (Zhao et al., 2008); (Cheung et al., 2007); (Liu et al., 2011), little is known about the role of PRMT4 in normal or malignant hematopoiesis.
RUNX1 (also known as AML1) is a transcription factor that binds to a consensus binding sequence (CBS) -PyGpyGGTPy (Py = pyrimidine) in the regulatory regions of promoters and enhancers of genes that play important roles in hematopoiesis. RUNX1 knock out mice die between embryonic day [E] 11.5 - [E] 13.5 with a complete lack of fetal liver (i.e. definitive) hematopoiesis (Okuda et al., 1996), while conditional deletion of RUNX1 in adult mice results in profound lineage-specific abnormalities, including a block in lymphoid development and reduced megakaryocytic production, with little effect on adult hematopoietic stem cells (HSCs). RUNX1 is one of the most frequently altered genes in acute leukemia; either by chromosomal translocations such as the t(8;21) (Blyth et al., 2005) or by point mutations or deletions which occur in 4-10% of patients with sporadic or therapy-related MDS and AML (Osato, 2004). Furthermore, RUNX1 point mutations are found in affected individuals with the inherited FPD/AML syndrome (familial platelet disorder with propensity to AML) (Song et al., 1999). Post-translational modifications, including ubiquitination, phosphorylation, acetylation and methylation fine-tune RUNX1 function (Wang et al., 2009); for instance, arginine methylation of an RTAMR motif in RUNX1 by PRMT1 abrogates SIN3A binding, thereby potentiating RUNX1 dependent transcriptional activation of its target genes (Zhao et al., 2008). Similarly, microRNAs such as miR-17-5p, miR-20a and miR-106a can regulate RUNX1 protein expression and thereby control aspects of hematopoietic cell differentiation (Fontana et al., 2007).
The myeloid specific microRNA-223 (miR-223) has been shown to affect granulocytic differentiation. Loss of miR-223 impairs granulocytic maturation (Johnnidis et al., 2008), while miR-223 overexpression promotes myeloid differentiation (Fazi et al., 2005b). miR-223 expression has been shown to be transcriptionally regulated by NF-IA (Fazi et al., 2005b), by C/EBPs and PU.1 (Fukao et al., 2007) and by E2F1 (Pulikkan et al., 2010). Fazi et al. reported that the AML1-ETO fusion protein represses miR-223 expression by binding to a RUNX CBS located upstream of the pre-miR-223 (Fazi et al., 2007). They, and others, have found that miR-223 expression is downregulated in AML patient samples (Fazi et al., 2007); (Pulikkan et al., 2010); (Eyholzer et al., 2010).
In this study, we have identified PRMT4 as a novel regulator of the myeloid differentiation process in stem/progenitor human hematopoietic cells (HSPCs). PRMT4 forms a regulatory loop with miR-223, to reciprocally suppress the expression of each other during myeloid differentiation. We also show that RUNX1 is a substrate of PRMT4 and that methylation of RUNX1 by PRMT4 (on R223) leads to the recruitment of a DPF2-containing repressor complex that binds critical miR-223 transcriptional regulatory elements (that are distinct from the element identified by Fazi et al 2007), and represses miR-223 expression. A decrease in PRMT4 expression is critical for normal myeloid differentiation, but this normal event can be subverted by malignant hematopoietic cell transformation. Thus, targeting PRMT4 represents a potential therapeutic approach to promote the differentiation of AML blast cells.
Results
PRMT4 regulates myeloid differentiation
To examine its function in hematopoiesis, we knocked down PRMT4 in human cord blood (CB) derived, CD34+ hematopoietic stem/progenitor cells (HSPCs), using lentiviral vectors that express GFP and shRNAs directed against PRMT4. We assayed the extent of PRMT4 knockdown (KD) in the GFP positive transduced cells and found a 70%-80% decrease in PRMT4 expression for the KD1 and KD2 short hairpin RNAs, respectively (Figure 1A). The PRMT4-KD cells generated far fewer CFUs when plated in methylcellulose (Figure 1B) and showed enhanced myeloid differentiation after 7 days in myeloid- promoting liquid culture (which contains SCF, FLT-3 ligand, IL-3, IL-6, GM-CSF and G-CSF) with 60%-70% of the KD cells being CD11b positive and CD13 positive, vs. 40% (and 14%) of the control cells being CD11b (or CD13) positive (Figure 1C and S1B). Consistent with the immunophenotypic evidence, morphologic evidence also showed more mature myeloid cells following PRMT4 KD (Figure 1D), with the KD cells showing a condensed nucleus and clear nuclear lobulation. In addition, PRMT4 KD mildly impaired erythroid differentiation under erythroid - promoting culture conditions (SCF and EPO) (Figure S1E). Consistent with its effect on CFU generation, a decrease in the numbers and size of the cobblestone areas was seen at week 5, reflecting impaired HSPC self renewal when PRMT4 was knocked down (Figure S1D).
Figure 1. PRMT4 regulates myeloid differentiation of HSPCs.
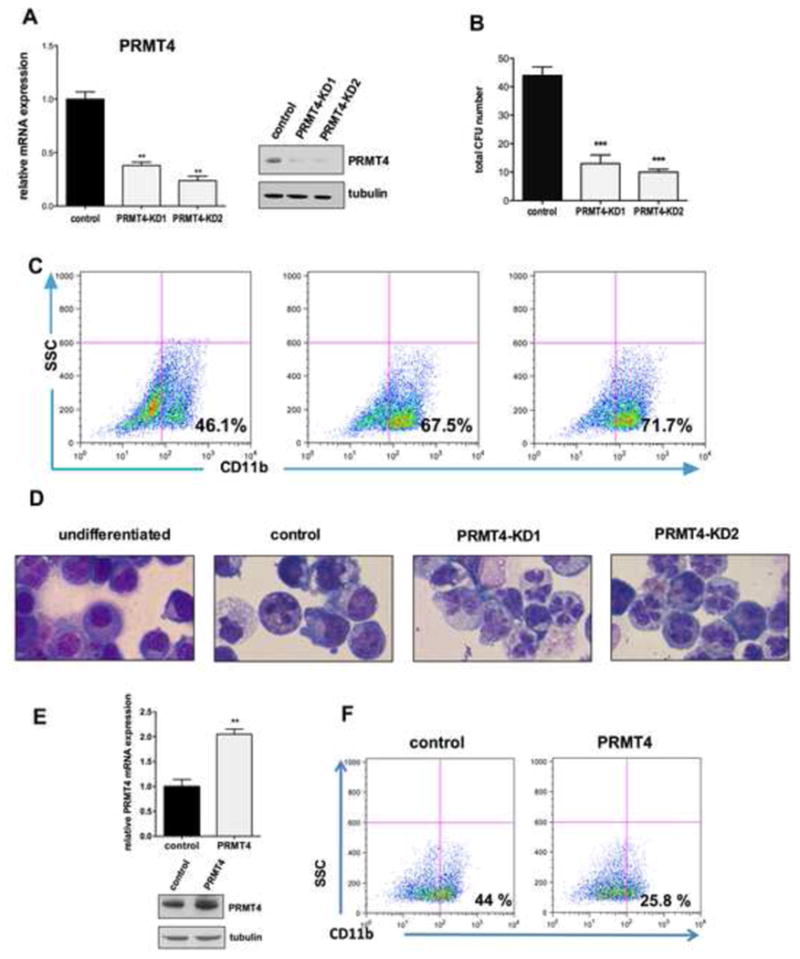
(A). Efficient knock down of PRMT4. Human CB CD34+ cells were transduced with lentiviruses expressing a control (scrambled) shRNA or one of two shRNAs directed against PRMT4. GFP-positive cells were sorted 3 days after transfection and collected to perform qRT-PCR and Western blot analyses. qRT-PCR data represents the mean ± SD of the three independent experiments. ** p < 0.01 by Student's t test. Tubulin served as the loading control.
(B). Downregulation of PRMT4 decreases CFU formation. 104 of the control or PRMT4 knock down cells were plated in methylcellulose. The total number of colony forming units (CFUs) was scored 2 weeks after the plating. The data represents the mean ± SD of the three independent experiments. *** p < 0.001 by Student's t test.
(C). Downregulation of PRMT4 promotes the myeloid differentiation of HSPCs. GFP+ CD34+ cells were cultured in myeloid-promoting cytokine containing medium for 7 days. Myeloid differentiation was determined by FACS analysis of CD11b expression.
(D). Downregulation of PRMT4 promotes the myeloid differentiation of HSPCs. Cellular morphology was evaluated after 7 days in myeloid-promoting cytokine containing medium. Cells growing in basic culture were used as the control for myeloid differentiation.
(E). Overexpression of PRMT4 was demonstrated at the mRNA and protein levels. Human CB CD34+ cells were transduced with retroviruses expressing either control (GFP alone) or GFP and HA-PRMT4. GFP+ cells were sorted after 3 days of transfection and collected to perform qRT-PCR and Western blot analyses. qRT-PCR data represents the mean ± SD of the three independent experiments. ** p < 0.01 by Student's t test.
(E). Overexpression of PRMT4 blocks the myeloid differentiation of HSPCs. GFP+ CD34+ cells were cultured in myeloid-promoting cytokine containing medium for 7 days. Myeloid differentiation was determined by FACS analysis of CD11b expression.
See also Figure S1.
We next examined whether PRMT4 overexpression blocks myeloid differentiation. Indeed, we found a marked reduction in CD11b positive cells generated from PRMT4 overexpressing CD34+ cells (compared to the control cells) after 7 days in myeloid differentiation promoting cultures (Figure 1E). Thus, PRMT4 appears to be an important negative regulator of normal myeloid differentiation.
PRMT4 is regulated post-transcriptionally by miR-223 during myeloid differentiation
Given the prominent effect of PRMT4 on myeloid differentiation, we assessed changes in PRMT4 expression during normal in vitro HSPC differentiation. We cultured human CB CD34+ cells in myeloid - promoting liquid culture and observed a significant decrease in PRMT4 protein levels over a seven-day period (Figure 2A). PRMT4 mRNA levels varied only slightly during this process, suggesting that PRMT4 is being regulated post-transcriptionally. MicroRNA target prediction programs (TargetScan release 5.2 (Figure S2A) and PITA) suggested that PRMT4 is a potential target of several microRNAs, including miR-223, a myeloid specific microRNA. Interestingly, a seed sequence for miR-223 is found in the 3′-UTR region of PRMT4, which is located adjacent to the stop codon of the PRMT4 open reading frame (ORF) (16-22 nt from the stop codon); this location could confer a strong, translational regulatory effect (Grimson et al., 2007); (Eulalio et al., 2008). Indeed, we found that miR-223 expression steadily increases during myeloid differentiation (Figure 2B), concomitant with decreasing PRMT4 protein expression. To determine whether miR-223 regulates PRMT4 expression, we transiently transfected CD34+ cells with a short hairpin encoding the mature miR-223 for 24 hours and monitored PRMT4 expression, using siPRMT4 as a short-hairpin positive control (and a scrambled short-hairpin as a negative control). PRMT4 protein levels decreased by 50% in the miR-223 overexpressing cells, similar to that seen in the cells expressing siRNA directed against PRMT4 (Figure 2C).
Figure 2. PRMT4 is a potential target gene of miR-223 during myeloid differentiation of HSPCs.
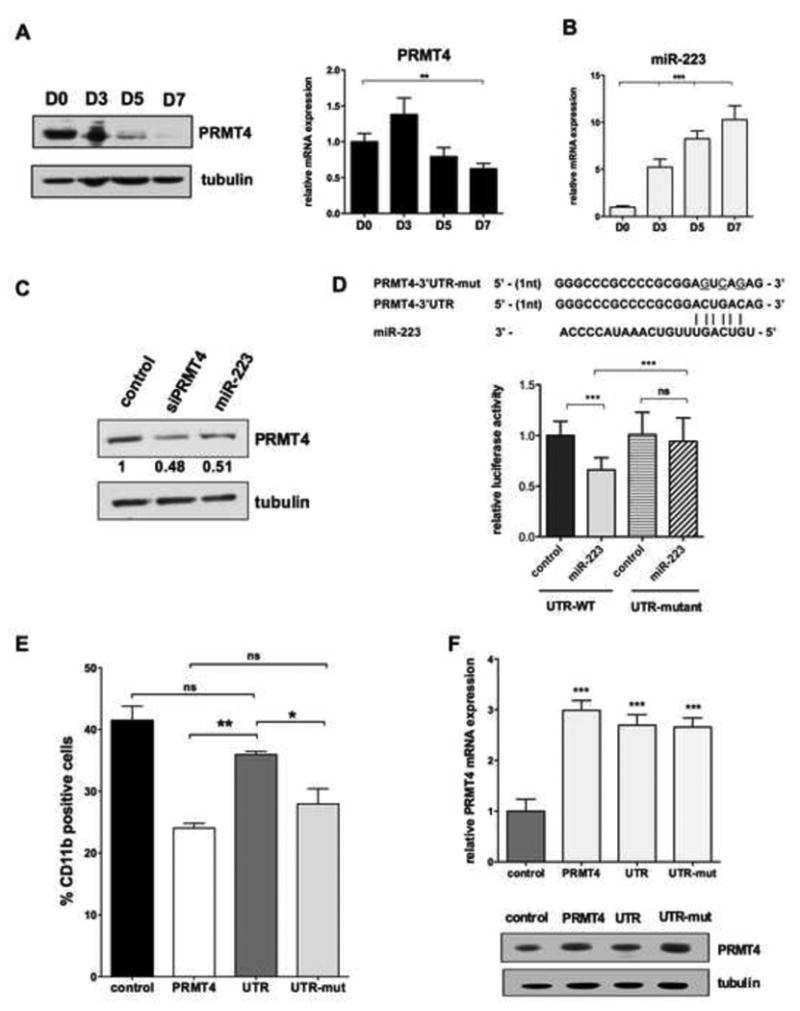
(A). PRMT4 protein expression is progressively downregulated during myeloid differentiation (left), while PRMT4 mRNA level decreases modestly during myeloid differentiation (right). Isolated CD34+ cells were cultured in myeloid – promoting cytokine containing medium and collected at sequential time points: day (D) 0, 3, 5 and 7. Western blot and qRT-PCR analyses were performed. qRT-PCR data represents the mean ± SD of three independent experiments. ** p < 0.01 by Student's t test. Tubulin served as the loading control.
(B). miR-223 expression steadily increases during myeloid differentiation. qRT-PCR data represents the mean ± SD of three independent experiments. *** p < 0.001 by Student's t test.
(C). Overexpression of miR-223 or siRNA directed against PRMT4 lowers PRMT4 protein levels in HSPCs. miR-223, siRNA against PRMT4, and control oligonucleotides were transiently expressed in CD34+ cells and 24 hours post-electroporation, the cells were collected and assayed for PRMT4 expression by western blot analyses.
(D). Putative miR-223 binding site in the PRMT4 3′UTR is shown at the top (based on TargetScan.org release 5.2). Luciferase activity in 293T cells co-transfected with a reporter plasmid containing either the wild type 3′-UTR-PRMT4 or the mutated 3′-UTR (3-UTR-mut, which lacks the seed miR-223 sequence) with or without miR-223. Renilla luciferase values are normalized based on the value of firefly luciferase. Mean ± SD from three independent experiments is shown. *** p < 0.001 by Student's t test.
(E). Control of PRMT4 expression by miR-223 is essential to regulate PRMT4 function during normal myeloid differentiation. Human CB CD34+ cells were transduced with retroviruses expressing control -GFP alone; GFP- PRMT4-ORF or GFP- PRMT4-3′-UTR or GFP-PRMT4- 3′-UTR-mut. Sorted GFP+ CD34+ cells were cultured in myeloid-promoting cytokine containing medium for 7 days. Myeloid differentiation was determined by FACS analysis of CD11b expression. The percentage of CD11b positive cells was quantified as mean ± SD based on three independent experiments. * p < 0.05; ** p < 0.01 by Student's t test.
(F).qRT-PCR and Western blot analyses of PRMT4 expression in control CD34+ cells or CD34+ cells expressing PRMT4-ORF, PRMT4-3′-UTR or PRMT4-3′-UTR-mut. Tubulin served as the loading control.
See also Figure S1.
To validate that PRMT4 is directly targeted by miR-223, we cloned its full length 3′-UTR into a luciferase reporter plasmid (UTR- WT), using the same construct with a mutated miR-223 targeting sequence (UTR-mut), which cannot bind miR-223, as negative control. We expressed these constructs in 293T cells, with either the miR-223 short-hairpin or the control short-hairpin, and found that miR-223 decreased the luciferase activity of the UTR-WT, with no effect on the UTR-mut reporter plasmid (Figure 2D and S2C). Thus, miR-223 directly targets PRMT4 by binding to its recognition sequence in the 3′-UTR; this suggests that PRMT4 and miR-223 form a regulatory loop to regulate myeloid differentiation.
To determine how important the regulation of PRMT4 expression by miR-223 is for the effects of PRMT4 on myeloid differentiation, we overexpressed the PRMT4-ORF with either the WT or the mutant PRMT4 3′-UTR, in human HSPCs. While the WT 3′-UTR abrogated the PRMT4 imposed block in myeloid differentiation, when PRMT4 was expressed without the 3′-UTR, or with the mutant 3′-UTR, the block in myeloid differentiation was seen(Figure 2E). These effects correlate with PRMT4 protein levels (Figure 2F), and demonstrate that the regulation of PRMT4 by miR-223 is important to its function during myeloid differentiation.
PRMT4 represses miR-223 expression
Given the known transcription regulatory role of PRMT4, to determine how PRMT4 controls myeloid differentiation, we first examined the expression level of several differentiation-specific “master” transcription factors, including PU.1, C/EBPα, KLF4 and GATA1 in PRMT4-KD cells. While we found no significant changes (Figure S3A), we observed a consistent increase in miR-223 expression in the PRMT4-KD cells (Figure 3A). Because upregulation of miR-223 has been reported to promote the myeloid differentiation of leukemia cells (Fazi et al., 2007), we overexpressed miR-223 in normal CB CD34+ cells, and saw a significant increase in CD11b positive cells (51.2% vs. 38.1% for the control cells) (Figure 3B) as well as decreased PRMT4 expression (Figure S2B). We observed decreased miR-223 expression, when PRMT4 is overexpressed (Figure 3C). When we knocked down miR-223 expression, we found a modest but consistent reduction in CD11b positive cells (38.6% ± 3.1 vs. 44.8% ±1.8, p <0.05) (Figure 3D and S3B), which suggests that other microRNAs may compensate for miR-223 during myeloid differentiation. This is consistent with the phenotype of miR-223 knockout mice, where miR-223 is important, but not essential, for granulocytic maturation and function (Johnnidis et al., 2008).
Figure 3. PRMT4 regulates miR-223 expression.
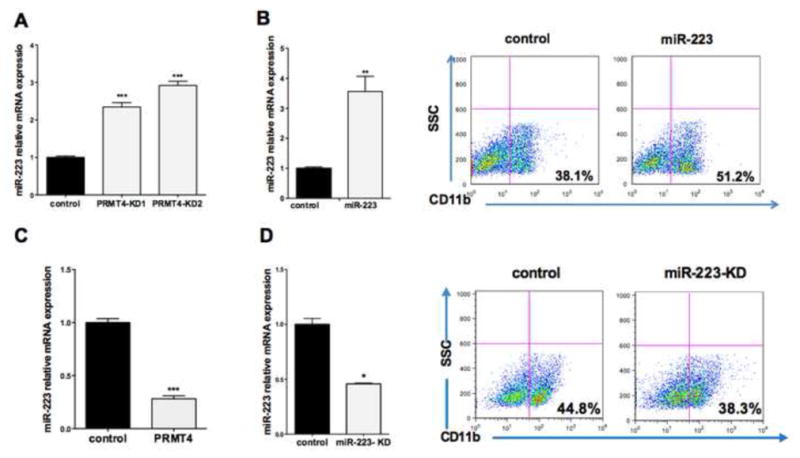
(A). PRMT4 downregulation leads to upregulation of miR-223 expression. qRT-PCR data represents the mean ± SD of three independent experiments. *** p < 0.001 by Student's t test.
(B). Overexpression of miR-223 in HSPCs enhances myeloid differentiation. (Left) qRT-PCR data represents the mean ± SD of three independent experiments. ** p < 0.01 by Student's t test. (Right) Cells were plated in myeloid differentiation promoting culture and assayed for CD11b expression after 7 days.
(C). Overexpression of PRMT4 diminishes miR-223 expression. qRT-PCR data represents the mean ± SD of three independent experiments. *** p < 0.001 by Student's t test.
(D). Knock down of miR-223 in HSPCS slightly impairs myeloid differentiation. (Left) qRT-PCR data represents the mean ± SD of three independent experiments. * p < 0.05 by Student's t test. (Right) Cells were plated in myeloid differentiation promoting culture and assayed for CD11b expression after 7 days.
See also Figure S3.
To determine whether PRMT4 regulates the transcription of miR-223, we quantified the level of miR-223 primary transcript (pri-miR-223) and found that PRMT4 expression does reciprocally regulate pri-miR-223 levels in CD34+ cells (Figure S3C). In addition, gene expression analysis of PRMT4 KD cells (GSE46056) revealed a gene signature consistent with an upregulation of myeloid differentiation (Figure S3D). Thus, PRMT4 regulates myeloid differentiation, at least in part, by modulating miR-223 expression.
RUNX1 is methylated by PRMT4 on arginine 223 (R223)
PRMT4 is generally thought to act as a transcriptional co-activator, however, we found that PRMT4 functions as a repressor of miR-223 expression in hematopoietic cells. To determine how this “co-activator” suppresses gene expression, we hypothesized that PRMT4 modulates the activity of a regulatory transcription factor. Based on the report that the leukemogenic AML1-ETO fusion protein represses miR-223 expression via a RUNX1 consensus-binding site (CBS) in a specific regulatory region (Fazi et al., 2007), we examined the miR-223 promoter and found another RUNX1 CBS close to the miR-223 promoter (Fukao et al., 2007). Given the presence of these CBS, we investigated whether wild type RUNX1 also regulates miR-223 transcription. We knocked down RUNX1 in CD34+ cells using two different shRNAs and observed up-regulation of pri-miR-223 and pre-miR-223 expression (Figure S4A), indicating that RUNX1 can transcriptionally inhibit miR-223 expression in HSPCs. Given that both RUNX1 and PRMT4 repress miR-223 transcription, we examined whether RUNX1 is arginine methylated by PRMT4. Using an in vitro methylation assay, we identified one specific site in RUNX1, R223, which is methylated by PRMT4. This arginine residue is located just C-terminal to the RUNX1 DNA binding domain (Figure S5A) and is conserved among vertebrates (Figure S5B).
To determine whether RUNX1 is methylated at R223 by PRMT4 in vivo, we generated a methylation specific anti-methyl-R223RUNX1 antibody, which recognizes an asymmetric di-methylated R223RUNX1 peptide, but not the unmethylated peptide (data not shown). We confirmed the specificity of the antibody by overexpressing Flag-RUNX1 or the Flag-RUNX1R223K mutant protein in 293T cells with or without HA-PRMT4, or with an enzymatically dead form of PRMT4 (PRMT4EQ). Methylation of RUNX1 at R223 was strongly detected when both RUNX1 and PRMT4 were overexpressed (Figure 4A. lane1) or when RUNX1 was overexpressed by itself (lane 5). No methylation was detected when either the mutant R223K-RUNX1 protein was expressed or when PRMT4EQ was overexpressed (Figure 4A, lanes 2,3 and 4). Thus, we conclude that PRMT4 methylates RUNX1 at residue R223 in vivo. The same experiment showed the physical interaction between RUNX1 and PRMT4, using an anti-HA antibody to detect PRMT4 in the RUNX1-containing immunoprecipitate; we detected an interaction between RUNX1 and PRMT4 when RUNX1 and PRMT4EQ were co-expressed or when the R223K-RUNX1 mutant was over expressed with PRMT4 (or PRMT4EQ) (Figure 4A. IP- lane 2, 3, 4). Minimal interaction was seen when RUNX1 overexpressed with WT-PRMT4 (Figure 4A.IP-lane1), suggesting that PRMT4 preferably associates with the non-methylated form of RUNX1.
Figure 4. RUNX1 is arginine methylated by PRMT4 on R223.
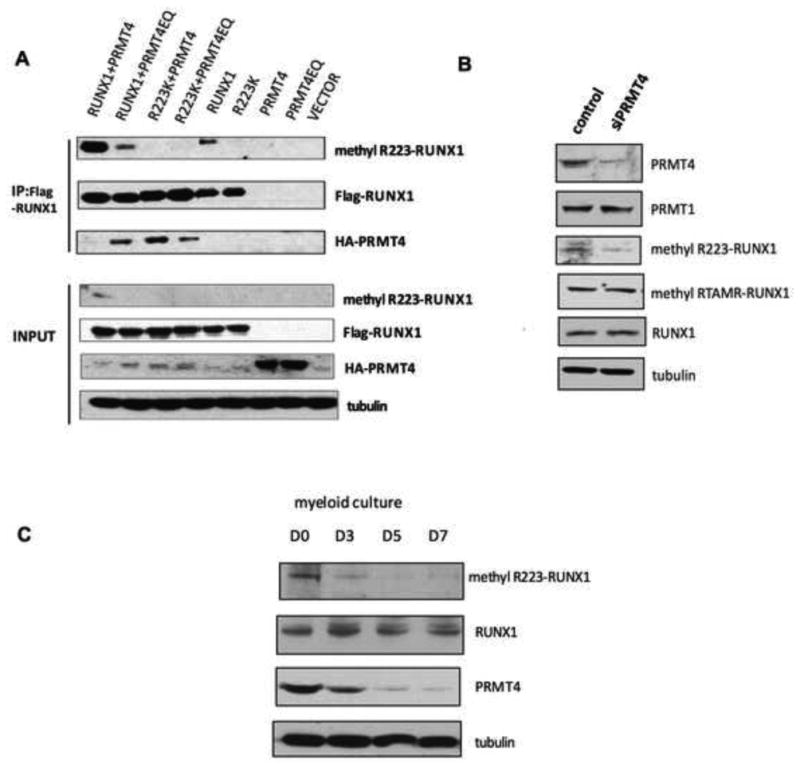
(A). In vivo methylation of RUNX1 by PRMT4 was detected using a methyl-RUNX1 specific antibody. The full-length Flag- RUNX1 or Flag- RUNX1-R223K mutant cDNAs were overexpressed in 293T cells, with or without HA-tagged-WT-PRMT4, or an enzymaticallly dead PRMT4 mutant (PRMT4EQ). Immunoprecipitation was performed using an anti-Flag antibody and immunoblotting with anti-FLAG or anti-HA antibodies. Wild type RUNX1 (lane 1 and 2), but not the R223K mutant protein (lane 3 and 4) is methylated by wild type PRMT4. The physical interaction between RUNX1 and PRMT4 is detected when RUNX1 is overexpressed with PRMT4EQ, but not WT-PRMT4, and when R223K is overexpressed with PRMT4 or PRMT4EQ (lanes 1-4 in the third row). Tubulin served as the loading control.
(B). Knock down of PRMT4 in HEL cells using siRNA reduces the level of endogenous methylR223-RUNX1, without altering total RUNX1 levels or the methylation of RUNX1 at the RTAMR motif.
(C). The level of RUNX1R223 methylation decreases during myeloid differentiation, without changes in total RUNX1 expression but concordant with changes in PRMT4 protein levels.
See also Figure S4.
To determine whether PRMT4 is the major enzyme catalyzing RUNX1R223 methylation in vivo, we knocked down PRMT4 using siRNA in HEL cells; we observed a marked reduction in RUNX1 R223 methylation, but no change in RUNX1 methylation at the RTAMR site, which we previously showed is methylated by PRMT1 (Figure 4B). Thus, PRMT4 appears to be the major methyltransferase that asymmetrically methylates RUNX1 at R223 in vivo.
Given the downregulation of PRMT4 protein levels during myeloid differentiation, we examined whether RUNX1R223 methylation is similarly down regulated and indeed, as human CD34+ cells differentiate and PRMT4 expression diminishes, the level of methylR223-RUNX1 decreases, even though total RUNX1 protein levels are unchanged (Figure 4C). To determine whether R223 methylation is important for RUNX1 transcriptional activity, we compared the transcriptional regulatory activity of RUNX1 R223K vs.WT-RUNX1 on the pri-mir-223 expression in CB CD34+ cells. While RUNX1 overexpression repressed pri-miR-223 transcript levels, the RUNX1 R223K mutant did not (Figure S4B). Thus, the repression effects of PRMT4 on miR-223 expression appear to relate to its ability to post-transcriptionally modify RUNX1 on R223.
Methylation of RUNX1 at R223 regulates its interaction with DPF2
We hypothesized that methylation of R223 in RUNX1 by PRMT4 affects its protein-protein interactions, and performed peptide pull-downs using both a methyl-R223 RUNX1 peptide and an unmodified RUNX1 peptide as bait, followed by mass spectrometry, to identify proteins that interact with unmethylated or R223 methylated RUNX1 protein. We identified several proteins that interact with the R223 methyl peptide (Table S1), including DPF2 (double PhD Finger 2), a widely expressed member of the d4 protein family that is characterized by the presence of a tandem plant-homodomain (PHD domain). DPF2 was recently reported to interact with lysine-acetylated histones and acts as a co-repressor (Matsuyama et al., 2010). We first verified the interaction of DPF2 with the methyl-R223 RUNX1 peptide in a peptide pull-down assay: DPF2 preferentially bound to the arginine methylated peptide, while PRMT4 preferred the non-methylated RUNX1 peptide, similar to the interaction of PRMT4 with full length RUNX1 (Figure 5A).
Figure 5. Methylation of RUNX1 at R223 regulates its interaction with DPF2.
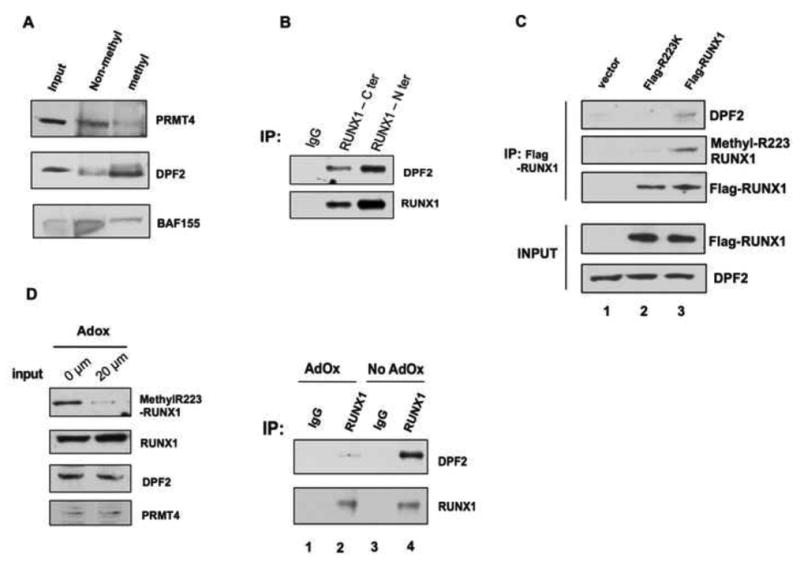
(A). DPF2 is preferably pulled down by a methylR223- RUNX1 peptide, while the unmodified R223-RUNX1 peptide interacts more strongly with PRMT4. BAF155 is used as a control, which shows no preference for binding to either the modified or the unmodified peptide. Input: ten percent of the used nuclear extract.
(B). The endogenous RUNX1 and DPF2 proteins physically interact in vivo. Two anti-RUNX1 antibodies were used to immunoprecipitate DPF2 from HEL cell nuclear extract. DPF2 was detected using an anti-DPF2 antibody. Pre-immune rabbit serum was used as control.
(C). The interaction between RUNX1 and DPF2 is dependent on the RUNX1 methylation status. Overexpressed Flag-RUNX1 is immunoprecipitated using an anti-Flag antibody. DPF2 is co-precipitated with Flag-RUNX1, but not the Flag-R223K mutant (compare lane 1 vs. lane 3).
(D). Treatment of cells with AdOx reduces the level of RUNX1 methylation (input), abrogating its interaction with DPF2 (compare lane 2 vs. lane 4).
See also Figure S5.
To determine whether RUNX1 and DPF2 associate in vivo, we performed co-IP assays using two different RUNX1 antibodies and consistently immunoprecipitated the endogenous DPF2 protein, confirming their in vivo interaction (Figure 5B). To show that this interaction depends on arginine methylation, we treated HEL cells with Adenosine-2′,3′-dialdehyde – AdOx (20 μM), a pan of methyltransferase inhibitor, which significantly reduced RUNX1 methylation after 16h (Figure 5D). This treatment abrogated the interaction of RUNX1 with DPF2 (Figure 5D, compare lane 2 and 4). To further demonstrate that DPF2 interacts specifically with R223methylated RUNX1, we overexpressed Flag-RUNX1 and Flag-RUNX1R223K and performed co-immunoprecipitation studies using anti-Flag beads. DPF2 associated with Flag-RUNX1, but not Flag-RUNX1R223K (Figure 5C, compare lanes 2 and 3), again demonstrating that the interaction of DPF2 with RUNX1 is dependent on the R223 methylation.
miR-223 expression is regulated by a RUNX1-methylation dependent repressor complex
Given the decrease in RUNX1R223 methylation during myeloid differentiation and the demonstrated interaction of R223- methylated RUNX1 with DPF2, we hypothesized that the DPF2-RUNX1 interaction regulates miR-223 expression. We examined the binding of RUNX1, PRMT4, and DPF2 to miR-223 regulatory regions by chromatin immunoprecipitation (ChIP) assays using HSPCs and in vitro differentiated myeloid cells, which correspond to the states where miR-223 is low and RUNX1 R223 methylation high (i.e. the HSPC stage), and where miR-223 is high, and RUNX1 R223 methylation low (i.e. in differentiated myeloid cells) (Figure 6A and 6B). Using primer pairs that cover much of the miR-223 putative regulatory regions (as depicted in Figure 6C and table S2), we detected RUNX1, methyl-R223 RUNX1 and DPF2 at the pre-miR-223 promoter region, when the cells were at the “stem/progenitor” stage and miR-223 was minimally expressed (Figure 6D). However, in the differentiated cells where miR-223 was actively transcribed, we found RUNX1 and not methyl-R223 RUNX1 (Figure 6E). PRMT4 is found throughout the miR-223 regulatory region in the HSPCs but not in the differentiated cells, with a slight peak at region 4. DPF2 protein is clearly expressed in the differentiated cells (Figure 6A), but it is not found at the miR-223 regulatory regions (Figure 6E), suggesting that its recruitment to the miR-223 promoter depends on the methylation status of RUNX1. Thus, recruitment of DPF2 by methyl-R223 RUNX1 may dictate the transcriptional effects of RUNX1 on the miR-223 locus.
Figure 6. The RUNX1 methylation dependent repressor complex regulates miR-223 expression.
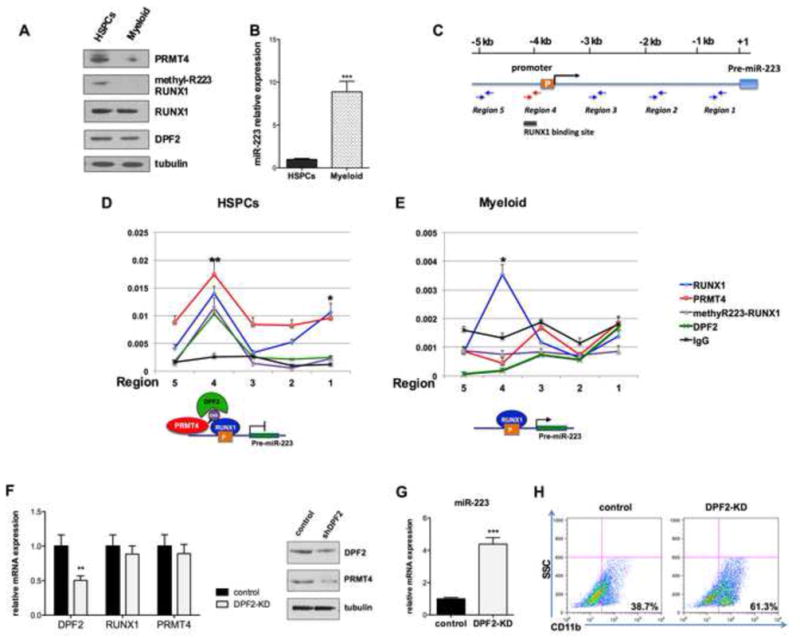
(A). Western blot analysis quantified the levels of expression of PRMT4, methyl R223-RUNX1, RUNX1 and DPF2 in CD34+ cells maintained in basic culture or in cells cultured in myeloid differentiation-promoting medium for 7 days. Tubulin served as the loading control.
(B). The level of miR-223 expression in the HSPCs and myeloid differentiated cells. qRT-PCR data represents mean ± SD of three independent experiments. *** p < 0.001 by Student's t test.
(C). A schematic diagram of the miR-223 promoter. The location of the RUNX1 consensus binding sites and the primers used for the ChIP assays is shown.
(D). (E). ChIP assays show the presence of a methyl-R223 RUNX1- dependent complex at the promoter of miR-223 in HSPCs (D), but not in myeloid differentiated cells (E). Upper panel: Enrichment of proteins of interest at miR-223 regulatory regions was assayed by qRT-PCR and shown as percentage of the genomic input DNA. * p < 0.5; ** p < 0.01 by Student's t test.
Lower panel: diagrams demonstrating the recruitment of RUNX1, methylR223-RUNX1 and DPF2 to the miR-223 promoter.
(F). Efficient knock down of DPF2. CD34+ cells were transduced with lentiviruses expressing a control (scrambled) shRNA or shRNA directed against DPF2. GFP-positive cells were sorted 3 days after transfection and collected to perform qRT-PCR and Western blot analyses. qRT-PCR data represents the mean ± SD of three independent experiments. ** p < 0.01 by Student's t test.
(G). Downregulation of DPF2 increases miR-223 expression. qRT-PCR analysis of miR-223 in control and DPF2 knock down CD34+ cells. The data represents the mean ± SD of three independent experiments. *** p < 0.001 by Student's t test.
(H). Downregulation of DPF2 accelerates the myeloid differentiation of HSPCs. GFP+ CD34+ cells were cultured in myeloid differentiation promoting medium for 7 days. Myeloid differentiation was determined by FACS analysis of CD11b expression.
See also Figure S6.
DPF2 inhibits miR-223 expression and myeloid differentiation
We next examined whether DPF2 can directly regulate miR-223 expression. We achieved a 50% knock down of DPF2 mRNA and protein using shRNA (Figure 6F) and found a 3-fold increase in miR-223 expression (Figure 6G) and a decrease in PRMT4 protein levels. The DPF2 KD cells also showed enhanced myeloid differentiation (based on CD11b expression, Figure 6H) and decreased clonogenic potential (Figure S6) similar to what occurred when we knocked down PRMT4. This places PRMT4 and DPF2 in a RUNX1 containing complex that down-regulates miR-223 expression, and impairs the myeloid differentiation of HSPCs.
Knockdown of PRMT4 is sufficient to induce myeloid differentiation in AML cell lines
As PRMT4 expression impairs the differentiation of human HSPCs, we examined the level of PRMT4 expression in a cohort of 318 AML patient samples (GSE24505) (Figueroa et al., 2010). PRMT4 levels were significantly upregulated in the AML samples, compared to the control group (n = 5) (Figure 7A). A high level of PRMT4 expression was seen in core-binding factor (CBF) AMLs (which express either AML1-ETO or CBFβ-SMMHC), which exhibit low-level miR-223 expression, but overall, about 70% of the AML patients had at least a 2-fold increase in PRMT4 expression (Figure S7A).
Figure 7. Downregulation of PRMT4 is sufficient to induce myeloid differentiation in leukemia cells.
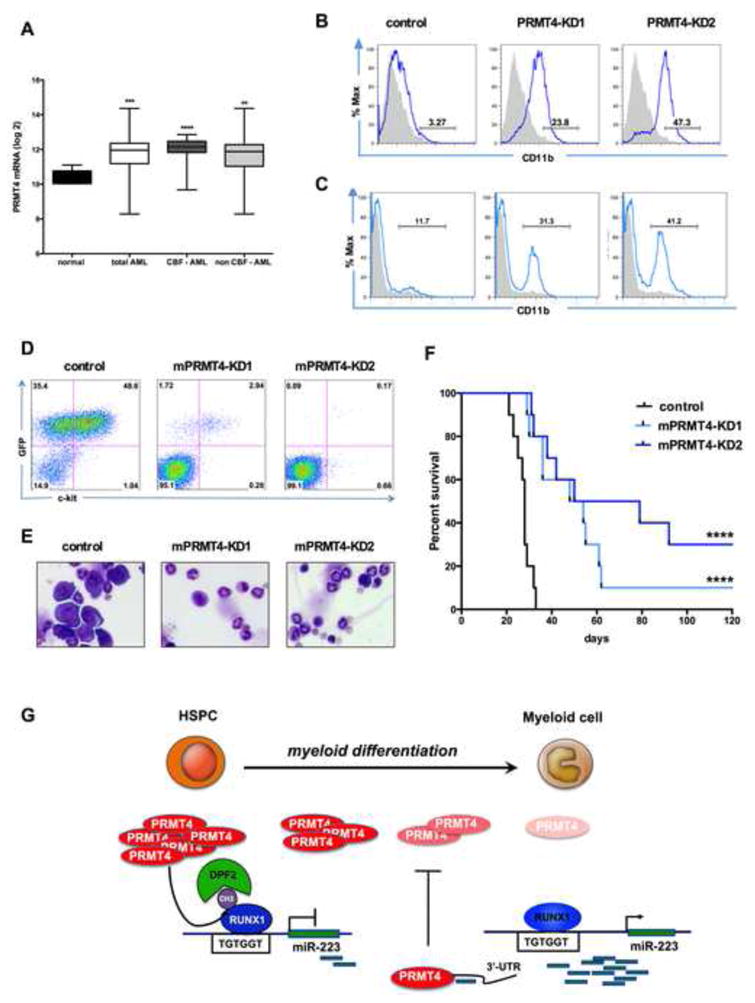
(A). PRMT4 expression is upregulated in AML patient samples. The graph shows the log2 expression of PRMT4 from transcript profiling of CD34+ bone marrow cells isolated from 5 healthy donors (normal) or 318 AML patients. CBF: core-binding factor. CBF-AML n=57. non CBF-AML n=261. **** p < 0.0001 *** p < 0.001 ** p < 0.01 by Student's t test.
(B). Knock down of PRMT4 triggers the myeloid differentiation of NB4 cells. NB4 cells were transduced with lentiviruses expressing control (scrambled) shRNA or shRNA directed against PRMT4. Sorted GFP positive cells were cultured for 3 days prior to FACS analysis of CD11b expression. Quantitative data in Figure S7D.
(C). Knock down of PRMT4 triggers the myeloid differentiation of Kasumi-1 cells. Kasumi-1 cells were transduced with lentiviruses expressing control (scrambled) shRNA or shRNA directed against PRMT4. Sorted GFP positive cells were cultured for 3 days prior to FACS analysis of CD11b expression. Quantitative data in Figure S7D.
(D). FACS analysis showed far fewer GFP+ ckit + cells in peripheral blood (PB) of the mice transplanted with AE9a – mPRMT4-KD cells in compare to AE9a – control cells at week 3. AE9a expressing mouse leukemia cells were transduced with shRNA scramble control (AE9a-control), or shRNAs against PRMT4 (AE9a-mPRMT4-KD1 and mPRMT4-KD2). Transduced cells were sorted for expression of RFP and the sorted cells were injected into recipient mice that had received sublethal irradiation.
(E). Bone marrow (BM) morphology shows marked reduction in the number of leukemic cells in mice transplanted with AE9a -mPRMT4-KD cells, compared to AE9a- control cells.
(F). Knock down of PRMT4 prolongs the survival of AE9a transplanted mice. The median survival was extended in the knock down groups, compared to the control group (54 days and 64.5 days vs. 28 days p <0.0001).
(G). A schematic model showing how PRMT4 regulates myeloid differentiation of human HSPCs. PRMT4 and miR-223 form a regulatory loop that is critical for myeloid differentiation. PRMT4 inhibits myeloid differentiation by assembling a methyl R223-RUNX1-DPF2 repressor complex that suppresses miR-223 expression. When HSPCs undergo myeloid differentiation, PRMT4 expression is downregulated, releasing the miR-223 locus from transcription repression, allowing it to be transcribed. At that stage, the higher expression of miR-223 targets the PRMT4 3′-UTR to further decrease PRMT4 expression, thereby reinforcing the myeloid differentiation process.
See also Figure S7.
Several studies have shown that overexpression of miR-223 can promote the granulocytic differentiation of NB4 acute promyelocytic leukemia (APL) cells (Fazi et al., 2005a). We knocked down PRMT4 in NB4 cells and triggered myeloid differentiation with a significant increase in the number of CD11b positive cells (3.3% control vs. 23.8% and 47.3%) (Figure 7B) as well as changes in cellular morphology and an upregulation of miR-223 expression (Figure S7B, C). A similar induction of differentiation was also observed in the ATRA-resistant NB4-R4 cells following PRMT4-KD (Rosenauer et al., 1996) (Figure S7D and S7E). Because therapeutic targeting of most leukemia fusion proteins, including AML1-ETO remains elusive, we tested whether targeting PRMT4 can promote the differentiation of AML1-ETO expressing cells (Figure 7A). We knocked down PRMT4 in t(8;21) positive Kasumi-1 cells and found increased differentiation (Figure 7C and S7E). We also saw significant apoptosis of all three cell lines when PRMT4 is knocked down (Figure S7F), suggesting that PRMT4 not only impairs the differentiation of these leukemia cells, but it is also critical for their survival.
Knockdown of PRMT4 reduces the leukemia burden in vivo
To investigate the in vivo role of PRMT4 in leukemogenesis, we used shRNA expressing lentiviruses to knock down PRMT4 in the AML1-ETO9a (AE9a) driven mouse AML model (Yan et al., 2006; Wang et al., 2011). Leukemia cells growing in culture were transduced with 2 different shRNAs and ≥ 70%-80% KD was achieved (Figure S7G). The transduced AE9a-mPRMT4KD and control cells were injected into sub-lethally irradiated C57Bl/6 mice (day 0). We observed decreased numbers of immature GFP+ c-kit+ blast cells in the peripheral blood of the AE9a-mPRMT4KD mice compared to the control mice at week 3 (Figure 7D). Morphological analysis of the bone marrow (Figure 7E) and peripheral blood (Figure S7I) also showed a marked reduction in blast cells at week 4 with lower white blood cell counts, and less anemia and thrombocytopenia (Figure S7H), compared to the AE9a-control mice. This translated to a significant increase in median survival, from 28 days for the control group, to 51 days and 64.5 days for the AE9a-mPRMT4-KD1 and AE9a-mPRMT4-KD2 groups, respectively (p<0.0001; Figure 7F). This demonstrates an important role for PRMT4 in leukemogenesis, and identifies it as an important therapeutic target.
Discussion
We have found that PRMT4 is highly expressed in HSPCs, where it functions as an inhibitor of myeloid differentiation (Figure 7G). In these cells, PRMT4 methylates RUNX1 at R223, promoting the assembly of a DPF2-containing transcriptional co-repressive complex, and repressing transcription at the miR-223 locus. As HSPCs undergo myeloid differentiation, PRMT4 expression decreases, reducing the amount of R223-methyl RUNX1, which in turn, decreases the presence of DPF2 at the miR-223 promoter region, thereby allowing miR-223 to be transcribed. The ability of miR-223 to target the 3′-UTR of PRMT4 allows the further upregulation of miR-223 expression, which further decreases PRMT4 and sustains the myeloid differentiation process. While PRMT4 promotes differentiation in several biological systems including T cell, adipocyte and muscle development, it blocks differentiation in the hematopoietic system, allowing HSPCs to maintain stemness. This role is consistent with its role in embryonic stem cells, where PRMT4 functions to maintain pluripotency (Torres-Padilla et al., 2007; Wu et al., 2009).
The changes in PRMT4 expression and miR-223 during myeloid differentiation allowed us to define a novel link between an arginine methyltransferase and the expression of a microRNA (miR-223), with PRMT4 and miR-223 forming a regulatory loop to influence myeloid differentiation. MicroRNAs that target enzymes involved in epigenetic regulation are being identified, and PRMT4 can join the list of targets, that thus far include Dnmt3A (miR-29) (Fabbri et al., 2007; Garzon et al., 2009) and EZH2 (miR-101) (Varambally et al., 2008). While we have identified the specific sequence in the PRMT4 3′-UTR that contributes to its regulation by miR-223, the 3′-UTR region of PRMT4 has binding sequences for other microRNAs that could also target its expression. Whether these microRNAs also contribute to the regulation of PRMT4 during hematopoiesis will require further study.
The initial signals that trigger the downregulation of PRMT4 expression, which helps drive the process of myeloid differentiation, remain to be determined. Once the decrease in PRMT4 activity occurs, increased expression of miR-223 (and likely other targets of the methyl-RUNX1 repressor complex) occurs, which can further downregulate PRMT4 protein levels and activity, and promote differentiation. This regulatory feedback loop therefore pushes the differentiation process forward. It is known that PRMT4 enzymatic activity can be regulated by post-translational modifications (Cheung et al., 2008; Higashimoto et al., 2007), thus upstream signaling pathways (such as PI3K/ AKT-supplemental results) could control PRMT4 enzymatic activity and expression.
A fundamental aspect of transcriptional regulation has been to define how a given protein can function either as an activator or a repressor. We have recently shown that AML1 (RUNX1)-ETO, a well-known leukemia-associated TF fusion protein generally thought to function as a transcriptional repressor, has activator functions as well, that are critical to its leukemogenic properties (Wang et al., 2011). Our study of PRMT4 provides further evidence for a flexible model of how proteins regulate gene expression. PRMT4 has been thought of as a “secondary” co-activator molecule, that helps activate transcription of its target genes via methylation of histone H3. We have identified a transcriptional repressor function for PRMT4, and provide a molecular basis for this function, which involves the methylation of a non-histone substrate, namely RUNX1. The interaction of PRMT4 with RUNX1 appears to be transient, i.e. a kind of “hit and run”. However, the recruitment of PRMT4 to the chromatin of its target genes could be more stable, either due to its binding histones or other chromatin associated factors.
It is clear that RUNX1 can assemble a variety of multi-protein complexes that affects its transcriptional regulatory functions. These complexes are regulated by various post-transcriptional modifications. The association of RUNX1 with mSIN3A is disrupted by the PRMT1-dependent methylation of RUNX1 on R206 and R210 (Zhao et al., 2008). Similarly, the methylation of C/EBPβ by PRMT4 interfered with its association with both the SWI/SNF and Mediator complexes (Kowenz-Leutz et al., 2010). In contrast to that model, we show that the methylation of RUNX1 by PRMT4 actually promotes protein-protein interactions.
We found the preferential binding of DPF2 to R223-methylated RUNX1 and that by recruiting DPF2, RUNX1 can repress miR-223 expression. This function of DPF2 is consistent with its ability to act as a co-suppressor of nuclear receptor- mediated transcription regulation, by binding HDAC1 (Matsuyama et al., 2010). Although DPF2 has been implicated in transcriptional regulation, little is known about its biological functions. Here, DPF2 appears to be another important regulator of myeloid differentiation that can cooperate with PRMT4 to maintain the “stemness” of HSPCs. As both PRMT4 (Torres-Padilla et al., 2007b; Wu et al., 2009) and DPF2 are expressed in ES cells (Ho et al., 2009), they may also cooperatively regulate gene expression in ES cells.
Targeting of the differentiation (and apoptotic) processes has become a promising therapeutic approach in the treatment of hematologic malignancies like AML, which are characterized by a block in differentiation. We were able to differentiate myeloid leukemic cells by knocking down PRMT4 and observed this effect even in ATRA-resistant cell lines. By utilizing the AML1-ETO driven leukemia model, we showed that knocking down of PRMT4 not only induced myeloid differentiation but also triggered apoptosis, leading to improved survival in an in vivo mouse AML model. These findings strongly suggest that targeting PRMT4 function could hold potential as a novel therapy of acute myelogenous leukemia.
Experimental Procedures
Purification, infection and culture of HSPC-CD34+ cells
CD34+ HSPCs were purified by positive selection using the Midi MACS (magnetic-activated cell sorting) LS+ separation columns and isolation Kit (Miltenyi) starting with mononuclear cells that were isolated from cord blood (CB) by Ficoll-Hypaque Plus density centrifugation. CD34+ cells were cultured in Iscove's modified Dulbecco's medium (IMDM, Cellgro) 20% BIT 9500 medium (Stem Cell Technologies) supplemented with SCF (100 ng/ml), FLT-3 ligand (10 ng/ml), IL-6 (20 ng/ml) and TPO (100 ng/ml) as the basic culture. CD34+ cells were infected with high-titer lentiviral concentrated suspensions, with 8µg/ml polybrene (Aldrich). To differentiate HSPCs, cells were cultured under the myeloid-promoting conditions: SCF (100 ng/ml), FLT-3 ligands (10 ng/ml), IL-3 (20 ng/ml), IL-6 (20 ng/ml), GM-CSF (20 ng/ml) and G-CSF (20 ng/ml) and the erythroid-promoting conditions: Epo (6 IU/ml) and SCF (100 ng/ml). Cytokines were purchased from Peprotech, NJ.
Peptide pull-down assay
Methylated (Acetyl-TPNPR (Asymmetric-dimethyl) ASLNHS-C-amide) and non-methylated (Acetyl-TPNPRASLNHS-C-amide) peptides were synthesized, quantified and conjugated to SulfoLink agarose (Pierce, Rockland, IL). For each pull down reaction, 10mg of HEL cell nuclear extract was used with 10μg peptide bound beads in H lysis buffer (20 mM Hepes pH 7.9, 150mM NaCl, 1mM MgCl2, 1% NP40, 10mM NaF, 0.2mM NaVO4, 10 mM β-glycerol phosphate, 5% glycerol) with freshly added 1mM DTT and proteinase inhibitor cocktail (Roche). After rotating overnight at 4°C, the beads were washed five times with the binding solution. The bound protein was then eluted with 1XSDS sample buffer and analyzed on 4-12% NUPAGE gels.
Chromatin Immunoprecipitation (ChIP) assays
Approximately 4×106 cells were used per ChIP reaction after crosslinking with 1% formaldehyde for 10 minutes at room temperature. ChIP assays were performed according the previously reported methodology (Zhao et al. 2000). After purification, the associated DNA was subjected to qRT-PCR to detect specific DNA sequences. Quantitative results are represented as percentages relative to 5% DNA input. Table S2 provides primer sequences.
In vivo transplantation of AE9a leukemia cells
AE9a expressing mouse leukemia cells were generated based on the work of Wang et al. 2011(Wang et al., 2011). These cells were transduced with lentiviruses expressing RFP and shRNAs against PRMT4 or a scrambled control shRNA. Transduced cells were sorted for RFP positivity and 105-sorted cells were injected into female C57Bl/6 recipient mice that has been sublethally irradiated with 475 cGy via tail vein.
Statistic
Statistical analyses were carried out using Prism 5.0 for Macintosh. All data are shown as mean ±SD. The mean values of each group were compared by Student's t-test.
Supplementary Material
highlights.
PRMT4 blocks myeloid differentiation of human hematopoietic stem/progenitor cells
PRMT4 is downregulated by miR-223 during normal myeloid differentiation
PRMT4 represses miR-223 expression by assembling a methyl-RUNX1 dependent complex
Knockdown of PRMT4 reduces the leukemia cell burden in an AML mouse model
Acknowledgments
We thank Michael Kharas, Minkui Luo, Ross Levine and the members of the Nimer laboratory for providing thoughtful suggestions and comments. We thank the MSKCC Flow Cytometry core facility for their assistance and the Eastern Cooperative Oncology Group (ECOG) bank for making their database of AML patient samples available. The mass spectrometry work was supported by NCI Cancer Center support grant P30 CA08748 to Microchemistry and Proteomics Core Laboratory, MSKCC. We would like to thank Elizabeth Chang for help with mass spectrometry sample preparation. This study was funded by a grant from the National Institutes of Health (R01CA166835.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Bedford MT, Clarke SG. Protein Arginine Methylation in Mammals: Who, What, and Why. Molecular cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyth K, Cameron ER, Neil JC. The runx genes: gain or loss of function in cancer. Nat Rev Cancer. 2005;5:376–387. doi: 10.1038/nrc1607. [DOI] [PubMed] [Google Scholar]
- Chen SL, Loffler KA, Chen D, Stallcup MR, Muscat GEO. The Coactivator-associated Arginine Methyltransferase Is Necessary for Muscle Differentiation. Journal of Biological Chemistry. 2002;277:4324–4333. doi: 10.1074/jbc.M109835200. [DOI] [PubMed] [Google Scholar]
- Cheung N, Chan LC, Thompson A, Cleary ML, So CWE. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9:1208–1215. doi: 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- Cheung WD, Sakabe K, Housley MP, Dias WB, Hart GW. O-linked beta-N-acetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. J Biol Chem. 2008;283:33935–33941. doi: 10.1074/jbc.M806199200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, Imhof R, Bedford MT, Natoli G, Hottiger MO. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J. 2005;24:85–96. doi: 10.1038/sj.emboj.7600500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Wilkinson CR, Waterman SR, Kok CH, Salerno DG, Diakiw SM, Reynolds B, Scott HS, Tsykin A, Glonek GF, et al. Genetic regulators of myelopoiesis and leukemic signaling identified by gene profiling and linear modeling. Journal of leukocyte biology. 2006;80:433–447. doi: 10.1189/jlb.0206112. [DOI] [PubMed] [Google Scholar]
- Daujat S, Bauer UM, Shah V, Turner B, Berger S, Kouzarides T. Crosstalk between CARM1 Methylation and CBP Acetylation on Histone H3. Current Biology. 2002;12:2090–2097. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- Eulalio A, Huntzinger E, Izaurralde E. Getting to the Root of miRNA-Mediated Gene Silencing. Cell. 2008;132:9–14. doi: 10.1016/j.cell.2007.12.024. [DOI] [PubMed] [Google Scholar]
- Eyholzer M, Schmid S, Schardt JA, Haefliger S, Mueller BU, Pabst T. Complexity of miR-223 regulation by CEBPA in human AML. Leukemia research. 2010;34:672–676. doi: 10.1016/j.leukres.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proceedings of the National Academy of Sciences. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazi F, Racanicchi S, Zardo G, Starnes LM, Mancini M, Travaglini L, Diverio D, Ammatuna E, Cimino G, Lo-Coco F, et al. Epigenetic Silencing of the Myelopoiesis Regulator microRNA-223 by the AML1/ETO Oncoprotein. Cancer Cell. 2007;12:457–466. doi: 10.1016/j.ccr.2007.09.020. [DOI] [PubMed] [Google Scholar]
- Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I. A Minicircuitry Comprised of MicroRNA-223 and Transcription Factors NFI-A and C/EBP± Regulates Human Granulopoiesis. Cell. 2005a;123:819–831. doi: 10.1016/j.cell.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 Mutations Result in†a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, Croce CM, Brunetti E, Grignani F, Peschle C. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol. 2007;9:775–787. doi: 10.1038/ncb1613. [DOI] [PubMed] [Google Scholar]
- Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y, Kawamura A, Nakamura K, Takeuchi T, Tanabe M. An Evolutionarily Conserved Mechanism for MicroRNA-223 Expression Revealed by MicroRNA Gene Profiling. Cell. 2007;129:617–631. doi: 10.1016/j.cell.2007.02.048. [DOI] [PubMed] [Google Scholar]
- Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CEA, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KKH, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Molecular cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashimoto K, Kuhn P, Desai D, Cheng X, Xu W. Phosphorylation-mediated inactivation of coactivator-associated arginine methyltransferase 1. Proceedings of the National Academy of Sciences. 2007;104:12318–12323. doi: 10.1073/pnas.0610792104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Ronan JL, Wu J, Staahl BT, Chen L, Kuo A, Lessard J, Nesvizhskii AI, Ranish J, Crabtree GR. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proceedings of the National Academy of Sciences. 2009;106:5181–5186. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, Camargo FD. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008;451:1125–1129. doi: 10.1038/nature06607. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee J, Yadav N, Wu Q, Carter C, Richard S, Richie E, Bedford MT. Loss of CARM1 results in hypomethylation of thymocyte cyclic AMP-regulated phosphoprotein and deregulated early T cell development. J Biol Chem. 2004;279:25339–25344. doi: 10.1074/jbc.M402544200. [DOI] [PubMed] [Google Scholar]
- Koh SS, Li H, Lee YH, Widelitz RB, Chuong CM, Stallcup MR. Synergistic Coactivator Function by Coactivator-associated Arginine Methyltransferase (CARM) 1 and β-Catenin with Two Different Classes of DNA-binding Transcriptional Activators. Journal of Biological Chemistry. 2002;277:26031–26035. doi: 10.1074/jbc.M110865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowenz-Leutz E, Pless O, Dittmar G, Knoblich M, Leutz A. Crosstalk between C/EBP[beta] phosphorylation, arginine methylation, and SWI/SNF/Mediator implies an indexing transcription factor code. EMBO J. 2010;29:1105–1115. doi: 10.1038/emboj.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YH, Bedford MT, Stallcup MR. Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes & Development. 2011;25:176–188. doi: 10.1101/gad.1975811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Zhao X, Perna F, Wang L, Koppikar P, Abdel-Wahab O, Harr MW, Levine RL, Xu H, Tefferi A, et al. JAK2V617F-Mediated Phosphorylation of PRMT5 Downregulates Its Methyltransferase Activity and Promotes Myeloproliferation. Cancer Cell. 2011;19:283–294. doi: 10.1016/j.ccr.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama R, Takada I, Yokoyama A, Fujiyma-Nakamura S, Tsuji N, Kitagawa H, Fujiki R, Kim M, Kouzu-Fujita M, Yano T, et al. Double PHD Fingers Protein DPF2 Recognizes Acetylated Histones and Suppresses the Function of Estrogen-related Receptor a through Histone Deacetylase 1. Journal of Biological Chemistry. 2010;285:18166–18176. doi: 10.1074/jbc.M109.077024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the Target of Multiple Chromosomal Translocations in Human Leukemia, Is Essential for Normal Fetal Liver Hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- Osato M. Point mutations in the RUNX1//AML1 gene: another actor in RUNX leukemia. Oncogene. 2004;23:4284–4296. doi: 10.1038/sj.onc.1207779. [DOI] [PubMed] [Google Scholar]
- Pulikkan JA, Dengler V, Peramangalam PS, Peer Zada AA, Muller-Tidow C, Bohlander SK, Tenen DG, Behre G. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood. 2010;115:1768–1778. doi: 10.1182/blood-2009-08-240101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenauer A, Raelson J, Nervi C, Eydoux P, DeBlasio A, Miller WJ. Alterations in expression, binding to ligand and DNA, and transcriptional activity of rearranged and wild-type retinoid receptors in retinoid-resistant acute promyelocytic leukemia cell lines. Blood. 1996;88:2671–2682. [PubMed] [Google Scholar]
- Schurter BT, Koh SS, Chen D, Bunick GJ, Harp JM, Hanson BL, Henschen-Edman A, Mackay DR, Stallcup MR, Aswad DW. Methylation of Histone H3 by Coactivator-Associated Arginine Methyltransferase 1Ć. Biochemistry. 2001;40:5747–5756. doi: 10.1021/bi002631b. [DOI] [PubMed] [Google Scholar]
- Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, Ratajczak J, Resende IC, Haworth C, Hock R, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23:166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- Torres-Padilla ME, Parfitt DE, Kouzarides T, Zernicka-Goetz M. Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature. 2007;445:214–218. doi: 10.1038/nature05458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al. Genomic Loss of microRNA-101 Leads to Overexpression of Histone Methyltransferase EZH2 in Cancer. Science. 2008;322:1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, Hatlen MA, Vu L, Liu F, Xu H, et al. The Leukemogenicity of AML1-ETO Is Dependent on Site-Specific Lysine Acetylation. Science. 2011;333:765–769. doi: 10.1126/science.1201662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Huang G, Zhao X, Hatlen MA, Vu L, Liu F, Nimer SD. Post-translational modifications of Runx1 regulate its activity in the cell. Blood Cells, Molecules, and Diseases. 2009;43:30–34. doi: 10.1016/j.bcmd.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, Hatlen MA, Vu L, Liu F, Xu H, et al. The Leukemogenicity of AML1-ETO Is Dependent on Site-Specific Lysine Acetylation. Science. 2011;333:765–769. doi: 10.1126/science.1201662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Bruce AW, Jedrusik A, Ellis PD, Andrews RM, Langford CF, Glover DM, Zernicka-Goetz M. CARM1 is required in embryonic stem cells to maintain pluripotency and resist differentiation. Stem Cells. 2009;27:2637–2645. doi: 10.1002/stem.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM, Montminy M, Evans RM. A Transcriptional Switch Mediated by Cofactor Methylation. Science. 2001;294:2507–2511. doi: 10.1126/science.1065961. [DOI] [PubMed] [Google Scholar]
- Yadav N, Cheng D, Richard S, Morel M, Iyer VR, Aldaz CM, Bedford MT. CARM1 promotes adipocyte differentiation by coactivating PPAR[gamma] EMBO Rep. 2008;9:193–198. doi: 10.1038/sj.embor.7401151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, Chen IM, Chen Z, Rowley JD, Willman CL, et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006;12:945–949. doi: 10.1038/nm1443. [DOI] [PubMed] [Google Scholar]
- Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S, Zhang J, Dunne R, Xiao A, Erdjument-Bromage H, et al. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes & Development. 2008;22:640–653. doi: 10.1101/gad.1632608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.