

Abstract
Fundamental research into bioinorganic catalysis of the kind presented at this Faraday Discussion has the potential to turn inspiration drawn from impressive natural energy and chemical transformations into artificial catalyst constructions useful to mankind. Creating bio-inspired artificial constructions requires a level of understanding well beyond simple description of structures and mechanisms of natural enzymes. To be useful, such description must be augmented by a practical sense of structural and energetic engineering tolerances of the mechanism. Significant barriers to achieving an engineering understanding of enzyme mechanisms arise from natural protein complexity. In certain cases we can surmount these barriers to understanding, such as natural electron tunneling, coupling of electron tunneling to light capture and proton exchange as well as simpler bond breaking redox catalysis. Hope for similar solutions of more complex bioinorganic enzymes is indicated in several papers presented in this Discussion. Armed with an engineering understanding of mechanism, the current serious frustrations to successful creation of functional artificial proteins that are rooted in protein complexity can fall away. Here we discuss the genetic and biological roots of protein complexity and show how to dodge and minimize the effects of complexity. In the best-understood cases, artificial enzymes can be designed from scratch using the simplest of protein scaffolds.
The pie chart of Fig. 1 gathers together the principal speakers at this 148th Faraday Discussion on mechanisms in bioinorganic chemistry. They are grouped into general topic areas or approaches along with the metal central to the enzyme that they study. The papers presented in this Faraday Discussion will be a fine reference source for anyone interested in finding out from the bioinorganic chemistry perspective how electron, proton and molecular transfer and metal-catalyzed oxidation–reduction chemistry is currently studied and the challenges which are facing the field of metalloenzmes more generally.
Fig. 1.
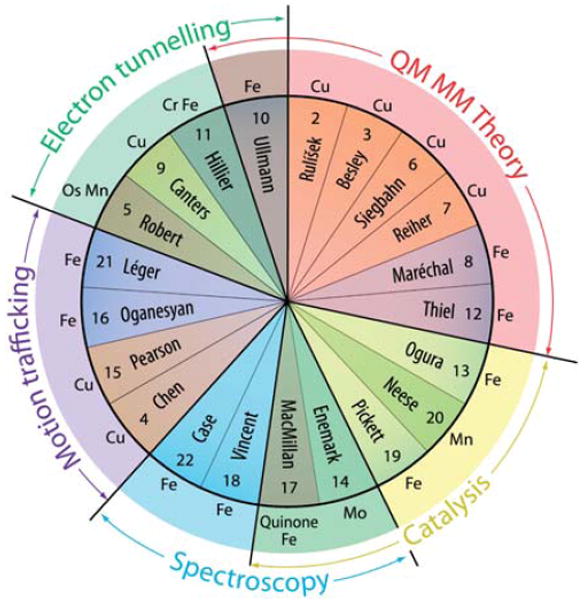
Pie chart of the principal Faraday speakers grouped into topics.
A recurring theme throughout the papers is the challenge of complexity faced by experimentalists and theorists in revealing mechanisms at catalytic metal ligand and protein levels. In this regard it is most cogently argued by Reiher et al.1 in his QM/MM studies on copper enzymes that there is the need, despite complexity, to take basic knowledge gained regarding mechanisms further than just description and to broaden it with the hope of revealing underlying engineering principles that will point to rational ways to modify enzymes or design new ones. It is also something that I have had an interest in for more than a decade and so in these concluding reflections on this Faraday Discussion, I shall air some of my own views and describe an alternative approach to overcoming the frustrations of biological complexity, whether it affects the study of metalloenzymes or the efforts to reproduce them in man-made scaffolds. I have included my colleague Christopher C. Moser as a coauthor in these concluding remarks because the discussions rekindled ideas that we share.
We are all impressed by the speed and efficiency of catalysis promoted and retarded in natural enzymes. This is for good reason because natural enzymes generally beat man-made catalysts hands down for speed, selectivity, performance at ambient temperatures and pressures and, not least, their readiness to be regulated even remotely. The catalysis that natural enzymes perform occurs by means currently beyond our understanding if Richard Feynman’s maxim (1988), “What I cannot create I do not understand”, is to be taken seriously. If our modest progress in creating enzymes is anything to go on, whether by redesign by biologists, de novo design by biochemists or nanoscale material assembly by physicists (see ref. 2 for citations), we still have a long way to go. But as I have already mentioned, an encouraging characteristic of several of the discussion papers spread across the groups in Fig. 1 is that they aspire not only to describe the catalytic mechanism, but also to understand the mechanism’s engineering and construction tolerances which include energetic and distance scales, structural motion, dynamics and flexibility of the bioinorganic cofactor and the supporting protein. This is surely the kind of understanding Feynman considered necessary before we can reasonably expect to create enzymes in man-made scaffolds.
Oxidoreductase enzymes constitute about 25% of the Earth’s enzymes as depicted in Fig. 2. The capability for us to design and build oxidoreductases selected from this family, to-date unrealized in any substantive way, is of increasing importance for medicine, ecology, agriculture and the production of sustainable clean energy. Enzymes that capture light energy and drive the oxygen, nitrogen and carbon cycles have become prime candidates for inspiring catalysis development. While they occupy the relatively slim electron transfer slice of the pie, they exert a profound influence on the Earth. Over eons these enzymes evolved with the Earth and helped craft an atmosphere and biosphere of which humans have become a part relatively recently. And in short order we are facing the possibility that we may be about to alter the character of the Earth as catastrophically as when 3 billion years ago photosynthesis split water and released the then-poisonous by-product oxygen into a reducing atmosphere. With carbon and nitrogen oxide greenhouse gas accumulation we may be moving inexorably toward what could be an irreversible transition toward a new and hostile climate. One of the few practical options that have a hope of reversing this uncomfortable trend lies in creating solar light harvesting arrays and electronic charge separating reaction centers that can be put to use to yield powerful reductants from substrate water to drive man-made enzymes to catalyze the reduction of protons, carbon dioxide and nitrogen for chemical sources and fuels. It is a tall order, but as Thomas Edison said in conversation with his friend Henry Ford in 1931: “I’d put my money on the sun. What a source of power! I hope we don’t have to wait until oil and coal run out until we tackle that!”
Fig. 2.
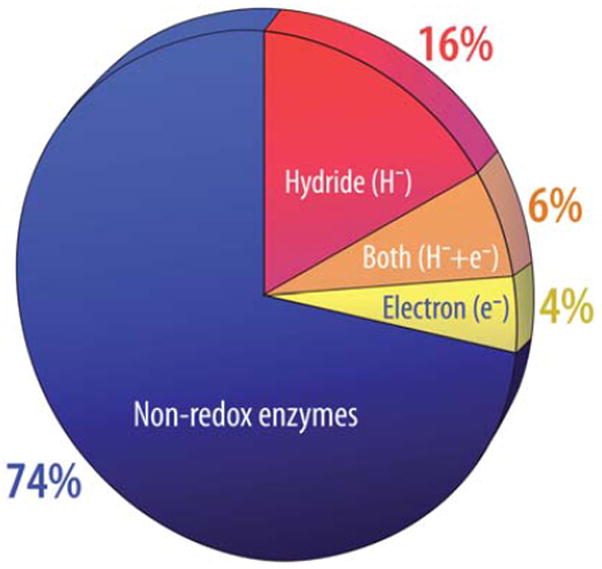
Pie chart of oxidoreductases taken from the IUBMB Enzyme Classification resource. The data was abstracted by inspection of the enzyme classification in 2009. Shown is the 74% population of different enzymes in nature engaged catalysis that does not involve net oxidation and reduction of substrates (blue); the remaining 26% are oxidoreductases of different types. These are sectioned into those known to transfer hydride to/from substrates and generally called dehydrogenases which are common in intermediary metabolism (red slice), and those that promote single electron tunneling through extended chains of cofactors common in photosynthesis and respiration and oxidative and reductive metabolism terminated by dehydrogenase activity (orange slice) or terminated by sites containing clusters of cofactors catalyzing multiple electron chemistry (yellow slice). See ref. 2.
Why is it a tall order? One reason is surely the immense complexity of protein structure. All of us who study biological mechanisms fail in one way or another to escape the obfuscations of biological complexity. It seems reasonable to suggest that progress toward both the understanding and the creation sides of biological catalysis is frustrated less by the intricacies of the chemistry and physics of the catalytic mechanism under study than by the confounding impact of uncharted complexities of enzymes derived from genetic roots and multiple activities intrinsic to natural proteins. Years ago Muller3 made it clear that over time genetic systems acquire interdependency and irreversible mutational load. The resulting gene product proteins under natural selection accumulate complexity and fragility—this includes individual amino acids becoming irreversibly dependent on each other. Fig. 3 shows a self-explanatory example of Muller’s ‘ratchet’ in action.
Fig. 3.
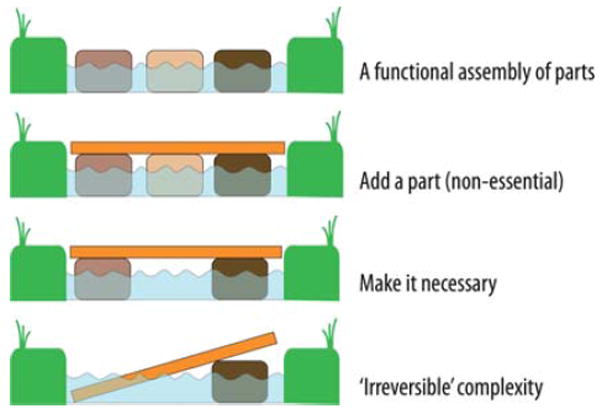
Muller’s ratchet explains the source of fragility, amino acid interdependence and irreversible complexity in proteins.
Another source of biological complexity arises from Darwin’s principle of multiple utility.4 The principle originally applied to organisms recognizes that any one trait in an organism is subject to be selected by multiple forces. At a molecular level, as shown in Fig. 4, the principle indicates that any one amino acid will pick up multiple structural and functional roles during the protein development; twelve of many possible utilities are shown on the pie chart. While such shoals of complexity challenge every approach applied to resolve biological mechanisms, they do not usually prevent molecular descriptions from being ultimately wrested from this complexity, as is amply demonstrated by experimental and theoretical papers at this Faraday Discussion. Mullerian and Darwinian complexities, when combined as they inevitably are in natural systems (Fig. 5), take their toll on efforts that intend to go beyond mechanistic description to obtain the kind of structural and engineering information important for a hoped-for systematic navigation from understanding to creation.
Fig. 4.
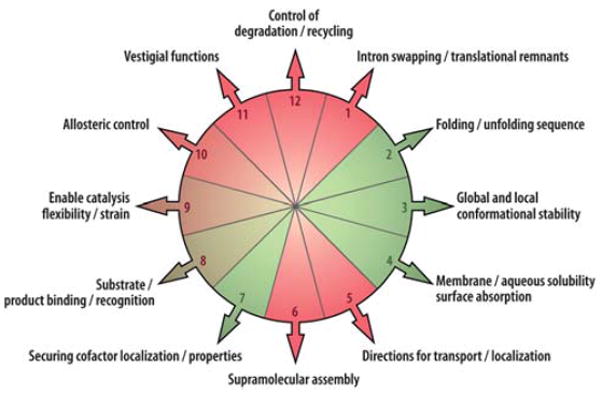
Darwin’s principle of multiple utility at the protein level. In maquette design red segments 1, 11 and 12 would be excluded, as initially would 5 and 6. Green segments 2, 3, 4 and 7 represent minimal utilities for the simplest maquette incorporating a bioinorganic cofactor; green/red segments 8, 9 and 10 represent catalytic utilities.
Fig. 5.
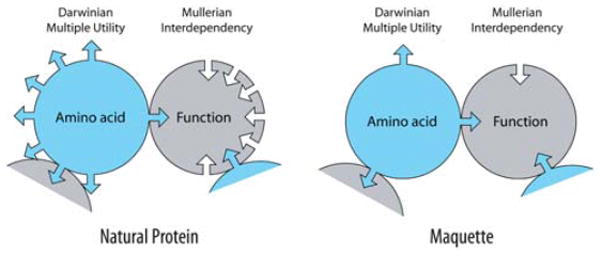
Complexity derived from Mullerian and Darwinian sources in natural protein and in simple proteins engineered from scratch (maquettes). The figure explains that any amino acid serves a number of utilities in a protein. It also demonstrates that an amino acid or a function in a protein displays interdependencies, close and far, with a number of other amino acids. The maquette approach to protein construction diminishes both the number of utilities and interdependencies.
A different approach to this problem asserts that complexity is not essential for enzyme activity.2 It essentially starts the design process from scratch and aims to use the simplest of protein scaffolds to accomplish a selected functional task. We call these protein scaffolds maquettes since they are working models. To date, we have made use of bundles of three, or more commonly, four α-helices, with each helix typically including 30–35 amino acids and hence long enough to span a natural membrane. Elements understood from experimentation or suggested from theory as critical for the selected function of the natural enzyme are introduced into the scaffold by progressive, iterative and entirely reversible design steps. Characterization is continuous. Mimicry of natural enzymes structures is avoided. The design process is maintained independently of natural selection.
Thus, as depicted in Fig. 5, the maquette approach minimizes Mullerian amino acid interdependencies and dodges irreversible complexity. The approach also minimizes and organizes Darwinian multiple utilities. With the combined effects observed in natural proteins greatly diminished in maquettes it becomes feasible to reconcile every amino acid introduced into the maquette with their prescribed purposes.
Maquette simplicity and proven general applicability has been already utilized with the engineering and construction firstly of electron transfer proteins with prescribed characteristics,5,6 as confirmed by Bombarda and Ullmann’s paper;7 secondly in the field of proton coupled electron transfer8,9 in a way analogous to the osmium and manganese biomimetics of Robert et al.;10 and thirdly in the field of the hemoglobin family of oxygen transporters.2
These biological functions, although relatively elementary, are very widespread in nature and represent key functional components or states of many oxidoreductases. They also represent viable stages from which to progress to the design and construction of simpler light- and oxygen-activated oxidative catalysis that is common in natural enzymes. By the time of the next Faraday Discussion in the not too distant future, we can hope to hear the fruits of collaborations between theorist, spectroscopist and experimentalist groups about how enzymes that efficiently catalyze multi-electron bond breaking and forming chemistry can be engineered and constructed to prescribed characteristics.
Acknowledgments
This work was supported by grants from National Institutes of Health GM 41048 and Department of Energy grant DE-PSE02-08ER15944. We are also grateful to J. L. R. Anderson who constructed the pie chart highlighting oxidoreductases.
References
- 1.Podewitz M, Stiebritz MT, Reiher M. Faraday Discuss. 2010;148 doi: 10.1039/c004195e. [DOI] [PubMed] [Google Scholar]
- 2.Koder RL, Anderson JLR, Solomon LA, Reddy KS, Moser CC, Dutton PL. Nature. 2009;458:305–309. doi: 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muller HJ. Mutat Res. 1964;1:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- 4.Darwin C. Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life. 6. 1892. [PMC free article] [PubMed] [Google Scholar]
- 5.Page CC, Moser CC, Chen XX, Dutton PL. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 6.Moser CC, Anderson JL, Dutton PL. Biochim Biophys Acta, Bioenerg. 2010;1797:1573–1586. doi: 10.1016/j.bbabio.2010.04.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bombarda E, Ullmann GM. Faraday Discuss. 2010;148 doi: 10.1039/c003905e. [DOI] [PubMed] [Google Scholar]
- 8.Chen X, Discher BM, Pilloud DL, Gibney BR, Moser CC, Dutton PL. J Phys Chem B. 2002;106:617–624. [Google Scholar]
- 9.Discher BM, Noy D, Strzalka J, Ye S, Moser CC, Lear J, Blasie JK, Dutton PL. Biochemistry. 2005;44:12329–12343. doi: 10.1021/bi050695m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anxolabéhère-Mallart E, Costentin C, Policar C, Robert M, Savéant J-M, Teillout A-L. Faraday Discuss. 2010;148 doi: 10.1039/c004276e. [DOI] [PubMed] [Google Scholar]