

The ability to generate large numbers of bone- and cartilage-forming cells from induced pluripotent stem cells (iPSCs) would mark a major advance in tissue engineering. A number of protocols exist, but the overall quality and consistency of this type of differentiation are under-reported. In this study, the authors analyzed differentiated iPSCs in vitro and in vivo by stringent criteria, and found that in vitro analysis does not predict in vivo differentiation.
Keywords: Induced pluripotent stem cells, Bone, Osteoblast, Chondrogenesis, Transplantation
Abstract
The ability to differentiate induced pluripotent stem cells (iPSCs) into committed skeletal progenitors could allow for an unlimited autologous supply of such cells for therapeutic uses; therefore, we attempted to create novel bone-forming cells from human iPSCs using lines from two distinct tissue sources and methods of differentiation that we previously devised for osteogenic differentiation of human embryonic stem cells, and as suggested by other publications. The resulting cells were assayed using in vitro methods, and the results were compared with those obtained from in vivo transplantation assays. Our results show that true bone was formed in vivo by derivatives of several iPSC lines, but that the successful cell lines and differentiation methodologies were not predicted by the results of the in vitro assays. In addition, bone was formed equally well from iPSCs originating from skin or bone marrow stromal cells (also known as bone marrow-derived mesenchymal stem cells), suggesting that the iPSCs did not retain a “memory” of their previous life. Furthermore, one of the iPSC-derived cell lines formed verifiable cartilage in vivo, which likewise was not predicted by in vitro assays.
Introduction
There is no doubt that having a continuous source of induced pluripotent stem cell (iPSC)-derived autologous cells for skeletal tissue regeneration would mark a major step forward in skeletal tissue engineering. There are reports of attempts to generate osteo- and chondroprogenitors (also known as bone marrow-derived mesenchymal stem cells [MSCs]) from embryonic stem cells (ESCs) [1–10] and iPSCs [11–14]. However, there are three broad limitations in performing these types of studies. First, the definition of osteo- and chondroprogenitor cells is often less than rigorous [15]; it usually relies on cell surface markers, in vitro osteogenic, chondrogenic, and adipogenic assays [16], and/or immunohistochemistry and polymerase chain reaction (PCR) analysis of differentiation markers. However, these criteria alone cannot discriminate between results that are potentially artifactual in nature [15] and those with physiological and clinical relevance. Second, differentiation methods may include ex vivo steps such as embryoid body (EB) formation [2, 4, 12, 17]. Studies reliant on EBs are limited by variability and substitute a black box for a true mechanistic understanding of differentiation processes. Third, although bone formation can be assayed with great specificity by in vivo transplantation, many studies that do so are limited by an incomplete collection or presentation of data or analysis of the results [5, 10, 13, 18–24]. Thus, reports on the formation of bone or cartilage from donor cells are sometimes premature based on the evidence presented.
In the first case, precise and exclusive molecular-based definitions of mature bone-forming cells are fairly well established, but that is not the case for MSCs or even for more narrowly classified bone marrow stromal stem cells (BMSCs) [15, 25]. The accepted markers are nonspecific, do not isolate a uniform population of cells, and do not define any self-replicating stem cell from any connective tissue. The only rigorous definition of bone marrow-derived MSCs is functional and retrospective: they are plastic-adherent, fibroblastic cells that make bone, hematopoiesis-supportive stroma, and marrow adipocytes of donor origin in vivo, and cartilage in pellet cultures in vitro, based on the subset of skeletal stem cells within the population [15, 25]. Confusingly, the definition of MSCs has been expanded to include plastic-adherent, fibroblastic cells derived from other tissues that share cell surface characteristics with BMSCs, due solely to their fibroblastic nature. In any case, although a molecular definition is lacking, the only relevant proof of BMSC potency is shown by functional assays, that is, formation of a bone/marrow organ in vivo and cartilage in vitro [15, 25].
Second, although EB derivatives can lead to discovery of physiologically relevant and clinically useful products, EBs are restricting in several ways. EB heterogeneity requires enzymatic digestion and isolation of cells by means of cell surface character and/or plating under specific culture conditions. With respect to bone regeneration, this would preclude generation of sufficient numbers of cells for direct clinical applications. Also, lack of mechanistic data on how cells underwent differentiation limits the ability to optimize differentiation in a manner consistent with scalability and purity requirements for the U.S. Food and Drug Administration and current good manufacturing processes. Therefore, iPSC derivation (preferably xeno-free) and defined monolayer differentiation are highly desirable methods, which indeed are becoming standardized [26].
To address the third limitation, an in vivo bone-forming assay, usually xenografts in immunocompromised mice (“ectopic ossicles”), must be performed and the results rigorously analyzed for the presence of true bone before claims of osteogenic differentiation can be made. Frequently, microcomputed tomography analysis is used to demonstrate bone formation; however, it cannot distinguish between dystrophic calcification induced by dead and dying cells versus matrix mineralization. Ideally, assaying bone cell transplants begins with histology (H&E stain). Promising bone-like structures that include embedded cells (osteocytes) can be further observed under polarized light for the detection of a collagenous matrix [27] and by UV light for bone autofluorescence [28]. In conjunction with positive histology, the polarized/UV light assays strongly indicate the presence of true bone and are simple to perform on standard microscopes fitted with a polarizer and/or with UV capability. Bone should be further analyzed for donor origin. In addition, transplants should be harvested at several time points to ensure consistency. Lastly, to determine definitively whether the cells have been differentiated into a BMSC- (“MSC”)-like phenotype, in vivo transplants should demonstrate not only bone formation, but also support of hematopoiesis, a critical function of BMSCs [15]. Taken together, these assays ensure a strong probability of true bone-forming potential by transplanted cells.
In this study, we attempted to differentiate iPSCs from various lines into bone-forming cells using a monolayer approach. To test the effectiveness of various methods, we tested the ability of differentiated iPSC lines to form bone in vitro and in vivo. Bone-like structures found in our in vivo transplants were carefully analyzed for histology, matrix, and species of origin. We also attempted to correlate the in vitro assays with the functional in vivo results to define successful predictive methods and elucidate any mechanisms that correlated with the formation of true bone.
Materials and Methods
Human Skin Fibroblast and Bone Marrow Stromal Cell Cultures
Two human skin fibroblast (SF) cultures were obtained from the Coriell Institute for Medical Research (Camden, NJ, http://www.coriell.org) from patients with Parkinson’s disease (AG20443, 71-year-old male; AG08395, 85-year-old female) and expanded as described previously [29]. Human BMSCs were harvested from surgical waste or bone marrow aspirates under institutionally approved protocols (NIH exemption #3113; NIH protocol #94-D-0183, respectively). For reprogramming, BMSCs obtained from deidentified surgical waste from a 7-month-old female with polydactyly were used. Cells were established in culture as previously described [30] (supplemental online data).
Human iPSC Derivation
The two SF lines used in this study were reprogrammed to generate NIHi2 and NIHi7 using individual retroviral vectors (kindly provided by Shinya Yamanaka, Kyoto, Japan) for hOCT4, hSOX2, hKLF4, and hMYC [29] (supplemental online data). BMSCs were reprogrammed using the StemCCA lentiviral reprogramming kit (SCR-531; Millipore, Billerica, MA, http://www.millipore.com) according to the manufacturer’s instructions (supplemental online data), and three human iPSC lines (SCUi1, SCUi8, and SCUi9) are reported here and were characterized for their pluripotential nature as described below.
Human ESC and iPSC Cultures
Human ESC (HSF-6) and iPSC lines were grown as colonies on irradiated mouse embryonic fibroblast (MEF) feeder cells and passaged as described previously [30] (supplemental online data).
Pluripotency Assays
All lines were assessed for pluripotency markers by fluorescence-activated cell sorting (FACS) and for their ability to differentiate into representatives of the three germ layers in vitro and in vivo (supplemental online data).
Quantitative Real-Time PCR Primers and Conditions
Levels of mRNAs for pluripotency and mesodermal and osteogenic markers in undifferentiated and differentiated iPSCs were assessed by quantitative PRC (qPCR) in comparison with ESCs and BMSCs (supplemental online data), using primers found in supplemental online Table 1.
Human iPSC Differentiation
All differentiation schemes were performed in monolayer cultures. Undifferentiated cells were harvested from six-well plates using a “scraping” method [30]. Cells from one overgrown well were scraped, collected using a plastic pipette tip, and the fragments (including irradiated MEFs) were harvested without trituration, pelleted for 3 minutes at 800 rpm, and plated into a T-75 flask in medium according to the program and schedule. Cells that adhered to the T-75 plastic were called “differentiation P0.” For all cultures, differentiation medium consisted of Knockout D-MEM (Life Technologies, Burlington, Ontario, Canada, http://www.lifetechnologies.com), 10% fetal bovine serum, GlutaMAX, Pen/Strep, and nonessential amino acids. Other factors such as dexamethasone (10−8 M; Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com) with ascorbic acid-2-phosphate (Dex+AscP = A1; Wako Chemical, Osaka, Japan, http://www.wako-chem.co.jp/english); retinoic acid (10−6 M = A2; Sigma-Aldrich); rapamycin (10−9 M = A3; Sigma-Aldrich); and basic fibroblast growth factor (bFGF; 6 ng/ml) and bone morphogenetic protein (BMP4; 10 ng/ml, both from Invitrogen, Carlsbad, CA, http://www.invitrogen.com) (FGF + BMP4 = A4) supplemented the basic mixture. Passaging was performed using 1 mg/ml Collagenase Type IV for 30 minutes followed by 0.05% trypsin/EDTA for 20 minutes (with one additional trypsinization if necessary). Extra cells were frozen when available from each passage using a 1:1 mixture of basic growth medium and 2× iPSC freezing medium (supplemental online data). Differentiated cells were seeded onto particles of hydroxyapatite/tricalcium phosphate (HA/TCP, particle size 0.5–1 mm; Zimmer, Warsaw, IN, http://www.zimmer.com) for regular transplantation, or to form a “carpet” in medium A1 (first priority), or used for in vitro assays after incubation in mineralization medium (second priority), as described below.
In Vitro Osteogenic Assay
We seeded and expanded 5–10 × 104 differentiated cells per well of a six-well dish in BMSC medium (supplemental online data) until nearly confluent. BMSC medium was then supplemented with Dex+AscP+beta-glycerophosphate (GP) (mineralization medium) that was changed two or three times per week for 4–6 weeks, when signs of mineralization were visible under bright-field microscopy. Wells were fixed with fresh 4% formaldehyde for 1 hour, rinsed in double-distilled H2O (ddH2O), then incubated with 1% alizarin red S (weight per volume, with 97% ddH2O and 2% ethanol [volume per volume]) for 5 minutes. Excess stain was rinsed away with 5 changes in ddH2O. Each line except one was analyzed in triplicate.
Karyotyping
Karyotyping was performed to ensure that reprogramming and redifferentiation did not introduce gross chromosomal abnormalities that could influence our results. An aliquot of differentiated cells from the same cells that were used for in vivo transplantation was shipped live for analyses, per the company’s instructions (Cell Line Genetics, Madison, WI, http://www.clgenetics.com).
In Vivo Transplantation of Differentiated Human iPSCs
Differentiated cells were transplanted using either the “regular” method or the “carpet” method. For the regular method, differentiated cells (2 × 106 cells per 1 ml of differentiation medium) were mixed for 90 minutes at 37°C with 40 mg of HA/TCP. After cell attachment, each tube was spun for 1 minute at 300g and the supernatant was aspirated [31, 32]. For the carpet method, 0.5 × 106–2.5 × 106 cells were allowed to grow on a one-particle-thick layer (300 mg) of HA/TCP in differentiation medium A1 in one well of a nontissue culture six-well plate for 14 days, forming a dense carpet that could be picked up with forceps and cut into transplant-sized pieces [30]. Both types of constructs were transplanted subcutaneously into immunocompromised mice (either Crl:NIH-Lystbg Foxn1ν Btkxid [Charles River Laboratories, Wilmington, MA, http://www.criver.com] or Nod.Cg-PrkdcscidIL2RGtm1wjl/SzJ [Jackson Laboratories]), under National Institute of Dental and Craniofacial Research Animal Care and Use Committee-approved surgical protocols. Between 3 and 10 transplants were generated from each line, depending on the number of cells available. Transplants were harvested at 6, 8, 12, 16, and 22 weeks, fixed for 48 hours at 4°C in 4% formaldehyde in phosphate-buffered saline (PBS), rinsed in PBS, and demineralized in 0.25 M EDTA. Demineralization was verified using x-ray imaging, and transplants were paraffin-embedded for sectioning and H&E staining.
Analysis of Osteogenesis In Vivo
H&E-stained sections of transplants were viewed with a Zeiss AX-10 microscope fitted with a polarizer and equipped with AxioCam HRc and AxioCam MR cameras (Carl Zeiss, Jena, Germany, http://www.zeiss.com). Candidate bone areas were observed using polarized light under phased optics to verify the presence of organized collagen fibers [27] and using UV light for bone autofluorescence [28].
In previous publications, the following scoring system from 0 to 4 for bone formed within in vivo transplants was developed and validated [30]:
score 0 no bone formation;
score 1 minimal bone formation, a single or a few bone trabeculae, in one or a few sections;
score 2 low bone formation, bone in several parts of several sections, occupying only a small portion of each section;
score 3 moderate bone formation, bone occupies a significant portion but less than one half of most sections; and
score 4 abundant bone formation, bone occupies greater than one half of each section.
This scoring system was applied to semiquantitate the amount of bone formed by differentiated iPSCs.
To detect human cells in transplanted tissue, we performed in situ hybridization for human-specific ALU repetitive DNA sequences. A Rembrandt/Pan Path Digoxigenin label kit (Invitrogen A001K.9905) was used according to the kit instructions. Sections of mouse and human bone were used as negative and positive controls, respectively.
Analysis of Chondrogenesis In Vitro and In Vivo
For cartilage formation by differentiated NIHi2-A1 cells in vitro, 4 × 105 (protocol 1 [33]) or 2 × 106 (protocol 2 [14]) cells were treated as described in the supplemental online data. Sections of pellet cultures and in vivo transplants were stained with H&E, or Alcian blue counterstained with Nuclear Fast Red, or Toluidine Blue, and imaged by bright-field microscopy. Detection of human cells in cartilage formed in vivo was assessed by in situ hybridization for human-specific ALU repetitive DNA sequences as described above. Cartilage in the in vivo transplants was further confirmed using antibodies against aggrecan (Millipore AB1031 [1:100]) (all described in supplemental online data).
Statistical Analyses
Data are shown as the mean ± standard deviation.
Results
Creation of Human iPSCs From SFs and BMSCs
iPSCs lines derived from human SFs (NIHi2 and NIHi7) and BMSCs (SCUi1, SCUi8, and SCUi9) were comparable to hESCs, both in appearance (colonies of tightly packed cells with large nuclei, prominent nucleoli, and sharply defined edges) (supplemental online Fig. 1A; SCUi1, for example), rapid proliferation, and by the expression patterns of characteristic genes and cell surface markers. Like hESCs, they did not express SSEA1, but did express the pluripotency markers SSEA4 and TRA-1-81, as measured by FACS analysis (Fig. 1A). Furthermore, they expressed pluripotency transcription factors Nanog and Oct4 comparable to hESCs based on qPCR (Fig. 1B) and immunohistochemistry (supplemental online Fig. 1A). In contrast, BMSC expression of Oct4 and Nanog was between 0% and 0.1% relative to HSF-6 set at 1.0 [30] (Fig. 1B).
Figure 1.

Evidence for the reprogramming of induced pluripotent stem cell lines. (A): Expression of characteristic surface markers as determined by flow cytometry is displayed for NIHi2, NIHi7, and SCUi1 compared with that of a hESC control line. The isotype control antibody for each antibody tested is in black, and the gray line represents the specified antibody results. (B): Normalized fold expression of Nanog and Oct4 mRNA levels is displayed for NIHi2, NIHi7, and SCUi1 compared with the level found in human embryonic stem cells (ESC column). Virtually identical results were found for SCUi8 and SCUi9 by fluorescence-activated cell sorting and by quantitative polymerase chain reaction (data not shown). Abbreviations: BMSCs, bone marrow stromal stem cells; ESC, embryonic stem cell; hESC, human embryonic stem cell.
These iPSC lines were tested for pluripotency in vitro and in vivo. In vitro differentiation protocols (e.g., NIHi2 cells) yielded cells representative of all three lineages (hepatocytes, neurons, and cardiomyocytes (supplemental online Fig. 1B) and produced large teratomas containing cells representative of all germ layers in vivo (supplemental online Fig. 1C).
Osteogenic Differentiation of Human iPSCs
All lines were differentiated toward an osteogenic fate with a method that we previously reported using hESCs [30] and by methods reported by others. iPSCs scraped from one well of a six-well plate were allowed to attach to regular T-75 flasks in differentiation media, and were grown to confluence in the various differentiation media (Fig. 2). These cultures were labeled “passage 0.”
Figure 2.

Representation of the methods used to differentiate iPSC lines into a bone fate. Each of the three sections represents all the differentiation methods used. At the top of each section is a differentiation timeline. Underneath the timeline, the lengths and positions of the colored arrows represent the amount of time the cells in a particular program were exposed to each condition. The color of each arrow indicates that different additives were used as shown (e.g., RA [orange], R [green], F [red], B [pink], and F + B [yellow]). Light blue arrows always indicate the presence of D in the medium. Where carpet culture arrows are not shown, carpets were not made. Dashed lines indicate significant events in each program (such as differentiation, passaging, and transplant). Abbreviations: B, bone morphogenetic protein; bFGF, basic fibroblast growth factor; BMP4, bone morphogenetic protein; D, dexamethasone and ascorbic acid phosphate; F, basic fibroblast growth factor; iPSC, induced pluripotent stem cell; Pass, passage; R, rapamycin; RA, retinoic acid.
Passage 0 was maintained past confluence, according to a short schedule (line NIHi2 only, 16 days) or a long schedule (all lines, 23–44 days), and subjected to various differentiation additives such as Dex+AscP (A1), retinoic acid (A2), rapamycin (A3), and bFGF+BMP4 (A4) (Fig. 2). Under these conditions, cells initially differentiated spontaneously into forms resembling neurons, fibroblasts, epithelial cells, and beating cardiomyocytes, among others (data not shown). The cells were passaged three times further, generally becoming more uniform and fibroblastic in appearance with time (data not shown). At passage 4 (P4), cells were used for in vivo (first priority) and in vitro (second priority) differentiation assays and for mRNA analyses (third priority), and any remaining cells were frozen. Because some differentiated lines grew slowly or became arrested, there were instances in which only the in vivo transplantation assay was performed.
qPCR Analysis of Differentiated Cell Lines
qPCR was used to verify that expression of genes associated with pluripotency were downregulated in the treated lines compared with human ESCs (positive control), and to determine whether the same differentiation methods produced similar results in the different lines. The differentiated cells were compared with human BMSCs representative of osteoprogenitors.
RNA was isolated from each differentiated line at P4 and analyzed by qPCR. Consistent with their differentiated appearance, levels of pluripotency markers Nanog and Oct4 were either strongly reduced or undetectable in derivatives of NIHi2-A1 and NIHi7-A4 (lines for which we possessed three independent biological samples) compared with undifferentiated controls.
The similarity of differentiated iPSCs to BMSCs was evaluated by measuring the relative expression levels of a variety of transcripts (CD44, PDGFRα [mesodermal markers], RUNX2, OSX, ALPL, COL1A1, BSP, OCN [osteogenic markers], and COL2A1 [chondrogenic marker]) when we possessed three independent biological samples. Under various conditions, levels of CD44 and PDGFRα in the iPSC-derived cells varied from ∼4 times lower to ∼4 times higher than in BMSCs, with no clear pattern (Fig. 3A). Levels of RUNX2, the master regulator of bone formation, were comparable across all lines and differentiation conditions studied. However, ALP was always lower than in BMSCs, whereas expression of COL1A1 ranged from 4 to 6 times higher than BMSCs (Fig. 3A). OSX, BSP, and OCN transcripts, expressed by osteogenic cells, were barely detectable or undetectable in the iPSC-derived lines (data not shown). Finally, expression of COL2A1 transcripts was much greater in NIHi2-A1 and NIHi7-A4 than in BMSCs (Fig. 3A). Undifferentiated NIHi2 and NIHi7 expressed lower levels of the markers compared with their differentiated progeny, with the exception of ALP, which was expressed by undifferentiated iPSCs (Fig. 3A).
Figure 3.
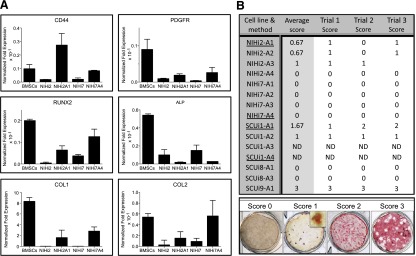
In vitro assays. (A): Quantitative PCR evidence for differentiation of induced pluripotent stem cell lines to a bone or cartilage fate. When available, three independent biological samples from cell lines and methods that successfully made bone in vivo were used to perform qPCR on mRNA harvested prior to cell transplantation. The results were compared with bone marrow stromal cell controls, which form bone in vivo and cartilage in vitro. (B): Results from in vitro mineralization assays. The in vitro mineralization assay was performed as described on all the cell lines and methods listed in the summary table, when available, in triplicate. After alizarin red S staining, each well was assigned a numerical score based on the amount of red staining visible, as shown in the scored samples across the bottom of the figure. For a score of 0, the well must have no significant red staining even under high magnification. For a score of 1, red staining must be visible only with magnification (inset), and wells scored as 2 must have red staining visible to the eye, but less than half the plate. To achieve a score of 3, the majority of the well must be stained. In the summary table, underlined cell lines and methods were those that made bone in vivo. The average score is the mathematical average of the three trial scores shown. Abbreviations: BMSCs, bone marrow stromal stem cells; ND, not determined because of a lack of cells (in vivo transplantation was the priority); PCR, polymerase chain reaction; qPCR, quantitative PCR.
In Vitro Cell Differentiation Assays
It is commonly presumed that in vitro mineralization assays predict in vivo outcomes of cell transplantation [16]. The in vitro assay is faster and more convenient than animal transplantation. Near-confluent differentiated cells in six-well plates were switched to mineralization medium containing 10 mM GP. After 3–4 weeks, the cells were fixed and stained with alizarin red S, a specific marker of calcium accumulation and thus potentially an early marker of bone formation (von Kossa staining, a marker of phosphate, was not used as it not specific for calcium [34]).
The results were varied, with some cells lacking any staining, whereas others showed significant and widespread calcium accumulation. Individual wells were scored on a scale of 0 to 3, and the average score per line is shown in Figure 3B. The average mineralization score was highest in SCUi9-A1. Of note, NIHi7 showed no in vitro staining under any culture condition.
In Vivo Bone Formation Assay
At the end of their respective differentiation schedules, the karyotype of each differentiated strain was analyzed and found to be normal (data not shown), and the cells were prepared for transplantation into immunocompromised mice. Unlike murine BMSCs that form bone in collagen sponges, it has been consistently found that the presence of mineral is essential for true bone formation in vivo when using human BMSCs [31, 32]. The assay was performed in two ways. First, differentiated cells in suspension were briefly incubated with HA/TCP (regular transplants) and transplanted directly. Second, differentiated cells were seeded into nonadhesive six-well plates containing a thin layer of granulated HA/TCP and grown to overconfluence over the course of 2 weeks with Dex+AscP. The cells grew in a thick, carpet-like layer, effectively connecting the HA/TCP particles. In many cases, this “carpet” became so dense that it could be picked up as one piece using forceps. The carpet was cut into four to six pieces, which were transplanted subcutaneously into immunocompromised mice.
Transplants were harvested at intervals between 6 and 22 weeks and were processed for histology. Initially, sections were stained with H&E, which stains mineralized matrix of bone with a deep red color. In addition, bone can also be identified by the presence of osteocytes embedded in lacunae, with osteoblasts on the surface of the forming bone. By these initial criteria, small amounts of bone-like material were observed in the transplants of all lines, but most prominently in four (Fig. 4, H&E; supplemental online Fig. 2A–2D). However, the presence of true bone can only be determined with rigor by using additional assays, including detection of organized collagen bundles in the bone matrix visible under polarized light [27] and autofluorescence visible under UV light [28]. Taken together, positive results for these tests indicate the presence of bona fide bone in transplants. Accordingly, we analyzed the transplant sections that were histologically consistent with the presence of bone for further indications of bone compared with transplants made with BMSCs. Under polarized light, widespread collagen bundles were visible in the transplants from four lines, indicating that the structures were not formed by dystrophic calcification (Fig. 4). Also, the suspected bone fragments were autofluorescent under UV light (Fig. 4). Based on these results, it was verified that true bone had been formed in these in vivo transplants. To determine the species origin of the bone, we performed human ALU-specific in situ hybridization on the transplants (Fig. 4), with human bone as a positive control and mouse bone as a negative control (supplemental online Fig. 3A, 3B). The hybridization revealed that the bone was human in all samples tested.
Figure 4.

Evidence that true bone was formed in certain in vivo transplants. For each cell line listed on the left, H&E, polarized light, and fluorescent light views of a single transplant section are shown compared with transplants of BMSCs, which form abundant areas of bone (b) and support hematopoiesis (hp). For H&E staining, the bone appears red and the hydroxyapatite carrier particles stain purple. ALU-positive cells (osteocytes) are observed in lacunae. When illuminated with polarized light, the collagen bundles in the bone matrix become visible as orange or pink striata, and the hydroxyapatite remains dark. Under fluorescent conditions, the pieces of bone are clear and bright green, and the surrounding tissue and carrier particles remain dark. For NIHi2-A1 and SCUi1-A4 transplants, an additional picture of an adjacent serial section is shown, in which human-specific ALU in situ hybridization was performed. Few intact nuclei were present for in situ hybridization because these are small pieces of bone, but enough are present to indicate that the bone and surrounding fibrous tissues are of human origin. All panels are at the same magnification (scale bar = 50 μm). Human bone (positive control) and mouse bone (negative control) are shown in supplemental online Figure 3. Abbreviation: BMSC, bone marrow stromal cell.
According to a transplant bone scoring system that we previously developed, the bone formed in these experiments did not rise above a score of 1 (scale of 0–4). An additional measure of the precursor nature of the cells under evaluation in these transplants would be the ability to support the formation of marrow with stroma and marrow adipocytes of human origin and hematopoiesis of mouse origin, as we have previously demonstrated [35, 36] (supplemental online Fig. 3C, 3D). Unlike BMSC transplants, no hematopoiesis was observed in any of the transplants made from iPSC-derived cells, signifying the lack of early progenitors in the lines tested. However, it is possible that the small amounts of bone observed were not sufficient to support hematopoiesis.
Many other areas in the transplants superficially resembled bone. These areas were evaluated by the polarized light and UV autofluorescence tests (Fig. 5). Although these areas outwardly resembled bone by H&E histology, they did not contain bundled collagen and did not autofluoresce. Fibrous connective tissue was predominant in many transplants, especially in those devoid of bone (supplemental online Fig. 2E, 2F).
Figure 5.

H&E-stained histological sections are not reliable indicators of true bone. When stained with H&E, several promising formations appeared red, which is characteristic of bone matrix when stained with H&E. Note also the appearance of widely spaced nuclei contained in apparent lacunae. However, when the same sections were examined under polarized light, no organized collagen bundles were apparent. Likewise, under fluorescent light, the bone-like formations remained dark with little autofluorescence, again indicating the lack of organized collagen that indicates true bone. These areas also did not stain with Toluidine Blue. All panels are at the same magnification (scale bar = 50 μm). Abbreviations: Not bone 1, NIHi2-A3; Not bone 2, SCUi9-A1.
Cartilage Formation
Surprisingly, 3 of 10 transplants made using NIHi2 cultured in A1 conditions also contained cartilage (Fig. 6A). In nearly 20 years of in vivo transplantation experiments using a ceramic-based scaffold, this was our first observation of cartilage formation. To definitively determine the presence of cartilage in the NIHi2-A1 transplants, we applied histological staining and immunohistochemistry. Sections were stained with Toluidine Blue, and a characteristic metachromatic (purple) staining was observed because of the presence of glycosaminoglycans. Antiaggrecan immunohistochemistry revealed staining of matrix and cells. In situ hybridization using the human-specific ALU probe revealed a human origin for the observed cartilage. However, it must be noted that cartilage was found in only one differentiated line transplanted by either the regular method or the carpet method (Fig. 7).
Figure 6.

Differentiated NIHi2-A1 cells formed cartilage. (A): Serial sections from two pieces of cartilage are shown, one in each row. Results of H&E and Toluidine Blue staining indicate the presence of characteristic cartilage matrix containing proteoglycans and glycosaminoglycans. In addition, the last two panels in row 1 show the result of antiaggrecan immunohistochemistry (including nonimmune control). Positive aggrecan staining strongly indicates the presence of cartilage. In row 2, the final panel shows the result of an in situ hybridization directed against human-specific ALU repeats. (B): NIHi2-A1 was tested in an in vitro pellet assay (protocol 1 is shown here) for the ability to form cartilage in comparison with bone marrow stromal cells (BMSCs). The resulting pellets were sectioned and stained using either Alcian blue with Nuclear Fast Red or Toluidine Blue. Chondrocytes are seen surrounded with Alcian blue positive matrix in BMSC pellets, but not in NIHi2-A1 pellets. However, note that staining of matrix in the BMSC pellet with Toluidine Blue that becomes purple (metachromasia) is far more definitive. One can see bona fide chondrocytes lying in lacunae surrounded by purple-stained matrix. At the same time, pellets made with NIHi2-A1 cells did not stain prominently with Alcian blue and did not display the characteristic metachromatic (purple) color associated with binding to sulfated glycosaminoglycans but remained a dull blue, with no histological evidence of cartilage formation visible. All panels are at the same magnification (scale bar = 50 μm). Abbreviation: hBMSCs, human bone marrow stromal stem cells.
Figure 7.

Data summary. In the top panel, the cell lines and method used to successfully differentiate induced pluripotent stem cells into bone in vivo are shown on the left, with the results on the right. Both regular and carpet culture transplant results are shown. The results of in vivo transplantation are displayed on the right in table form. In the results table, blue signifies that cartilage was found in a transplant, and red indicates that true bone was found. The results are stated as the number of bone- or cartilage-producing transplants over the total number of transplants created. In the bottom panel, a summary table shows qPCR, in vitro mineralization scores, in vitro cartilage formation scores, in vivo bone formation scores, in vivo bone formation frequency as a percentage of all transplants made, and an indicator for the presence of in vivo cartilage. In vitro mineralization scores are averaged as described in Figure 4. For in vivo bone scores, the highest score achieved in any transplant for the corresponding cell type is shown. In vivo bone frequency refers to the number of times true bone appears in a transplant, calculated as a percentage of all transplants attempted for the corresponding cell type. In vivo cartilage is represented as present (+) or not present (0). Abbreviations: B, bone morphogenetic protein; bFGF, basic fibroblast growth factor; BMP4, bone morphogenetic protein; BMSC, bone marrow stromal stem cell; D, dexamethasone and ascorbic acid phosphate; F, basic fibroblast growth factor; hESC, human embryonic stem cell; qPCR, quantitative polymerase chain reaction; R, rapamycin; RA, retinoic acid.
Based on this surprising result, a cell pellet assay for cartilage formation in vitro was retrospectively performed using differentiated cells frozen at the time that the in vivo transplants were generated to determine whether this in vitro assay would have predicted the formation of cartilage in vivo. Using two different protocols (supplemental online data), we did not observe cartilage in any of the NIHi2-A1-derived pellets by either Alcian blue or Toluidine Blue staining, unlike BMSC-generated pellet cultures (Fig. 6B).
Summary of Results
The in vivo and in vitro data are summarized in Figure 7. Four of 15 differentiated iPSC populations produced small quantities of bone in vivo (score 1) at low frequency and inconsistent time points; one also produced cartilage at varying time points. The most common time point for bone formation in vivo was 12 weeks (BMSCs form bone by 4 weeks). Differentiated iPSC lines that made true bone in vivo (the gold standard) were either differentiated in medium containing Dex+AscP (A1) or in a scheduled program of bFGF+BMP4 treatment, followed by Dex+AscP (A4).
qPCR analysis of mRNA was performed when three independent samples were available. For lines that made bone, qPCR data showed a commonality of gene expression in several categories: (a) pluripotency markers were strongly downregulated compared with undifferentiated controls, (b) mesodermal markers were upregulated and were roughly comparable to expression in BMSCs, (c) expression of the master bone regulator gene RUNX2 was similar to levels found in BMSCs, and (d) levels of downstream bone-related transcripts did not match the expression found in BMSCs (either not expressed, expressed at low levels, or strongly overexpressed). Thus, the lines that made bone and cartilage in vivo could not be distinguished from those that did not based on the gene expression profiles of these selected targets.
Mineralization assays also produced results at odds with the observed in vivo results. For example, four of six (67%) of the differentiated lines that mineralized in vitro did not form bone in vivo; one of four lines (25%) that did form bone in vivo did not mineralize in vitro. One line made cartilage in vivo, but did not produce cartilage in pellet cultures in vitro.
Discussion
Herein we report the generation of iPSCs from human SFs and BMSCs, and describe methods to differentiate bone- and cartilage-forming cells from them based on our previous work in hESCs [30], and from others (retinoic acid [12, 37], rapamycin [9], bFGF [38], and BMP4 [4, 37]). The two different sources of iPSC lines were used to determine whether some form of cell memory survived reprogramming. Once differentiated, the cells were analyzed using in vitro and in vivo assays, and the end products were tested rigorously for the hallmarks of bone and cartilage, including donor origin in vivo. Our results indicate that in vitro PCR and mineralization assays, as currently conducted, lack predictive power for in vivo formation of true bone and cartilage.
In Vitro Versus In Vivo Assays
Development of in vitro assays with an in vivo predictive ability remains extremely desirable and would facilitate translational bone and cartilage research. Based on the fact that in vivo formation of bone is the most rigorous and clinically relevant endpoint available, it is imperative that in vitro assays reliably and specifically predict the in vivo results. However, it was found that qPCR-based assays performed at the conclusion of the iPSC differentiation programs did not predict bone formation in vivo based on any one gene or combination of genes that were tested. In addition, using data not shown here, the expression of any one gene or combination of the genes that were examined could not be used to successfully predict which differentiated iPSC lines would not form bone in vivo. Any particular cell line tended to express a similar pattern regardless of the differentiation method used, that is, the starting cell line was more important to the outcome than the differentiation method.
Similarly, in vitro mineralization and cartilage pellet assays did not result in reliable or specific prediction of in vivo bone and cartilage formation. The lines with the greatest in vitro mineralization (alizarin red S staining) did not make bone in vivo. At the same time, one line (NIHi7-A4) that did not mineralize in vitro made bona fide bone in vivo. The lone iPSC line that made cartilage in vivo failed to make any cartilage in pellet cultures. Taking these evidences together, we conclude that the results of in vitro assays as described need to be corroborated with other substantial evidence (such as an in vivo assay) before strong claims of bone and cartilage formation are made.
A caveat of our study is that the in vitro sample size was relatively small. Therefore, it remains possible that in vitro results may help to enrich differentiation methods that produce bone and cartilage in vivo without absolutely defining them. Further experimentation may help tease out the degree of predictive power, if any.
Lack of iPSC Memory
Although it has been suggested that donor cell types influence differentiation potentials of iPSC lines [23, 39], our current in vivo results do not reflect this. Bone of equal quality and frequency was made from both SF- or BMSC-derived iPSCs. In a larger series of samples, it remains possible that some level of bias would be apparent. However, this determination must wait for future experiments. Also, it is worth noting that the donor age of cells used for the iPSC derivation (71 years old and 85 years old [SFs] vs. 7 months old [BMSCs]) had no influence on iPSC differentiation potential. Furthermore, the NIH lines were generated using retroviruses, whereas the SCU lines were generated using a polycistronic, excisable lentivirus, but this does not seem to have influenced the outcome of osteogenic differentiation.
Performance of hESCs Versus iPSCs in Bone Differentiation Assays
Slightly higher quality bone was made in vivo at a slightly higher frequency when derived from hESCs than from iPSCs (compare results reported here with those of Kuznetsov et al. [30]). The areas of bone made by differentiated hESCs were larger and more widespread than those made by human iPSCs. Some hESC-based bone scored 2 on the 0–4 scale, but none of the iPSC-based bone scored above 1. In addition, neither hESC- nor human iPSC-derived bone supported hematopoiesis, which suggests that the cells generated by our differentiation schemes were more osteogenically committed than BMSCs.
During hESC differentiation into bone, carcinomas or teratomas spontaneously appeared in some in vivo transplants, but an alternative differentiation method resulted in a decrease in the number of transplants with tumors [30]. In the work presented here, no tumors were found, suggesting that either iPSC-derived cells are less likely than hESC-derived cells to form spontaneous tumors or the growth conditions used are successful in preventing tumor growth and induced more differentiation, or a combination of these factors.
Conclusion
The potential usefulness of an unlimited supply of autologous bone-forming cells is well documented. Rather than generating mature osteogenic cells, it would be preferable to generate precursors with extensive proliferation capacity, such as BMSCs. However, during development, bone marrow stroma emanates from identifiable osteoblastic cells [40–42], that is, bone marrow stroma develops after bone. Although BMSCs are the prototype MSCs, the current definition of MSCs is less than rigorous. An attempt was made to establish a common definition, but this relies on nonspecific cell surface markers and in vitro differentiation assays that are prone to artifact [16], leading to unsubstantiated conclusions in many cases. More rigorously defined criteria, such as what we have described here, which demonstrate in vivo performance and clinical relevance will therefore benefit the entire field by reducing spurious claims and allowing the direct comparison of different methods.
The rate-limiting step in our work is the in vivo assay, the most clinically relevant standard for identifying an osteogenic phenotype. We continue to work toward developing rapid and convenient in vitro assays useful for predicting in vivo performance. However, it must also be noted that in vivo performance in immunocompromised mice may not be predictive of performance in immunocompetent recipients. Studies are clearly needed in which iPSCs are derived from an individual donor, differentiated, and transplanted back into that donor. Having said that, we have successfully differentiated iPSCs to bone-forming cells by incubation with bFGF+BMP4 and/or Dex+AscP. Future attempts will entail the use of a more developmental approach, along with use of cell surface markers for cell sorting. The resulting purified cells could be expanded and profiled using qPCR and in vivo assays. We expect that these improvements will increase performance. As we learn which subsets of differentiated cells perform best, we can change the differentiation conditions to emphasize the successful fraction of cells and obtain a faster readout and, therefore, greater progress.
Supplementary Material
Acknowledgments
We thank Dr. Linda Wolff (National Cancer Institute, NIH, U.S. Department of Health and Human Services) for kindly providing the gp293 cells. We are also indebted to Zimmer, Inc. for its gift of HA/TCP, and to Li Li (National Institute of Dental and Craniofacial Research, NIH, U.S. Department of Health and Human Services) for her excellent histotechnical assistance. This study was supported by the Division of Intramural Research of the National Institute of Dental and Craniofacial Research, the Division of Intramural Research of the National Institute of Neurological Disorders and Stroke, in the Intramural Research Program, NIH, U.S. Department of Health and Human Services, and by an award from the NIH Center for Regenerative Medicine in 2010.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
Author Contributions
M.D.P.: collection and assembly of data, data analysis and interpretation, manuscript writing; S.A.K.: collection and assembly of data, data analysis and interpretation; N.C.: collection and assembly of data, data analysis and interpretation; K.P.: provision of study material; K.G.C.: provision of study material; B.N.M.: collection of data; R.S.H.: collection and assembly of data; R.D.G.M.: provision of study material; J.G.C.: provision of study material; B.S.M.: provision of study material, collection and assembly of data, data analysis and interpretation; P.G.R.: conception and design, financial support, assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript.
References
- 1.Buttery LD, Bourne S, Xynos JD, et al. Differentiation of osteoblasts and in vitro bone formation from murine embryonic stem cells. Tissue Eng. 2001;7:89–99. doi: 10.1089/107632700300003323. [DOI] [PubMed] [Google Scholar]
- 2.Ahn SE, Kim S, Park KH, et al. Primary bone-derived cells induce osteogenic differentiation without exogenous factors in human embryonic stem cells. Biochem Biophys Res Commun. 2006;340:403–408. doi: 10.1016/j.bbrc.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 3.Kärner E, Unger C, Sloan AJ, et al. Bone matrix formation in osteogenic cultures derived from human embryonic stem cells in vitro. Stem Cells Dev. 2007;16:39–52. doi: 10.1089/scd.2006.0010. [DOI] [PubMed] [Google Scholar]
- 4.Toh WS, Yang Z, Liu H, et al. Effects of culture conditions and bone morphogenetic protein 2 on extent of chondrogenesis from human embryonic stem cells. Stem Cells. 2007;25:950–960. doi: 10.1634/stemcells.2006-0326. [DOI] [PubMed] [Google Scholar]
- 5.Kim S, Kim SS, Lee SH, et al. In vivo bone formation from human embryonic stem cell-derived osteogenic cells in poly(d,l-lactic-co-glycolic acid)/hydroxyapatite composite scaffolds. Biomaterials. 2008;29:1043–1053. doi: 10.1016/j.biomaterials.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 6.Mateizel I, De Becker A, Van de Velde H, et al. Efficient differentiation of human embryonic stem cells into a homogeneous population of osteoprogenitor-like cells. Reprod Biomed Online. 2008;16:741–753. doi: 10.1016/s1472-6483(10)60490-7. [DOI] [PubMed] [Google Scholar]
- 7.Karner E, Backesjo CM, Cedervall J et al. Dynamics of gene expression during bone matrix formation in osteogenic cultures derived from human embryonic stem cells in vitro. Biochim Biophys Acta 2009;1790:110–118. [DOI] [PubMed]
- 8.Lee EJ, Lee HN, Kang HJ, et al. Novel embryoid body-based method to derive mesenchymal stem cells from human embryonic stem cells. Tissue Eng Part A. 2010;16:705–715. doi: 10.1089/ten.tea.2008.0596. [DOI] [PubMed] [Google Scholar]
- 9.Lee KW, Yook JY, Son MY, et al. Rapamycin promotes the osteoblastic differentiation of human embryonic stem cells by blocking the mTOR pathway and stimulating the BMP/Smad pathway. Stem Cells Dev. 2010;19:557–568. doi: 10.1089/scd.2009.0147. [DOI] [PubMed] [Google Scholar]
- 10.Arpornmaeklong P, Wang Z, Pressler MJ, et al. Expansion and characterization of human embryonic stem cell-derived osteoblast-like cells. Cell Reprogram. 2010;12:377–389. doi: 10.1089/cell.2009.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kao CL, Tai LK, Chiou SH, et al. Resveratrol promotes osteogenic differentiation and protects against dexamethasone damage in murine induced pluripotent stem cells. Stem Cells Dev. 2010;19:247–258. doi: 10.1089/scd.2009.0186. [DOI] [PubMed] [Google Scholar]
- 12.Li F, Bronson S, Niyibizi C. Derivation of murine induced pluripotent stem cells (iPS) and assessment of their differentiation toward osteogenic lineage. J Cell Biochem. 2010;109:643–652. doi: 10.1002/jcb.22440. [DOI] [PubMed] [Google Scholar]
- 13.Li F, Niyibizi C. Cells derived from murine induced pluripotent stem cells (iPSC) by treatment with members of TGF-beta family give rise to osteoblasts differentiation and form bone in vivo. BMC Cell Biol. 2012;13:35. doi: 10.1186/1471-2121-13-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villa-Diaz LG, Brown SE, Liu Y, et al. Derivation of mesenchymal stem cells from human induced pluripotent stem cells cultured on synthetic substrates. Stem Cells. 2012;30:1174–1181. doi: 10.1002/stem.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–319. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 17.Hwang YS, Cho J, Tay F, et al. The use of murine embryonic stem cells, alginate encapsulation, and rotary microgravity bioreactor in bone tissue engineering. Biomaterials. 2009;30:499–507. doi: 10.1016/j.biomaterials.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 18.Jukes JM, Both SK, Leusink A, et al. Endochondral bone tissue engineering using embryonic stem cells. Proc Natl Acad Sci USA. 2008;105:6840–6845. doi: 10.1073/pnas.0711662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tremoleda JL, Forsyth NR, Khan NS, et al. Bone tissue formation from human embryonic stem cells in vivo. Cloning Stem Cells. 2008;10:119–132. doi: 10.1089/clo.2007.0R36. [DOI] [PubMed] [Google Scholar]
- 20.Undale A, Fraser D, Hefferan T, et al. Induction of fracture repair by mesenchymal cells derived from human embryonic stem cells or bone marrow. J Orthop Res. 2011;29:1804–1811. doi: 10.1002/jor.21480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim MJ, Park JS, Kim S, et al. Encapsulation of bone morphogenic protein-2 with Cbfa1-overexpressing osteogenic cells derived from human embryonic stem cells in hydrogel accelerates bone tissue regeneration. Stem Cells Dev. 2011;20:1349–1358. doi: 10.1089/scd.2010.0311. [DOI] [PubMed] [Google Scholar]
- 22.Domev H, Amit M, Laevsky I, et al. Efficient engineering of vascularized ectopic bone from human embryonic stem cell-derived mesenchymal stem cells. Tissue Eng Part A. 2012;18:2290–2302. doi: 10.1089/ten.TEA.2011.0371. [DOI] [PubMed] [Google Scholar]
- 23.de Peppo GM, Marcos-Campos I, Kahler DJ, et al. Engineering bone tissue substitutes from human induced pluripotent stem cells. Proc Natl Acad Sci USA. 2013;110:8680–8685. doi: 10.1073/pnas.1301190110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin GZ, Kim TH, Kim JH, et al. Bone tissue engineering of induced pluripotent stem cells cultured with macrochanneled polymer scaffold. J Biomed Mater Res A. 2013;101A:1283–1291. doi: 10.1002/jbm.a.34425. [DOI] [PubMed] [Google Scholar]
- 25.Bianco P, Cao X, Frenette PS, et al. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19:35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen KG, Mallon BS, Hamilton RS, et al. Non-colony type monolayer culture of human embryonic stem cells. Stem Cell Res (Amst) 2012;9:237–248. doi: 10.1016/j.scr.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Modis L. The organic bone matrix. In: Modis L, editor. Organization of the Extracellular matrix: A Polarization Microscopic Approach. Boca Raton, FL: CRC Press; 1991. pp. 99–122. [Google Scholar]
- 28.Prentice AI. Autofluorescence of bone tissues. J Clin Pathol. 1967;20:717–719. doi: 10.1136/jcp.20.5.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mallon BS, Chenoweth JG, Johnson KR, et al. StemCellDB: The human pluripotent stem cell database at the National Institutes of Health. Stem Cell Res (Amst) 2013;10:57–66. doi: 10.1016/j.scr.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuznetsov SA, Cherman N, Robey PG. In vivo bone formation by progeny of human embryonic stem cells. Stem Cells Dev. 2011;20:269–287. doi: 10.1089/scd.2009.0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krebsbach PH, Kuznetsov SA, Satomura K, et al. Bone formation in vivo: Comparison of osteogenesis by transplanted mouse and human marrow stromal fibroblasts. Transplantation. 1997;63:1059–1069. doi: 10.1097/00007890-199704270-00003. [DOI] [PubMed] [Google Scholar]
- 32.Robey PG. Cell sources for bone regeneration: The good, the bad, and the ugly (but promising) Tissue Eng Part B Rev. 2011;17:423–430. doi: 10.1089/ten.teb.2011.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoo JU, Barthel TS, Nishimura K, et al. The chondrogenic potential of human bone-marrow-derived mesenchymal progenitor cells. J Bone Joint Surg Am. 1998;80:1745–1757. doi: 10.2106/00004623-199812000-00004. [DOI] [PubMed] [Google Scholar]
- 34.Bonewald LF, Harris SE, Rosser J, et al. von Kossa staining alone is not sufficient to confirm that mineralization in vitro represents bone formation. Calcif Tissue Int. 2003;72:537–547. doi: 10.1007/s00223-002-1057-y. [DOI] [PubMed] [Google Scholar]
- 35.Kuznetsov SA, Krebsbach PH, Satomura K, et al. Single-colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J Bone Miner Res. 1997;12:1335–1347. doi: 10.1359/jbmr.1997.12.9.1335. [DOI] [PubMed] [Google Scholar]
- 36.Dieudonné SC, Xu T, Chou JY, et al. Immortalization and characterization of bone marrow stromal fibroblasts from a patient with a loss of function mutation in the estrogen receptor-alpha gene. J Bone Miner Res. 1998;13:598–608. doi: 10.1359/jbmr.1998.13.4.598. [DOI] [PubMed] [Google Scholar]
- 37.Kawaguchi J, Mee PJ, Smith AG. Osteogenic and chondrogenic differentiation of embryonic stem cells in response to specific growth factors. Bone. 2005;36:758–769. doi: 10.1016/j.bone.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 38.Woei Ng K, Speicher T, Dombrowski C, et al. Osteogenic differentiation of murine embryonic stem cells is mediated by fibroblast growth factor receptors. Stem Cells Dev. 2007;16:305–318. doi: 10.1089/scd.2006.0044. [DOI] [PubMed] [Google Scholar]
- 39.Kim K, Zhao R, Doi A, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Streeter GL. Developmental horizons in human embryos (fourth issue): A review of the histogenesis of cartilage and bone. Contr Embryol Carneg Instn 1949;33:151–167. [PubMed]
- 41.Bianco P, Riminucci M, Kuznetsov S, et al. Multipotential cells in the bone marrow stroma: Regulation in the context of organ physiology. Crit Rev Eukaryot Gene Expr. 1999;9:159–173. doi: 10.1615/critreveukargeneexpr.v9.i2.30. [DOI] [PubMed] [Google Scholar]
- 42.Maes C, Kobayashi T, Selig MK, et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell. 2010;19:329–344. doi: 10.1016/j.devcel.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.