

Abstract

Hollow and concave nanocrystals find applications in many fields, and their fabrication can follow different possible mechanisms. We report a new route to these nanostructures that exploits the oxidation of Cu2–xSe/Cu2–xS core/shell nanocrystals with various etchants. Even though the Cu2–xSe core is encased in a thick Cu2–xS shell, the initial effect of oxidation is the creation of a void in the core. This is rationalized in terms of diffusion of Cu+ ions and electrons from the core to the shell (and from there to the solution). Differently from the classical Kirkendall effect, which entails an imbalance between in-diffusion and out-diffusion of two different species across an interface, the present mechanism can be considered as a limiting case of such effect and is triggered by the stronger tendency of Cu2–xSe over Cu2–xS toward oxidation and by fast Cu+ diffusion in copper chalcogenides. As the oxidation progresses, expansion of the inner void erodes the entire Cu2–xSe core, accompanied by etching and partial collapse of the shell, yielding Cu2–xSySe1–y concave particles.
Introduction
Hollow and concave nanocrystals (NCs) are an interesting class of materials with potential applications in catalysis,1−8 energy storage,9−13 plasmonics,14 and medicine.8,15−19 They can be synthesized by implementing the Kirkendall effect at the nanoscale,20−27 by combining the Kirkendall effect with anion exchange,28,29 via Ostwald ripening,30 by selective etching of a core region initially used as a template for the growth of a shell of another material,8,21,31−33 or by selective oxidative etching of initial NCs.31,34 Also, there are several examples of hollow crystals of noble metals synthesized via galvanic replacement reactions,35−38 an approach that was recently extended to transition-metal oxides.36 Considerable work has been done on understanding the different mechanisms involved in the creation of voids: for example, the role of defects and dislocations in the formation of hollow nanocrystals was investigated by Tang et al.39 Often, hollow particles prepared via templating routes presented pores and cracks due to the large lattice mismatch between the template and the shell material,21,32 and the permeability of these NCs was exploited for the encapsulation of various organic and inorganic species. Also, reversible switching from “solid” to hollow particles was recently demonstrated by Ha et al.40 An interesting route to hollow metal oxide nanoparticles, recently discovered by Jen-La Plante and Mokari, is based on a melt fracture mechanism.41
We report in this work an approach to obtain colloidal hollow and concave NCs that exploits several characteristics of two copper chalcogenides of interest here, namely Cu2Se and Cu2S. Henceforth, we refer to them as Cu2–xSe and Cu2–xS, because even in as-synthesized NCs with nominal 2:1 Cu:chalcogen ratio, we expect and observe a slight copper deficiency, due to the ease of formation of Cu vacancies in these materials. The relevant aspects are (i) Cu+ ions (and electrons) can be easily extracted from both Cu2–xSe and Cu2–xS NCs upon reaction with various oxidizing agents;42,43 (ii) Cu2–xSe NCs are more prone to oxidation (i.e., to formation of additional Cu vacancies) than Cu2–xS NCs, which we demonstrate here via competitive etching experiments on mixtures of Cu2–xSe NCs and Cu2–xS NCs; (iii) Cu+ ions have a high diffusivity in both Cu2–xSe and Cu2–xS.44−46
These aspects helped us to rationalize the oxidative etching behavior of NCs composed of a core of Cu2–xSe buried in a shell of Cu2–xS (Scheme 1). The initial oxidation must occur at the outer Cu2–xS shell, with transfer of electrons from the NCs to the oxidizing agent and release of Cu+ ions to the solution. However, the formation of a void in the Cu2–xSe core (Scheme 1b) supports the hypothesis that this loss of Cu+ ions from the shell is counterbalanced by the diffusion of Cu+ ions from the Cu2–xSe core to the shell, with concomitant oxidation of the core, see Scheme 1a. In turn, Se species diffuse from the initial core toward the Cu2–xSe–Cu2–xS interface, forming a ternary Cu–S–Se alloy there. Prolonged oxidation leads to enlargement of the void, etching of the shell, and partial shell collapse around the core region (Scheme 1c). After the region initially occupied by the Cu2–xSe core is vacated, etching continues on the Cu2–xS shell. Eventually, a ternary Cu2–xSySe1–y porous nanocage is left (Scheme 1d). The mechanism of void formation can be classified as a limiting case of the nanoscale Kirkendall effect:26 in fact, different from the classical Kirkendall effect, here we are seeing the sole out-diffusion of components from the core of a core/shell NC triggered by the oxidation of the particles.
Scheme 1. Various Stages Involved in the Formation of Hollow/Concave NCs.
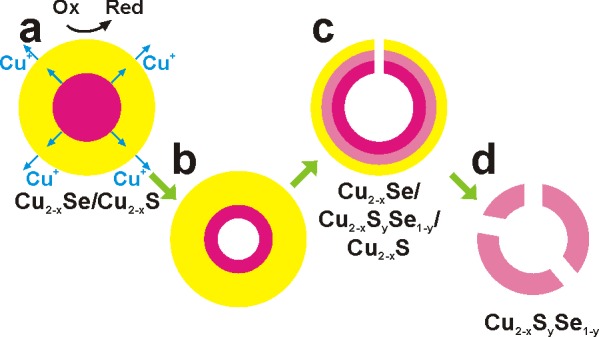
(a) Beginning of the oxidative process: electrons and Cu+ ions start being extracted from the Cu2–xS shell of an initial Cu2–xSe/Cu2–xS core/shell NC. The loss of Cu+ ions from the shell is counterbalanced by the diffusion of Cu+ ions from the Cu2–xSe core to the shell. (b) Early stage of etching: vacancy coalescence creates a void in the core. (c) Later stage: the core has been dismantled, etching of the shell begins and part of the shell collapses. A ternary alloy is being formed at the core/shell interface. (d) Eventually, a porous Cu2–xSySe1–y nanocage remains.
Experimental Section
Chemicals
Cadmium oxide (CdO, 99.99%), copper(I) chloride (CuCl, 99.999%), trioctylphosphine oxide (TOPO, 99%), trioctylphosphine (TOP, 97%), and selenium powder (Se, 99.99%) were purchased from Strem Chemicals. Cadmium chloride (CdCl2, 99.99%), iodine (I2, 99.99%), iron(III) chloride (FeCl3, 98%), oleylamine (70%), tetrakis(acetonitrile)copper(I) hexafluorophosphate, and sulfur powder (S, 99.9%) were purchased from Sigma-Aldrich. Copper(II) chloride (CuCl2, 99%) was purchased from Alfa Aesar. Octadecylphosphonic acid (ODPA) and hexylphosphonic acid (HPA) were purchased from Polycarbon Industries. Cerium(IV) ammonium nitrate (>98.5%), nitric acid (HNO3, 67−69%), anhydrous toluene and methanol were purchased from Carlo Erba reagents.
Synthesis of Bullet-Like Wurtzite CdSe Seeds for Bullet-in-Rod NCs
CdO (50 mg), ODPA (280 mg), HPA (80 mg), and 3 g of TOPO were loaded in a reaction flask and then heated to 120 °C under vacuum for 1 h. Nitrogen was pumped into the reaction mixture, and the temperature was raised to 380 °C, after which 2 mL of TOP were added to the flask. The temperature was allowed to recover to 380 °C, then 1 g of a stock solution of Se:TOP (prepared by dissolving 120 mg of Se in 10 mL of TOP) was injected rapidly into the flask. The reaction was allowed to run for 10 min with the temperature controller set at 380 °C. The heating mantle was then removed to allow the solution to cool down, and the resulting particles were transferred into a glovebox, washed by addition of methanol, centrifuged, and finally redissolved in 5 mL of toluene. The concentration of NCs in the solution (5.2 × 10–8 M) was estimated by a combination of the geometrical parameters of the crystals extracted from transmission electron microscopy with inductively coupled plasma optical emission spectroscopy (ICP-OES).
Synthesis of CdSe(core)/CdS(shell) Bullet-in-Rod NCs
CdO (50 mg), CdCl2 (12 mg), ODPA (280 mg), HPA (80 mg), and TOPO (3 g) were loaded in a reaction flask and then heated to 120 °C under vacuum for 1 h. Nitrogen was then pumped into the reaction mixture, and the temperature was raised to 380 °C, after which 2 mL of TOP was added to it, and the temperature was allowed to recover to 380 °C. A solution containing CdSe NCs in TOP and S:TOP was prepared in the following way: 4.3 × 10–11 moles of wurtzite bullet-like CdSe seeds dissolved in toluene (prepared in the previous stage, roughly 1/6 of volume of the mother solution was taken) were precipitated and redispersed in 1 mL TOP with assistance of ultrasonication. The resulting solution was then mixed with 0.5 g of a stock solution of S:TOP (previously prepared by mixing 960 mg of S in 10 mL of TOP). This mixture was swiftly injected into the reaction flask at 380 °C. The reaction was allowed to run for 10 min with the temperature controller set at 380 °C. The final product was purified by precipitation via addition of methanol, centrifugation, and redispersion in toluene.
Synthesis of Reversed CdS (core)/CdSxSe1–x (shell) Rod-in-Rod NCs by Sequential Anion Injection
In a 25 mL round-bottom flask, CdO (60 mg), CdCl2 (6 mg), HPA (80 mg), ODPA (290 mg) and TOPO (3 g) were mixed and degassed under vacuum at a temperature of 120 °C for a period of 1 h. The flask was then backfilled with nitrogen, and the temperature was raised to 380 °C at which point the red suspension transformed into a transparent solution. Two mL of TOP was then added, and the temperature was allowed to recover. S:TOP (0.25 g; prepared at a concentration of 90 mg of S per ml of TOP in a glovebox) solution was then injected swiftly, and the reaction was allowed to run for 10 min, which led to formation of CdS cores, characterized by darkening of the solution. The temperature of the reaction vessel was then reduced to 350 °C, and 0.25 g of Se:TOP (prepared at a concentration of 72 mg of Se per ml of TOP in a glovebox) was introduced all at once. The reaction mixture was allowed to stir at this temperature for a further 5 min to allow the growth of the CdSxSe1–x shell around the CdS cores, and afterward the heating mantle was removed to let the reaction mixture to cool down. The final brown product was then extracted in toluene and washed using methanol as the antisolvent. The NCs were then redispersed in toluene.
Synthesis of Monoclinic Cu2–xS Platelets
CuCl (100 mg) and oleylamine (10 mL) were loaded in a reaction flask and then kept at 130 °C for 2 h under vacuum. Then, nitrogen was fluxed into the reaction mixture, and the temperature was raised up to 240 °C, followed by addition of 32 mg of S dissolved in 2 mL of degassed oleylamine. The reaction time was 10 min (at 240 °C). The final product was transferred to the glovebox, precipitated by addition of methanol, centrifuged, and redispersed in toluene.
Cation Exchange on Cd-Based NCs
This was performed in an argon-filled glovebox following previously published protocols.47 In a standard procedure, a stock solution of the Cu(I) complex was prepared by dissolving 40 mg of tetrakis(acetonitrile)copper(I) hexafluorophosphate in 5 mL of methanol. Then 200 μL of the core/shell NC stock solution in toluene (the estimated concentration of NCs was 8.6 × 10–9 M) was injected in 3 mL of the Cu+ solution, under stirring. After 5 min, the particles were precipitated and resuspended in 3 mL of toluene, followed by addition of the remaining 2 mL of the Cu(I) solution. The particles were precipitated again and redissolved in toluene, with the addition of 25 μL of previously degassed oleylamine, to further stabilize them.
Etching Experiments Performed on Cu-Based NCs
The etching experiments were run using a 2 × 10–2 M solution of CuCl2 in methanol. The total amount of Cu atoms in the initial solution of NCs dispersed in toluene was instead determined by elemental analysis, via ICP-OES (see later). The samples were prepared by adding different amounts of the CuCl2 solution (to reach the desired etching stage) to 250 μL of the NCs solution in a vial. In the last step, additional few tens of μL of methanol were added to precipitate the particles, and the vial was then centrifuged at 5000 rpm for 10 min. The precipitate was finally redispersed in toluene.
TEM Analyses
Bright-field transmission electron microscopy (BF-TEM) images and selected area electron diffraction (SAED) patterns were acquired with a JEOL JEM-1011 microscope operating at 100 kV. The SAED patterns were acquired at constant camera length after mechanically adjusting the height of the sample to the eucentric height and after carefully focusing the NC images. In the same session, the diffraction camera length and the system distortions were calibrated using a nanocrystalline Au sputtered film on a standard C-covered Cu grid. The elaboration of SAED patterns (beam-stop removal, centering, azimuthal integration, and background subtraction) was carried out using the PASAD software.48 Energy-filtered TEM (EFTEM), high resolution TEM (HRTEM), high-angle annular dark-field scanning TEM (HAADF-STEM), and energy-dispersive X-ray spectroscopy (EDS) analyses were carried out by a JEOL JEM-2200FS instrument operating at 200 kV, equipped with a CEOS image aberration corrector, an in-column energy filter (Ω type), and a Bruker Quantax 400 system with a 60 mm2 silicon-drift detector (SDD). For octahedron-in-octapod and bullet-in-rod NCs, EFTEM maps were acquired with the three-window method at the S-L core-loss edge (165 eV onset energy, 20 eV slit width) and at the Se-L edge (1436 eV onset energy, 100 eV slit width). Due to higher residual amount of organics for the diamond-in-diamond NCs and for the reversed rod-in-rod NCs, elemental maps were acquired on these NCs by EDS mapping. For BF-TEM and SAED analyses, 50 μL of the NC suspensions were deposited on carbon-coated Cu grids, for HRTEM and EFTEM analyses on commercial ultrathin C-film-coated Cu grids, while for EDS analyses carbon-coated Ni grids and an analytical Be cup holder were used.
In view of the acquisition of the tilt series for tomography reconstruction, 50 μL of each NC solution were deposited onto a 1.5 × 1.5 mm2 C-coated copper grid, mounted on a sample holder for tomography (FISCHIONE model 2030). A single-tilt series of HAADF-STEM images in the widest possible angular range (maximum allowed in the employed system: −70° to +70°) with a 2° step was acquired for each sample, using the JEOL JEM-2200FS microscope (at 200 kV). Alignment based on cross-correlation and sample tomograms (without fiducial markers) was applied to the tilt series using the IMOD software package.49 The volume reconstruction was then performed starting from the aligned tilt series via a combination of weighted back-projection (WBP) and simultaneous iterative reconstruction technique (SIRT) using the plug-in TomoJ of ImageJ.50,51 After the initial WBP, a few tens of SIRT iterations were carried out until convergence was reached. The reported isosurface rendering was performed using the UCSF Chimera package.52
X-ray Diffraction (XRD)
XRD patterns were recorded on a Rigaku SmartLab 9 kW diffractometer. The X-ray source was operated at 40 kV and 150 mA. The diffractometer was equipped with a Cu rotating anode source and a Göbel mirror to obtain a parallel beam and to suppress Cu Kβ radiation (1.392 Å). To acquire data a 2θ/Ω scan geometry was used. Competitive etching experiments were performed using a 3:1 mixture of Cu2–xS (9.0 μM) and Cu2–xSe (1.3 μM) NCs solutions, which were drop cast onto a zero background silicon substrate. After a first XRD acquisition on the pristine sample, the so-obtained film was immersed for 1 min in a 0.02 M solution of CuCl2 in methanol and thoroughly rinsed with ethanol prior to XRD characterization of the etched NCs. The PDXL software of Rigaku was used for phase identification.
Elemental Analysis on NC Solutions
This was carried out via ICP-OES, using a iCAP 6500 Thermo spectrometer. Samples were dissolved in HCl/HNO3 3:1 (v/v) (Carlo Erba superpure grade) and left overnight at room temperature, in order to completely digest the NCs. Afterward, Milli-Q grade water (18.3 MΩcm) was added (7 mL) to the sample. The solution was then filtered using a 0.45 μm pore size filter. All chemical analyses performed by ICP-OES were affected by a systematic error of about 5–10%.
X-ray Photoelectron Spectroscopy (XPS) Measurements
They were performed on a Kratos Axis Ultra DLD spectrometer, using a monochromatic Al Kα source (15 kV, 20 mA). High-resolution narrow scans were performed at constant pass energy of 10 eV and steps of 0.1 eV. The photoelectrons were detected at a takeoff angle of Φ = 0° with respect to the surface normal. The pressure in the analysis chamber was maintained below 7 × 10–9 Torr for data acquisition. The data were converted to VAMAS format and processed using CasaXPS software, version 2.3.15. The binding energy (BE) scale was internally referenced to the C 1s peak (BE for C–C = 284.8 eV).
Results and Discussion
The Cu2–xSe/Cu2–xS core/shell NCs studied in this work were prepared from CdSe/CdS core/shell NCs by complete exchange of the Cd2+ cations with Cu+ cations (see Experimental Section). We started from three different geometries for the CdSe core and the CdS shell and consequently for the corresponding Cu-based NCs: bullet-in-rod, diamond-in-diamond, and octahedron-in-octapod. The bullet-in-rod NCs are reported here for the first time (see Experimental Section for details on synthesis), while the other two sample structures were prepared following procedures published by us and by other groups.47,53−57 As extensively reported in literature, cation exchange from CdSe/CdS NCs to Cu2–xSe/Cu2–xS NCs preserves the anion sublattice as well as the size and the shape of the parent NCs.47,54−56 Representative BF-TEM images (and sketches) of the various Cu2–xSe/Cu2–xS NCs are shown in Figure 1a–c. We tested different etching agents on these NCs (Cu2+, Fe3+, Ce4+, I2, and HNO3). The oxidizing effect of these agents on copper chalcogenide NCs is evident through the emergence of a surface plasmon absorption in the NIR region, due to collective excitation of free holes in the valence band, created by extraction of Cu(I) ions and electrons from the NCs upon oxidation (see Figure S1).42,43 Our starting NCs have already a weak NIR absorption, due to slight deviation from the 2:1 copper:chalcogen stoichiometry in the cation-exchanged particles. However, this absorption intensifies and shifts to shorter wavelengths as the density of free carriers increases following progressive oxidation of the particles.
Figure 1.

Shapes and geometries for Cu2–xSe/Cu2–xS core/shell NCs before and after moderate etching. (a–c) Overview BF-TEM images of Cu2–xSe/Cu2–xS core/shell NCs having various shapes and arrangements of both the inner core and the outer shell, namely (a) bullet-in-rod, (b) diamond-in-diamond, and (c) octahedron-in-octapod, schematic sketches shown in the respective lower right insets. The upper right insets in (a–c) give the combinations of elemental maps of Se and S within selected NCs, evidencing the location of the Cu2–xSe core (see the additional EDS line scan in Figure S3 for diamond-in-diamond NCs). (d–f) Corresponding samples after moderate etching (η = 2.5 for d, η = 0.7 for e, and η = 3.2 for f). Lower density regions are visible in all NCs. Scale bars are 100 nm for (a) (50 nm in the inset) and (d), 20 nm for (b) and (e), 50 nm for (c) and (f).
In all the experiments, the NCs were suspended in toluene and remained stable in solution. We discuss here the results of tests carried out using CuCl2, an oxidative etching agent typically used to extract Cu from various copper sulfides.58 Similar results were obtained using the other agents, as reported in Figure S2. The experiment consisted of room temperature dropwise additions of a solution of CuCl2 in methanol to the stirred solution of NCs under inert atmosphere (see Experimental Section for details). In the following, the reaction conditions will be identified in terms of ratio (η) of moles of Cu(II) ions added as CuCl2 (nCuCl2) to the moles of Cu(I) ions present in the NCs in the solution (nCu), therefore η = nCuCl2/nCu. After the addition of CuCl2, the solution quickly turned from brown to a color that eventually evolved to light yellow/green at a later stage of etching. The resulting mixture was left stirring for 5 min before the NCs were isolated by precipitation and redispersion in toluene. Figure 1d–f reports BF-TEM images of samples obtained in experiments run at mild etching conditions (η < 4). They evidence a decreased mass density in the regions where the Cu2–xSe core was initially located, suggesting that the core was preferentially dissolved.
In all these experiments on Cu2−xSe/Cu2−xS core-shell NCs, the cores were always preferentially etched, regardless of the size, shape, and crystal structure of the core as well as shape and crystal structure of the shell. For instance, the Cu2–xS shell in all samples had a chalcocite structure, in the form of hexagonal close-packed (β-chalcocite) for octahedron-in-octapod and diamond-in-diamond NCs or exhibiting slight structural displacements with respect to it in the bullet-in-rod NCs (α-chalcocite).55 The Cu2–xSe cores, on the other hand, had cubic structure in the octapods,55 but the other two samples most likely exhibited a hexagonal β-chalcocite-like structure, as found by us in a previous work (see also discussion later).59 Despite these differences among the various core/shell samples, they all behaved similarly when etched.
A deeper understanding of the processes induced by the exposure of the NCs to oxidizing environments was attained by compositional and structural analyses of the NCs upon progressive addition of CuCl2. In the following, we will focus on bullet-in-rod samples. Their peculiar design, with the relatively large Cu2–xSe core and the thick (about 3 nm around the core), uniform and elongated Cu2–xS shell, as well as their high stability under the electron beam, facilitated the study of their chemical and structural transformations. For these NCs, two consecutive stages can be clearly distinguished in the etching process: (i) an initial stage (η = 2), when only the core region of the starting particles is consumed (compare panels a–c in Figure 2); (ii) a second stage, which sets in after the initial core is emptied, upon further addition of oxidizing agent (between η = 4 and 9). At this stage the central cavity does not appear to expand further, while the extremities of the NCs start being severely consumed (see Figure 2d). An analogous evolution is found for the octahedron-in-octapod NCs (see Figures S4 and S5), while a less clear behavior is observed in the diamond-in-diamond NCs, probably due to the comparatively much smaller size of the Cu2–xSe domain in this type of NCs.
Figure 2.

Compositional and structural analyses of Cu2–xSe/Cu2–xS bullet-in-rod NCs upon progressive etching by CuCl2. (a–d) BF-TEM images (scale bars 50 nm) of groups of NCs taken from (a) a pristine sample; (b) a sample at initial stage of etching (η = 2); (c) a sample at moderate etching stage (η = 4) and at (d) a late etching stage (η = 9). (e–g) HAADF-STEM images of individual NCs taken from the samples in (a,c,d) (scale bars 20 nm): longitudinal EDS line scans along the axis of individual NCs for Cu (cyan), S (yellow), and Se (magenta) are superimposed on the corresponding images. The compositional change in the central region of the NCs evolved from Cu:S:Se = 1.9(±0.1):0.7(±0.1):0.3(±0.1) (sample in a) to Cu:S:Se = 2.0(±0.3):0.7(±0.1):0.3(±0.1) (sample in c) to Cu:S:Se = 0.8(±0.1):0.8(±0.1):0.2(±0.1) (sample in d) and in the extremities from Cu:S = 2.0(±0.2) (sample in a) to Cu:S = 2.1(±0.3) (sample in c) to Cu:S = 0.9(±0.1) (sample in d). (h) Azimuthally integrated SAED patterns of groups of NCs from a pristine sample, and from samples at moderate (η = 4) and late (η = 9) etching stage. These are compared with powder XRD patterns for α-chalcocite (monoclinic Cu2S, PDF card No. 01–073–6145), covellite (hexagonal CuS, PDF card no. 03-065-3556) and β-chalcocite-like Cu2Se (hexagonal Cu2Se, see Figure S6 and related discussion for details).59 The original SAED patterns are reported in Figure S7. (i) HRTEM image from the tip of a NC before etching and (j) magnified region of the image; this area, being away from the Cu2–xSe core has Cu2S composition, with the (k) corresponding Fourier transform (FT) indicating a α-chalcocite structure; (l) HRTEM image of a survival nanocage at a late stage of etching and (m) magnified region of the image; (n) corresponding FT, matching with covellite.
Longitudinal EDS line scans along single bullet-in-rod NCs are displayed in Figure 2e–g, which refer to three NCs, presented in Figure 2a,c, and d, respectively. In NCs from the pristine sample (Figure 2a), a Cu1.9S0.7Se0.3 stoichiometry (the standard deviations on the average EDS quantification of each element are reported in the caption of Figure 2) was obtained from the EDS quantification for the central region. This makes sense since both the Cu2–xSe core and the surrounding Cu2–xS shell are probed in that case. A Cu2S stoichiometry was found instead in the tip regions. Therefore, the overall Cu:chalcogen ratio is around 2:1 throughout the NC, as it should be for a pristine NC. The line scans of the other two samples (Figure 2f,g) clearly show that, as soon as a lower density region is formed in the region initially occupied by the Cu2–xSe core, a dip is visible in both the Se and Cu signals from the corresponding region (for instance, compare panels e and f of Figure 2). Note that a lower density region appears darker in a HAADF-STEM image. This low density region was encased by a “contour” region with slightly higher density than the rest of the rod in the NC sample shown in Figure 2f which was obtained after moderate etching (η = 4). At this stage, however, the overall Cu:chalcogen ratio had not changed much (within the experimental error) with respect to the pristine NCs: it was still Cu1.9S0.7Se0.3 when averaged over the central regions and Cu2.1S at the tips. At a late stage of etching (around η = 9, Figure 2g), the higher density contour surrounding the void had evolved into a thicker, cage-like region.
Nanocages are the only objects surviving at later stages of etching (see also Figure S5). The composition in these severely etched NCs had changed drastically with respect to the previous two samples: it was Cu0.8S0.8Se0.2 in the center and Cu0.9S at the tips, i.e., the NCs had lost a considerable fraction of the initial Cu but also S and Se species, as found by elemental analysis (see Experimental Section). Indeed, such analysis performed on solutions of the starting core/shell NCs and on purified solutions of the nanocages indicated a significant loss of copper (approximately 40%) and sulfur (approximately 20%) in the cages with respect to the starting sample, while the loss of selenium was within the experimental error (i.e., <10%).
Average structural information on the most significant samples was given by SAED patterns from groups of NCs (Figures 2h and S7). The SAED of the pristine sample (η = 0, Figure 2h, labeled as “pristine”) was consistent with a α-chalcocite structure (monoclinic Cu2S). In this pattern, as in all the others recorded on etched samples, the diffraction signal from the Cu2–xSe cores in the core/shell NCs was not really appreciable. These cores should have a β-chalcocite-like Cu2Se structure, as reported by us in a previous work.59 When the bullet-shaped CdSe NCs used for growing the bullet-in-rod CdSe/CdS NCs were indeed directly exchanged to Cu2–xSe NCs, their resulting XRD and SAED patterns could be matched to such β-chalcocite-like structure (see Figure S6 and related caption). At the initial stages of etching of the Cu2–xSe/Cu2–xS bullet-in-rod NCs (from η = 0 to 4), SAED indicated an evolution from the initial α-chalcocite structure to a structure similar to α-chalcocite, but with a shift to smaller lattice plane spacing (η = 4 in Figure 2h). This lattice contraction can be rationalized on the basis of out-diffusion of a fraction of Cu+ ions from the NCs. The SAED pattern observed on NCs at later stages of etching (η = 9 in Figure 2h) when almost only nanocages were left, presents much broader diffraction peaks, most likely due to the defective structure of the strongly etched NCs at this stage and the consequent small size of the crystallites. In addition, the pattern exhibited combined features of substoichiometric α-chalcocite and covellite (CuS).
The transformation from the initial NCs, dominated by a Cu2–xS shell with α-chalcocite structure, to nanocages having a covellite-like structure was further corroborated by HRTEM analyses (see Figures 2i–k and 2l–n, respectively). In particular, the identification of the {20–4} planes of α-chalcocite as the transversal ones (i.e., perpendicular to the elongation direction of the NCs, see Figure 2i–k) substantiates the relatively high-intensity peak at about 3 nm–1 observed in SAED patterns for η = 0 and 4 (Figure 2h), due to the strong anisotropy and the observed orientation of NCs, lying with their length parallel to the support film. It is known from previous works that the oxidation of chalcocite in dilute chloride solutions could lead to the formation of covellite.58 In the present case instead, most of the Cu2S α-chalcocite shell of the NCs evolved to a substoichiometric Cu2–xS phase and subsequently dissolved, with the exception of a region surrounding the initial Cu2–xSe core. Apparently, this interfacial region had resisted complete dissolution due to its mixed Cu/S/Se composition. Similarly to bullet-in-rod NCs, mixed Cu/S/Se hollow nanocages were also observed in the final etching stage for octahedron-in-octapod NCs (see Figures S4 and S5).
Compositional mapping and structural monitoring of the oxidation process do not provide an unequivocal description of the void formation process in the Cu2–xSe core region. One could argue whether, during etching, channels are formed from the Cu2–xSe core to the outer surface of the NCs, which can facilitate the escape of Cu species (and eventually of Se species). In order to get insights into the morphological evolution of the NCs, the volume of several bullet-in-rod NCs was reconstructed by means of HAADF-STEM-tomography. A series of samples collected at different stages of etching were analyzed, as reported in Figure 3. For each column in the figure, the upper panel is a HAADF-STEM image of a representative NC (these are labeled from “i” to “iv”). Then panel (a) is the reconstructed volume of the same NC in the isosurface representation, whereas panel (b) is a longitudinal cut-through, and finally panels (c and d) are two transversal cut-through from the core region of the reconstructed volume of the same NC. The NC of Figure 3i, (a–d) was collected at an initial stage of etching (η = 2): the reconstructed volume indicated the presence of a void inside the particle, surrounded by a thick continuous shell. The NC of Figure 3ii, (a–d) was collected at the same stage of etching. In this specific NC, a small central void was connected to the outer surface of the NC via a channel. Apart from this case which was quite rare at this stage of etching, most NCs had central voids with no channels. The channel formation is probably occurring in NCs in which the misfit strain in the Cu2–xS shell region around the Cu2–xSe core has partially or totally been released by insertion of misfit dislocations. Such a more defective structure, under exposure of the NCs to oxidizing environments, probably presents preferential migration avenues, which could facilitate the nucleation of voids and their coalescence.
Figure 3.
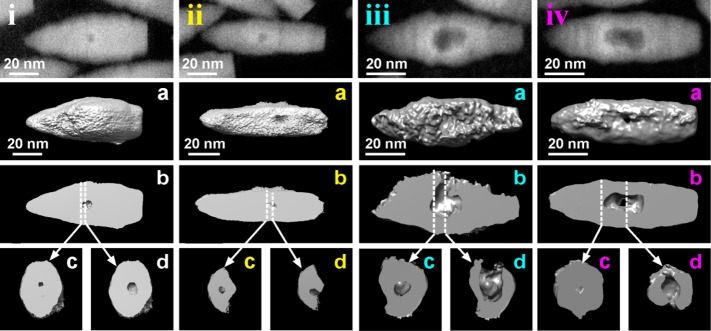
Results of HAADF-STEM tomography analyses on initial and intermediate etching stages. (i–iv, top): HAADF-STEM images of selected Cu2–xSe/Cu2–xS bullet-in-rod NCs observed at different stages of etching by CuCl2. The NCs in i and ii and the ones in iii and iv are from samples prepared with η = 2 and 4, respectively. For each stage, the insets present the following: (a) the isosurface rendering of the corresponding reconstructed volume of the particle; (b) a longitudinal cut-through; (c,d) transversal cut-through, with planes perpendicular to the elongation direction, and corresponding to the positions indicated in (b) by dashed lines.
In representative NCs collected at moderate etching stages (η = 4), for example, those of Figure 3iii, a–d) and Figure 3iv, a–d), the size of the central void was considerably enlarged, and most particles had one or multiple channels connecting the void to the outer surface of the NC (see the cross sections images of Figure 3iv, c,d). In many cases, the channels were so large (Figure 3iii, d) that the NCs could be considered as concave in shape (see also Figure S8). Tomography experiments on the diamond-in-diamond NCs (Figure 1b,e) were not successful, as the particle structure was altered after few minutes of electron beam irradiation, while we did not attempt them on the octapods (Figure 1c,f), due to their more complex geometry which would make it more difficult to disentangle the steps of void/channel formation. However, judging from the BF-TEM images of Figure 1, it is unlikely that these two NC types would follow a different evolution from the bullet-in-rod geometry.
The initial formation of voids in all the NCs studied in this work, upon exposure to oxidizing agents, is therefore compatible with a mechanism of vacancy coalescence following the preferential creation or accumulation of Cu vacancies in the central region of the particles, where the Cu2–xSe core was located. As discussed earlier, oxidation in copper chalcogenides leads to the release of Cu ions in solution, with concomitant decrease of their Cu content, i.e., formation of Cu vacancies.42,43 However, in the core/shell NCs the Cu2–xSe core is not directly exposed to the external environment, and obviously the direct oxidation of the NCs must start at the surface of the NCs, which is made of Cu2–xS. A plausible mechanism operative here could be the following: as soon as the Cu2–xS shell starts releasing copper ions in solution due to oxidation, thus creating a Cu gradient through the NC, a diffusion of copper ions (and of electrons) from the central Cu2–xSe core tries to counterbalance this loss in the shell, giving origin to the empty central region. Indeed, as reported in Figure S9, we observed via XPS measurements that, at an early etching stage, the Cu2–xSe core is already oxidized (Se species present as both selenides, Se2–, and short polyselenides, Se0),60 while the Cu2–xS shell is not yet affected by the oxidation process (S species present in the form of sulfides, S2–). This evidence implies that Cu2–xSe is more prone to oxidation than Cu2–xS.
A further evidence in favor of the stronger tendency of Cu2–xSe over Cu2–xS to oxidation was demonstrated by oxidation experiments on core/shell NCs with reversed geometry, i.e., Cu2–xS (core)/Cu2–xSe (shell) NCs, in which instead the shell was preferentially etched, and no hollow or concave structure were observed (see Figure 4). For these experiments, the starting NCs were prepared by direct synthesis of Cd1.6S(core)/Cd0.97S0.19Se0.81(shell) rod-in-rod NCs (see Figure S10). The synthesis conditions, described in the Experimental Section, were the only suitable ones that we could identify for the growth of a continuous Se-rich shell on top of the starting CdS cores. We do not exclude the possibility to optimize this procedure in the future so as to obtain a pure CdSe shell, but for the purpose of these experiments we believe the ability to grow a Se-rich shell (atomic ratio Se/S = 4 by EDS) was sufficient to prove our mechanism. The as-prepared rod-in-rod NCs were transformed into Cu-based counterparts via cation exchange, in analogy to what reported above for Cu2–xSe (core)/Cu2–xS (shell) NCs. BF-TEM micrographs (Figure 4a) in combination with the EDS compositional maps and quantification of the cation-exchanged NCs (Figure 4c) indicate a Cu3.1S/Cu1.8S0.17Se0.82 rod-in-rod structure with a Se/S atomic ratio of 1.2:1 (as determined by EDS). These NCs were then subjected to etching, using the same procedure as for the Cu2–xSe (core)/Cu2–xS (shell) NCs. In this case, no hollow structures were found. Instead, at an etching stage corresponding to η =4, the NCs appeared thinner than the starting particles (compare BF-TEM images of Figure 4a,d). Most importantly, EDS mapping clearly shows that the shell is almost completely missing, with the exception of thin residues observed at one end of some of the sulfide rods (Figure 4f). Correspondingly, a Se/S atomic ratio of 0.14 is found in the etched sample. The results on the reversed NCs show that, whatever the arrangement in a core/shell heterostructure is, the Se-rich phase is etched preferentially with respect to the S-rich one.
Figure 4.
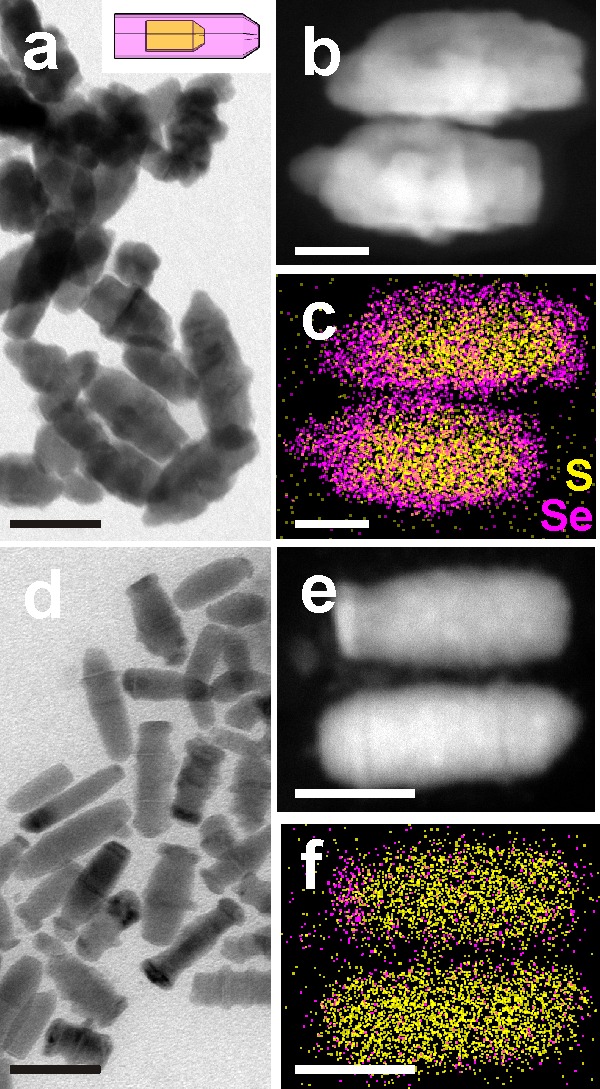
Results of etching experiments on Cu2–xS/Cu2–xSySe1–y rod-in-rod NCs. (a) Overview BF-TEM image of the NCs, (b) HAADF-STEM image and (c) corresponding EDS map for S and Se for two NCs. (d) Overview BF-TEM image after etching (η = 4), (e) HAADF-STEM image and (f) corresponding EDS map for S and Se for two NCs in the sample. Scale bars are 50 nm in (a,d) and 20 nm in (b,c,e,f).
Both types of core/shell structures described in this work, i.e., Cu2–xSe(core)/Cu2–xS(shell) and Cu2–xS(core)/Cu2–xSySe1–y(shell), were obtained by cation exchange from the corresponding Cd-based NCs. Elemental analysis (both by ICP-OES and EDS) indicates that there are residual Cd ions in the cation-exchanged NCs, although the Cd/Cu atomic ratio was low (around 1% or less). As the NCs are not entirely Cd-free, one might argue if the residual Cd ions are preferentially located in one region of the NCs (for example in the core or in the shell), and consequently if this would explain the higher rate of dissolution of the Se-rich regions. However, the unambiguous localization of Cd in a specific region of the NCs (core or shell) was not possible due to the low signal/noise ratio for Cd in the corresponding EDS spectra. Therefore, in order to rule out any possible role of residual Cd on the differential etching behavior observed here, we carried out competitive etching experiments on a simpler system, i.e. a mixture of Cu2–xSe and Cu2–xS NCs, each prepared following standard literature protocols (with minor modifications, see Experimental Section)43,61 and not obtained by cation exchange. In these experiments, the as-synthesized Cu2–xSe NCs had cubic structure, while the Cu2–xS NCs had monoclinic structure, both materials exhibiting slightly substoichiometric compositions. Fine details on the shape or crystal structure as well as exact initial composition of these NCs should have little relevance in the etching process, similarly to the case of the core/shell NCs discussed earlier. In the competitive etching experiments, the mixtures of Cu2–xSe and Cu2–xS NCs were subjected to the same oxidizing treatment as the various core/shell samples discussed above. The main results are summarized in Figure 5.
Figure 5.
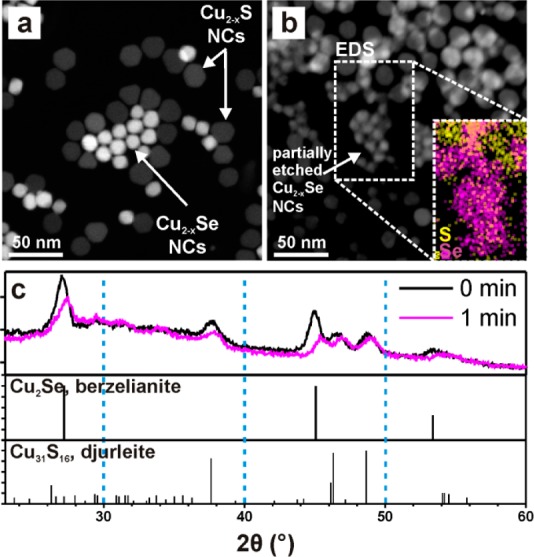
Results of competitive etching experiments on mixtures of Cu2–xSe and Cu2–xS NCs. (a,b) HAADF-STEM images of a mixture of Cu2–xSe and Cu2–xS NCs (a) as-synthesized and (b) after exposure of the same grid to CuCl2 methanol solution for 10 s (the inset represents EDS mapping of Se (magenta) and S (yellow) for the region within the box). (c) Comparison of XRD spectra for a mixture of Cu2Se and Cu2–xS NCs before and after 1 min immersion in a CuCl2 solution, compared with berzelianite Cu2Se (PDF card no. 01-088-2043) and djurleite Cu31S16 (PDF card no. 230959) reference patterns.
Starting from the non-oxidized samples and at any stage of the etching, combination of HAADF-STEM imaging and EDS compositional mapping allowed us to unambiguously distinguish the Cu2–xSe NCs from the Cu2–xS NCs in a mixture of them. Upon oxidation, the initial Cu2–xSe NCs, much brighter than Cu2–xS NCs, due to both the higher thickness and the higher atomic weight of Se over S (Figure 5a), were dissolved, while Cu2–xS NCs did not undergo an appreciable size variation (Figure 5b), as additionally supported by EDS elemental mapping (inset of Figure 5b). A rather unequivocal proof was additionally provided by XRD analyses of a film containing a mixture of Cu2–xS and Cu2–xSe NCs before and after immersion of the film in a solution of CuCl2 in methanol for 1 min (Figure 5c). Two main effects are evident from the comparison of the pristine sample and the one immersed in the oxidizing solution: (i) the diffraction peaks corresponding to both phases shift to higher 2θ values, due to cell contraction, which can be ascribed to Cu+ extraction; and (ii) the intensity of the features related to Cu2–xSe NCs decreases more than the one of the Cu2–xS-related features.
One reason for the stronger tendency of Cu2–xSe NCs toward oxidation with respect to Cu2–xS NCs might reside in a lower electron affinity of Cu2–xSe NCs over Cu2–xS NCs. This is supported by XPS investigation of the valence band (VB) in films of Cu2–xSe and Cu2–xS NCs, reported in Figure S11: the VB edge for Cu2–xSe NCs lies at slightly higher energy (+0.3 eV) than that of Cu2–xS NCs, in agreement with previous works on films of the same materials.62 This is indeed expected for copper chalcogenides, considering that in these materials the top of the valence band has a strong contribution from the chalcogen p orbitals,62 and that their energy increases following the S 3p → Se 4p trend. On the other hand, even in pristine core/shell Cu2–xSe/Cu2–xS NC structures, the free carriers already present should quickly equilibrate the Fermi levels of the two domains, leveling off differences in electron affinities between the two domains.
A convincing explanation for preferential Cu depletion in Cu2–xSe over Cu2–xS might be sought in the lower average energy of the Cu–Se bond over that of the Cu–S bond, which should entail a lower formation energy of Cu vacancies in Cu2–xSe than in Cu2–xS. In diatomic gaseous Cu-E (E = S, Se) species, the bond dissociation energy (i.e., for the Cu–E → Cu + E reaction) is 255 KJmol–1 for CuSe, and 275 KJmol–1 for CuS.63 Also, the standard enthalpies of formation at room temperature of Cu2S and Cu2Se are −79.5 and −59.4 kJmol–1, respectively.64 These data support our hypothesis. Direct calculations of vacancy formation energies in Cu2S and Cu2Se are made difficult by the many possibilities for both Cu and vacancy occupation sites in these materials and by the variety of crystal structures for both Cu2S and Cu2Se (recently, a Cu vacancy formation energy of 1.6 eV in Cu2S was experimentally derived by Bekenstein et al.).65 On the other hand, vacancy formation energies have been calculated for compound such as Cu2ZnSnS4 (CZTS) and Cu2ZnSnSe4 (CZTSe),66 for CuInS2, CuInSe2,67 and their corresponding S–Se alloys68 as well as for CuGaS2 and CuGaSe2.67 In all cases, Cu vacancies were always found to have lower formation energy in the Se-based (or Se-rich) semiconductors than in the S-based (or S-rich) ones.
Conclusions
We have demonstrated an approach to synthesize hollow and concave nanoparticles based on the preferential oxidation of the Cu2–xSe core region in colloidal core/shell Cu2–xSe/Cu2–xS NCs. Our experiments indicate that the initial effect of oxidation is the creation of a void in the core, due to diffusion of Cu+ ions and electrons from the core to the shell, in an effort to counterbalance the loss of Cu+ and electrons from the shell to the solution phase. This mechanism of void formation is rationalized in terms of a stronger tendency of Cu2–xSe over Cu2–xS towards oxidation and of the fast Cu+ diffusion in copper chalcogenides. Further oxidation of the NCs leads to an expansion of the inner void, until it erodes the initial Cu2–xSe core. At the same time, even the shell starts being etched and partially dismantled, leaving concave particles with Cu2–xSySe1–y composition. Future developments in this direction will include the study of this mechanism in other combinations of materials having a high diffusion cation in common. Apart from the synthesis of hollow and concave particles, another interesting extension could be the selective etching of a given domain in a segmented heterostructured NC/nanowire, with the consequence of disassembling it into smaller components at precisely defined “cutting” regions.
Acknowledgments
This work was supported in part by the European Union through the FP7 consolidator ERC Grant TRANS-NANO (contract no. 614897). We thank Francesco De Donato for help with the nanocrystal syntheses and Simone Nitti for support on elemental analysis.
Supporting Information Available
Optical characterization of the samples at different stages of etching, effect of different etching agents, BF-TEM characterization, EDS line scan on diamond-in-diamond NCs, etching evolution for octahedron-in-octapod NCs, XRD and SAED pattern for Cu-exchanged seeds for CdSe/CdS bullet-in-rod NCs, SAED pattern evolution with etching for bullet-in-rod NCs, tomographic reconstruction of concave bullet-in-rod NCs, TEM analysis of CdS/CdSxSe1–x rod-in-rod NCs, XPS analysis (comparison of the valence bands for S and Se anion sublattices, XPS analysis of a sample at intermediate stage of etching). This material is available free of charge via the Internet at http://pubs.acs.org/.
Author Contributions
§ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Joo J. B.; Zhang Q.; Lee I.; Dahl M.; Zaera F.; Yin Y. Adv. Funct. Mater. 2012, 22, 166–174. [Google Scholar]
- Kim S. M.; Jeon M.; Kim K. W.; Park J.; Lee I. S. J. Am. Chem. Soc. 2013, 135, 15714–15717. [DOI] [PubMed] [Google Scholar]
- Mahmoud M. A.; Saira F.; El-Sayed M. A. Nano Lett. 2010, 10, 3764–3769. [DOI] [PubMed] [Google Scholar]
- Wang A.-L.; Xu H.; Feng J.-X.; Ding L.-X.; Tong Y.-X.; Li G.-R. J. Am. Chem. Soc. 2013, 135, 10703–10709. [DOI] [PubMed] [Google Scholar]
- Ding L.-X.; Wang A.-L.; Li G.-R.; Liu Z.-Q.; Zhao W.-X.; Su C.-Y.; Tong Y.-X. J. Am. Chem. Soc. 2012, 134, 5730–5733. [DOI] [PubMed] [Google Scholar]
- Bai F.; Sun Z.; Wu H.; Haddad R. E.; Xiao X.; Fan H. Nano Lett. 2011, 11, 3759–3762. [DOI] [PubMed] [Google Scholar]
- Guo S.; Zhang S.; Wu L.; Sun S. Angew. Chem., Int. Ed. 2012, 51, 11770–11773. [DOI] [PubMed] [Google Scholar]
- George C.; Dorfs D.; Bertoni G.; Falqui A.; Genovese A.; Pellegrino T.; Roig A.; Quarta A.; Comparelli R.; Curri M. L.; Cingolani R.; Manna L. J. Am. Chem. Soc. 2011, 133, 2205–2217. [DOI] [PubMed] [Google Scholar]
- Keng P. Y.; Kim B. Y.; Shim I.-B.; Sahoo R.; Veneman P. E.; Armstrong N. R.; Yoo H.; Pemberton J. E.; Bull M. M.; Griebel J. J.; Ratcliff E. L.; Nebesny K. G.; Pyun J. ACS Nano 2009, 3, 3143–3157. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Li X.; Park K.; Song J.; Hong J.; Zhou L.; Mai Y.-W.; Huang H.; Goodenough J. B. J. Am. Chem. Soc. 2013, 135, 16280–16283. [DOI] [PubMed] [Google Scholar]
- Zheng G.; Zhang Q.; Cha J. J.; Yang Y.; Li W.; Seh Z. W.; Cui Y. Nano Lett. 2013, 13, 1265–1270. [DOI] [PubMed] [Google Scholar]
- Koo B.; Xiong H.; Slater M. D.; Prakapenka V. B.; Balasubramanian M.; Podsiadlo P.; Johnson C. S.; Rajh T.; Shevchenko E. V. Nano Lett. 2012, 12, 2429–2435. [DOI] [PubMed] [Google Scholar]
- Yao Y.; McDowell M. T.; Ryu I.; Wu H.; Liu N.; Hu L.; Nix W. D.; Cui Y. Nano Lett. 2011, 11, 2949–2954. [DOI] [PubMed] [Google Scholar]
- De Angelis F.; Malerba M.; Patrini M.; Miele E.; Das G.; Toma A.; Zaccaria R. P.; Di Fabrizio E. Nano Lett. 2013, 13, 3553–3558. [DOI] [PubMed] [Google Scholar]
- An K.; Hyeon T. Nano Today 2009, 4, 359–373. [Google Scholar]
- Piao Y.; Kim J.; Na H. B.; Kim D.; Baek J. S.; Ko M. K.; Lee J. H.; Shokouhimehr M.; Hyeon T. Nat. Mater. 2008, 7, 242–247. [DOI] [PubMed] [Google Scholar]
- Gao J.; Liang G.; Cheung J. S.; Pan Y.; Kuang Y.; Zhao F.; Zhang B.; Zhang X.; Wu E. X.; Xu B. J. Am. Chem. Soc. 2008, 130, 11828–11833. [DOI] [PubMed] [Google Scholar]
- Kang X.; Yang D.; Ma P. a.; Dai Y.; Shang M.; Geng D.; Cheng Z.; Lin J. Langmuir 2013, 29, 1286–1294. [DOI] [PubMed] [Google Scholar]
- Cheng K.; Sun S. Nano Today 2010, 5, 183–196. [Google Scholar]
- Yang Y.; Yang R. B.; Fan H. J.; Scholz R.; Huang Z.; Berger A.; Qin Y.; Knez M.; Gösele U. Angew. Chem., Int. Ed. 2010, 49, 1442–1446. [DOI] [PubMed] [Google Scholar]
- Fan H. J.; Gösele U.; Zacharias M. Small 2007, 3, 1660–1671. [DOI] [PubMed] [Google Scholar]
- Niu K.-Y.; Park J.; Zheng H.; Alivisatos A. P. Nano Lett. 2013, 13, 5715–5719. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Rioux R. M.; Erdonmez C. K.; Hughes S.; Somorjai G. A.; Alivisatos A. P. Science 2004, 304, 711–714. [DOI] [PubMed] [Google Scholar]
- Ibáñez M.; Fan J.; Li W.; Cadavid D.; Nafria R.; Carrete A.; Cabot A. Chem. Mater. 2011, 23, 3095–3104. [Google Scholar]
- Wang W.; Dahl M.; Yin Y. Chem. Mater. 2012, 25, 1179–1189. [Google Scholar]
- Jin fan H.; Knez M.; Scholz R.; Nielsch K.; Pippel E.; Hesse D.; Zacharias M.; Gosele U. Nat. Mater. 2006, 5, 627–631. [DOI] [PubMed] [Google Scholar]
- Ha D.-H.; Moreau L. M.; Honrao S.; Hennig R. G.; Robinson R. D. J. Phys. Chem. C 2013, 117, 14303–14312. [Google Scholar]
- Ye X.; Reifsnyder Hickey D.; Fei J.; Diroll B. T.; Paik T.; Chen J.; Murray C. B. J. Am. Chem. Soc. 2014, 136, 5106–5115. [DOI] [PubMed] [Google Scholar]
- Park J.; Zheng H.; Jun Y.-w.; Alivisatos A. P. J. Am. Chem. Soc. 2009, 131, 13943–13945. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Pan F.; Li H.; Zhao D.; Liu L.; Xu G. Q.; Chen W. J. Phys. Chem. C 2013, 117, 21718–21723. [Google Scholar]
- Wang Z.; Luan D.; Li C. M.; Su F.; Madhavi S.; Boey F. Y. C.; Lou X. W. J. Am. Chem. Soc. 2010, 132, 16271–16277. [DOI] [PubMed] [Google Scholar]
- Gao P. X.; Wang Z. L. J. Am. Chem. Soc. 2003, 125, 11299–11305. [DOI] [PubMed] [Google Scholar]
- Lou X. W.; Archer L. A.; Yang Z. Adv. Mater. 2008, 20, 3987–4019. [Google Scholar]
- Sui Y.; Fu W.; Zeng Y.; Yang H.; Zhang Y.; Chen H.; Li Y.; Li M.; Zou G. Angew. Chem., Int. Ed. 2010, 49, 4282–4285. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Xia Y. Science 2002, 298, 2176–2179. [DOI] [PubMed] [Google Scholar]
- Oh M. H.; Yu T.; Yu S.-H.; Lim B.; Ko K.-T.; Willinger M.-G.; Seo D.-H.; Kim B. H.; Cho M. G.; Park J.-H.; Kang K.; Sung Y.-E.; Pinna N.; Hyeon T. Science 2013, 340, 964–968. [DOI] [PubMed] [Google Scholar]
- Macdonald J. E.; Bar Sadan M.; Houben L.; Popov I.; Banin U. Nat. Mater. 2010, 9, 810–815. [DOI] [PubMed] [Google Scholar]
- González E.; Arbiol J.; Puntes V. F. Science 2011, 334, 1377–1380. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Ouyang M. Nat. Mater. 2007, 6, 754–759. [DOI] [PubMed] [Google Scholar]
- Ha T.-L.; Kim J. G.; Kim S. M.; Lee I. S. J. Am. Chem. Soc. 2012, 135, 1378–1385. [DOI] [PubMed] [Google Scholar]
- Jen-La Plante I.; Mokari T. Small 2013, 9, 56–60. [DOI] [PubMed] [Google Scholar]
- Luther J. M.; Jain P. K.; Ewers T.; Alivisatos A. P. Nat. Mater. 2011, 10, 361–366. [DOI] [PubMed] [Google Scholar]
- Dorfs D.; Härtling T.; Miszta K.; Bigall N. C.; Kim M. R.; Genovese A.; Falqui A.; Povia M.; Manna L. J. Am. Chem. Soc. 2011, 133, 11175–11180. [DOI] [PubMed] [Google Scholar]
- Etienne A. J. Electrochem. Soc. 1970, 117, 870–874. [Google Scholar]
- Takahashi T.; Yamamoto O.; Matsuyama F.; Noda Y. J. Solid State Chem. 1976, 16, 35–39. [Google Scholar]
- Miller T. A.; Wittenberg J. S.; Wen H.; Connor S.; Cui Y.; Lindenberg A. M. Nat. Commun. 2013, 4, 1369. [DOI] [PubMed] [Google Scholar]
- Li H.; Brescia R.; Krahne R.; Bertoni G.; Alcocer M. J. P.; D’Andrea C.; Scotognella F.; Tassone F.; Zanella M.; De Giorgi M.; Manna L. ACS Nano 2012, 6, 1637–1647. [DOI] [PubMed] [Google Scholar]
- Gammer C.; Mangler C.; Rentenberger C.; Karnthaler H. P. Scr. Mater. 2010, 63, 312–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer J. R.; Mastronarde D. N.; McIntosh J. R. J. Struct. Biol. 1996, 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Abramoff M. D.; Magalhães P. J.; Ram S. J. Biophot. Int. 2004, 11, 36–42. [Google Scholar]
- Messaoudi C.; Boudier T.; Sanchez Sorzano C. O.; Marco S. BMC Bioinform. 2007, 8, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- García-Santamaría F.; Chen Y.; Vela J.; Schaller R. D.; Hollingsworth J. A.; Klimov V. I. Nano Lett. 2009, 9, 3482–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P. K.; Amirav L.; Aloni S.; Alivisatos A. P. J. Am. Chem. Soc. 2010, 132, 9997–9999. [DOI] [PubMed] [Google Scholar]
- Miszta K.; Dorfs D.; Genovese A.; Kim M. R.; Manna L. ACS Nano 2011, 5, 7176–7183. [DOI] [PubMed] [Google Scholar]
- Bertoni G.; Grillo V.; Brescia R.; Ke X.; Bals S.; Catellani A.; Li H.; Manna L. ACS Nano 2012, 6, 6453–6461. [DOI] [PubMed] [Google Scholar]
- Christodoulou S.; Vaccaro G.; Pinchetti V.; De Donato F.; Grim J.; Casu A.; Genovese A.; Vicidomini G.; Diaspro A.; Brovelli S.; Manna L.; Moreels I. J. Mater. Chem. C 2014, 2, 3439–3447. [Google Scholar]
- Miki H.; Nicol M.; Velásquez-Yévenes L. Hydrometallurgy 2011, 105, 321–327. [Google Scholar]
- Li H.; Zanella M.; Genovese A.; Povia M.; Falqui A.; Giannini C.; Manna L. Nano Lett. 2011, 11, 4964–4970. [DOI] [PubMed] [Google Scholar]
- Riha S. C.; Johnson D. C.; Prieto A. L. J. Am. Chem. Soc. 2010, 133, 1383–1390. [DOI] [PubMed] [Google Scholar]
- Du Y.; Yin Z.; Zhu J.; Huang X.; Wu X.-J.; Zeng Z.; Yan Q.; Zhang H. Nat. Commun. 2012, 3, 1177. [DOI] [PubMed] [Google Scholar]
- Kashida S.; Shimosaka W.; Mori M.; Yoshimura D. J. Phys. Chem. Solids 2003, 64, 2357–2363. [Google Scholar]
- CRC Handbook of Chemistry and Physics, 94th ed.; CRC Press: Boca Raton, FL, 2013. [Google Scholar]
- Wagman D. D.; Evans W. H.; Parker V. B.; Schumm R. H.; Halow I.; Bailey S. M.; Churney K. L.; Nuttall R. L.. J. Phys. Chem. Ref. Data 1982, 11 (supplement no. 2). [Google Scholar]
- Bekenstein Y.; Vinokurov K.; Keren-Zur S.; Hadar I.; Schilt Y.; Raviv U.; Millo O.; Banin U. Nano Lett. 2014, 14, 1349–1353. [DOI] [PubMed] [Google Scholar]
- Maeda T.; Nakamura S.; Wada T. Jpn. J. Appl. Phys. 2011, 50, 6. [Google Scholar]
- Pohl J.; Albe K. J. Appl. Phys. 2010, 108, 023509. [Google Scholar]
- Kumar M.; Zhao H. Y.; Persson C. Thin Solid Films 2013, 535, 318–321. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.