

Abstract
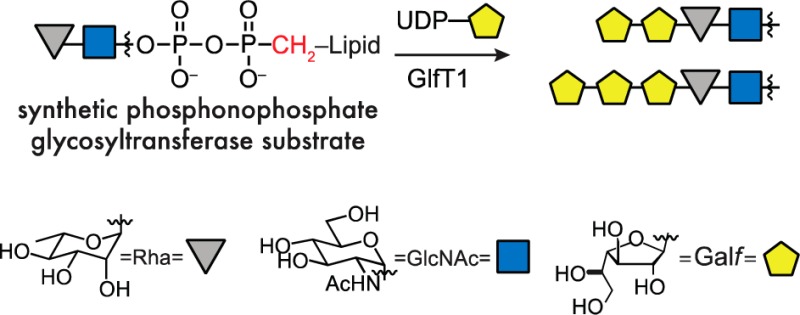
Glycosyltransferases that act on polyprenol pyrophosphate substrates are challenging to study because their lipid-linked substrates are difficult to isolate from natural sources and arduous to synthesize. To facilitate access to glycosyl acceptors, we assembled phosphonophosphate analogues and showed these are effective substrate surrogates for GlfT1, the essential product of mycobacterial gene Rv3782. Under chemically defined conditions, the galactofuranosyltransferase GlfT1 catalyzes the formation of a tetrasaccharide sequence en route to assembly of the mycobacterial galactan.
Lipid-linked oligosaccharide pyrophosphates serve as acceptors for a large class of glycosyltransferases.1 Progress in understanding these enzymes is hindered by the difficulty of isolating these biosynthetic acceptors from natural sources and the challenge of their chemical synthesis.2,3 Although some acceptors have been generated through astutely orchestrated syntheses,4−8 the lability of the allylic pyrophosphoryl group narrows the range of transformations that can be applied to their synthesis. We envisioned addressing these barriers by synthesizing a glycosyltransferase acceptor in which the pyrophosphoryl group was replaced with a phosphonophosphate group (Figure 1, top). This modification should mimic key features of the acceptor, while augmenting its hydrolytic stability and synthetic accessibility.9 The impetus for testing these surrogates arose from our investigation of galactan biosynthesis.
Figure 1.
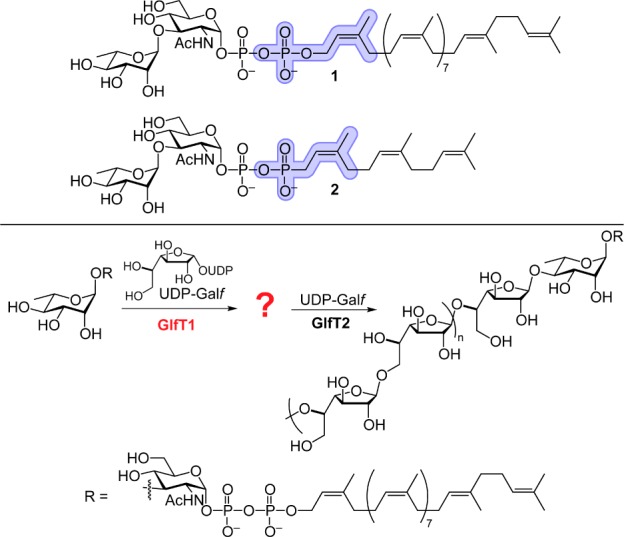
Top: Replacement of the allylic phosphate in 1 (blue) with a phosphonate (blue) and (2Z,6Z)-farnesyl lipid affords surrogate 2. Bottom: The product of GlfT1 (denoted with the question mark) is elongated by GlfT2 to afford the mycobacterial galactan (n = 10–20).
The galactan, a critical component of the mycobacterial cell wall, is composed of galactofuranose (Galf), the energetically disfavored 5-membered ring isomer of galactose.10,11 Galf has not been identified in any mammalian glycans, rendering the enzymes that mediate Galf incorporation potential antimycobacterial targets.12,13 The essential14Mycobacterium tuberculosis gene Rv3782 encodes a protein termed GlfT1. GlfT1 appears to be a galactofuranosyltransferase that primes galactan assembly. Its assigned role is to promote the elongation of decaprenyl-linked l-Rha-α-(1,3)-d-GlcNAc pyrophosphate 1 by one to three Galf residues (Figure 1, bottom).15 The resulting lipid-linked oligosaccharide is a substrate for the related galactofuranosyltransferase GlfT2.13 GlfT2 can process synthetic substrates bearing even a single galactofuranose residue.16 In principle, therefore, GlfT1 needs only to catalyze the addition of one Galf residue to its substrate (Figure 1). Still, experiments with membrane extracts or cell lysates suggested that GlfT1 could catalyze the addition of multiple Galf residues.15,17,18 We sought to assess the enzyme products under chemically defined conditions, which required obtaining active GlfT1 and a suitable acceptor.
The predicted GlfT1 acceptor 1 is a demanding synthetic target; therefore, we considered which of its features would be required for function. An analysis of literature suggested that the pyrophosphate group would be important.19−21 Consistent with this analysis, we found that O-alkyl disaccharides lacking a pyrophosphate linkage, such as 12-phenoxydodec-2-enyl Rha-α-(1,3)-GlcNAc S6, were not elongated by purified, recombinant GlfT1.22 Attempts to synthesize substrates analogous to 1 were complicated by the lability of the allylic pyrophosphate. We therefore considered a modification that would increase the stability of intermediates en route to the target glycosyl acceptors. Phosphonophosphates can serve as pyrophosphate analogues, and their increased stability suggested that they could be more readily synthesized. We therefore targeted surrogate 2 (Figure 1, top) with the goal of ascertaining whether this compound could function as a GlfT1 acceptor.
In selecting compound 2, we recognized the potential risk of generating a phosphonophosphate that is not isosteric with the pyrophosphate. Nevertheless, this risk by the increased synthetic accessibility of compound 2 and because enzymes, such as farnesyltransferases, bind lipid-substituted pyrophosphate and truncated phosphonophosphate derivatives similarly.23 Because the related glycosyltransferase GlfT2 is influenced by the acceptor lipid substituent,13 our synthetic route was designed to access acceptors with diverse lipid groups (Scheme 1). The decaprenol substituent was replaced with a (2Z,6Z)-farnesyl group, as this shorter lipid substituent can simplify substrate handling.24 Glycosyltransferases can be sensitive to the polyprenol lipid alkene and its geometry,4,7,25 so we preserved the isoprenyl alkene geometry closest to the oligosaccharide. We reasoned that if compound 2 was inactive, we could modify our route to identify a suitable substrate.
Scheme 1. Synthesis of GlfT1 Acceptor Surrogate 2.

The convergent route shown was also used to prepare 8 and 9.
The route to acceptor surrogate 2 began with the glycosylation of compounds 3 and 4. The stereochemical outcome of the glycosylation was controlled by using the α-selective silylated l-rhamnosyl donor 3.26 Activation of this “super armed” donor27 with N-iodosuccinimide and silver trifluoromethanesulfonate provided the desired α-glycoside. The silyl protecting groups were exchanged for acetate groups, as unmasking the former late in the synthetic route was problematic. This protecting group change facilitated purification and characterization of the acylated intermediates.
The α-phosphoryl group was installed using a phosphitylation–oxidation sequence.28 Hydrogenolysis afforded the anomeric lactol, which was exposed to dibenzyl N,N-diisopropylphosphoramidite in the presence of 1H-tetrazole.29 The intermediate glycosyl phosphite was oxidized in situ to yield a phosphotriester. The benzyl protecting groups were removed by reductive hydrogenation in the presence of an amine base to afford phosphomonoester 6 in good yield, with 9:1 α:β selectivity favoring the desired anomer.
A critical transformation was joining disaccharide 6 with lipid 7 to form the phosphonophosphate. Activation of the phosphoryl group of either coupling partner through formation of the phosphoryl chloride or phosphoryl imidazolide could result in self-coupling side products or product instability under the reaction conditions. Ultimately, Moffatt–Khorana conditions30 were effective. The electrophilic component, C15 isoprenyl phosphonomorpholidate 7, was prepared in four steps from (2Z,6Z)-farnesol (see Supporting Information). Disaccharide 6 and morpholidate 7 were exposed to 1H-tetrazole, and the product was saponified to afford phosphonophosphate 2. Though the yield was modest, the transformation was highly reproducible. We exploited this convergent synthetic route to produce acceptors to probe the structural features required for enzymatic activity. Surrogate 8 bearing a C10 isoprenyl (neryl) lipid carrier as well as surrogate 9 bearing only a monosaccharide were generated. The stability of the allylic phosphonophosphate derivatives facilitated their synthesis and subsequent analysis.
The ability of the acceptor surrogates to serve as substrates was tested using purified GlfT1. Recombinant His6-tagged Mycobacterium smegmatis GlfT1 was produced in M. smegmatis mc2155 and isolated by affinity chromatography. Exposure of disaccharide 2 to GlfT1 in the presence of donor sugar UDP-Galf31 afforded oligosaccharide products (Figure 2), indicating that compound 2 is an effective substrate. Mass spectrometry analysis indicated that the major product is extension of the acceptor by +2 Galf units. NMR data for the isolated product were consistent with those of a related tetrasaccharide32 (see Supporting Information). A product extended by +3 Galf units is also observed. GlfT1 consumes nearly all of the acceptor to produce these oligosaccharides.
Figure 2.

Top: Representative MALDI-TOF mass spectrum obtained from a reaction mixture of compound 2, UDP-Galf, and GlfT1. Masses corresponding to +2 and +3 Galf residues were observed. Bottom: Corresponding +2 and +3 products from elongation of acceptor 2. The linkage pattern shown is in agreement with that of endogenous galactan (see ref (10)).
The +1 product was not observed. This result is consistent with processing of the natural substrate by microsomal preparations of GlfT1.17 These results indicate that the phosphonophosphate acceptor emulates the natural substrate. The absence of the +1 product is notable given that the polymerase GlfT2 can elongate an acceptor with a single Galf residue.16 We postulate that the ability of GlfT2 to generate a polymer with faithfully alternating β-(1,5) and β-(1,6) linkages arises from GlfT1-catalyzed formation of the +2 Galf disaccharide. In this way, GlfT1 sets the register for polymerization by GlfT2.13,33,34 The +2 disaccharide product may also lead to more efficient polymerization by GlfT2. We previously showed that GlfT2 exhibits a kinetic lag phase when polymerizing a lipid-linked Galf disaccharide.13 The lag phase was abrogated with a substrate bearing a Galf tetrasaccharide, as this oligosaccharide presumably fills the monomer subsites during polymerization.33 Thus, the tetrasaccharide product generated by the action of GlfT1 should be processed rapidly by GlfT2.
The identity of the lipid carrier can affect a substrate’s ability to serve as a glycosyl acceptor. We therefore analyzed the efficiency of elongation of substrates bearing different lipids by quantifying the amount of UDP released upon GlfT1 addition. Specifically, the assay employed couples UDP production to the luciferase/luciferin reaction, wherein UDP production by GlfT1 is related linearly to increases in luminescence (Figure 3, top).35 As expected, no GlfT1 activity was observed with monosaccharide 9, highlighting that GlfT1 requires a substrate bearing the disaccharide l-Rha-α-(1,3)-d-GlcNAc (Figure 3, bottom). The difference between C10-linked 8 and C15-linked 2 was pronounced. GlfT1 treatment produces almost 5 times as much UDP in the presence of acceptor 2 than with 8. These data indicate that acceptor 2, with its longer (2Z,6Z)-farnesyl lipid, is a superior enzyme substrate. We used this assay to compare the kinetics of elongation by GlfT1 and determined an apparent Km of 86 ± 25 μM for acceptor 2 and an apparent Vmax of 1.53 ± 0.16 μM/min. We previously found that the lipid substituent is important for the binding and processing of acceptors by GlfT2,13 and our results suggest the lipid is also an important component for GlfT1 acceptor substrates.
Figure 3.
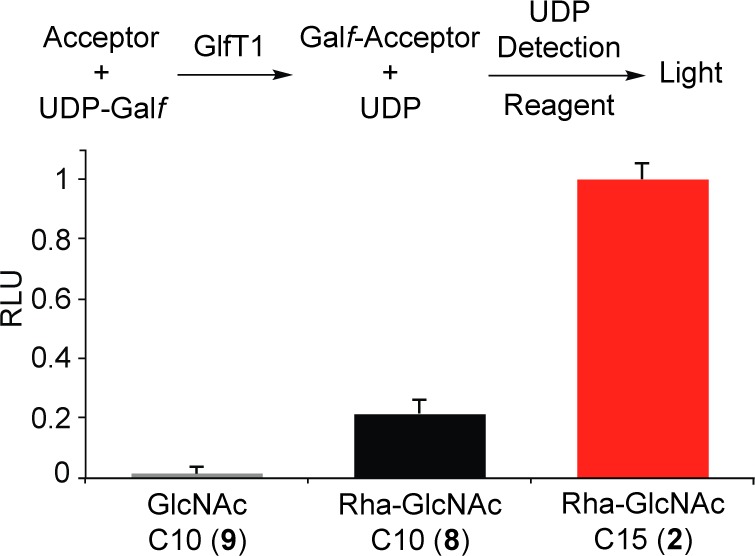
Top: Glycosyltransferase activity promotes UDP release, which is monitored using a luminescence assay. Bottom: Relative output of UDP-Galf turnover by GlfT1 with acceptors 2, 8, and 9.
The finding that phosphonophosphate analogues serve as effective substrates reveals fundamental features of GlfT1. These acceptors, the closest mimics of the natural substrate available, are processed to lipid-linked oligosaccharides by GlfT1. These observations indicate that the activity of GlfT1 outside of the endogenous mycobacterial milieu is robust—it does not require the presence of membranes or cell envelope components. Though similar GT-A domains and significant amino acid sequence identity (24% by ClustalO alignment) suggest an evolutionary link between GlfT1 and GlfT2, our data highlight their distinct roles in galactan assembly. Specifically, GlfT1 does not efficiently process O-alkyl disaccharides, but it does elongate lipid-linked pyrophosphate or phosphonophosphate acceptors substituted with the disaccharide l-Rha-α-(1,3)-d-GlcNAc by two to three Galf residues. Unlike the polymerase GlfT2, GlfT1 does not generate longer polymers and its substrate preference appears to be more narrowly defined. Indeed, GlfT2 is a relatively promiscuous carbohydrate polymerase that can act on varied truncated lipid-linked acceptors.16,36 The high specificity of GlfT1 and its ability to append at least two Galf residues should yield substrates that can be rapidly polymerized by GlfT2 to afford polysaccharides of defined sequence. These data highlight the key role of GlfT1 in controlling galactan biosynthesis.
Our studies of GlfT1 highlight the utility of the phosphonophosphate-containing acceptor substrates. The substitution of a phosphonophosphate for a pyrophosphate can facilitate the synthesis of complex glycosyltransferase acceptors. Glycosyltransferases in prokaryotes, eukaryotes, and archaea that act on pyrophosphate-linked acceptors are ubiquitous, including enzymes that mediate peptidoglycan assembly6,37 and that generate O-38,39 and N-linked5 glycans. We anticipate that the approach described herein will facilitate characterization of glycosyltransferase activity of the large family of enzymes that act on pyrophosphate-containing acceptors.
Acknowledgments
This research was supported by the NIH (R01-AI063596). M.A.M.F. thanks NIGMS for a fellowship (F31 GM101953), V.R.A. thanks NIGMS for a postdoctoral fellowship (F32 GM084475), and V.A.K. thanks the NSF (DGE-1256259). We thank S. A. Goueli and H. Zegzouti (Promega Co.) for reagents (Figure 3), M. R. Levengood for preliminary GlfT1 assays, D. A. Wesener for purified M. smegmatis mc2155 genomic DNA, and M. S. DeCanio for assistance generating the GlfT1 expression vector.
Supporting Information Available
Experimental procedures and 1H, 31P, and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lairson L. L.; Henrissat B.; Davies G. J.; Withers S. G. Annu. Rev. Biochem. 2008, 77, 521. [DOI] [PubMed] [Google Scholar]
- Warren C. D.; Konami Y.; Jeanloz R. W. Carbohydr. Res. 1973, 30, 257. [DOI] [PubMed] [Google Scholar]
- Lee Y. J.; Ishiwata A.; Ito Y. Tetrahedron 2009, 65, 6310. [Google Scholar]
- Chen L.; Men H.; Ha S.; Ye X.-Y.; Brunner L.; Hu Y.; Walker S. Biochemistry 2002, 41, 6824. [DOI] [PubMed] [Google Scholar]
- Weerapana E.; Glover K. J.; Chen M. M.; Imperiali B. J. Am. Chem. Soc. 2005, 127, 13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Fechter E. J.; Wang T.-S. A.; Barrett D.; Walker S.; Kahne D. E. J. Am. Chem. Soc. 2007, 129, 3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward R.; Yi W.; Zhao G.; Eguchi H.; Sridhar P. R.; Guo H.; Song J. K.; Motari E.; Cai L.; Kelleher P.; Liu X.; Han W.; Zhang W.; Ding Y.; Wang P. G. Nat. Chem. Biol. 2010, 6, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F.-C.; Chen K.-T.; Huang L.-Y.; Shih H.-W.; Chang H.-H.; Nien F.-Y.; Liang P.-H.; Cheng T.-J. R.; Wong C.-H.; Cheng W.-C. Org. Lett. 2011, 13, 5306. [DOI] [PubMed] [Google Scholar]
- Engel R. Chem. Rev. 1977, 77, 349. [Google Scholar]
- McNeil M.; Daffe M.; Brennan P. J. J. Biol. Chem. 1990, 265, 18200. [PubMed] [Google Scholar]
- Besra G. S.; Khoo K.-H.; McNeil M.; Dell A.; Morris H. R.; Brennan P. J. Biochemistry 1995, 34, 4257. [DOI] [PubMed] [Google Scholar]
- Dykhuizen E. C.; May J. F.; Tongpenyai A.; Kiessling L. L. J. Am. Chem. Soc. 2008, 130, 6706. [DOI] [PubMed] [Google Scholar]
- May J. F.; Splain R. A.; Brotschi C.; Kiessling L. L. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. Mol. Microbiol. 2003, 48, 77. [DOI] [PubMed] [Google Scholar]
- Mikušová K.; Beláňová M.; Korduláková J.; Honda K.; McNeil M. R.; Mahapatra S.; Crick D. C.; Brennan P. J. J. Bacteriol. 2006, 188, 6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splain R. A.; Kiessling L. L. Bioorg. Med. Chem. 2010, 18, 3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beláňová M.; Dianišková P.; Brennan P. J.; Completo G. C.; Rose N. L.; Lowary T. L.; Mikušová K. J. Bacteriol. 2008, 190, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltier P.; Beláňová M.; Dianišková P.; Zhou R.; Zheng R. B.; Pearcey J. A.; Joe M.; Brennan P. J.; Nugier-Chauvin C.; Ferrières V.; Lowary T. L.; Daniellou R.; Mikušová K. Chem. Biol. 2010, 17, 1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Men H. B.; Park P.; Ge M.; Walker S. J. Am. Chem. Soc. 1998, 120, 2484. [Google Scholar]
- Yi W.; Yao Q.; Zhang Y.; Motari E.; Lin S.; Wang P. G. Biochem. Biophys. Res. Commun. 2006, 344, 631. [DOI] [PubMed] [Google Scholar]
- Riley J. G.; Xu C.; Brockhausen I. Carbohydr. Res. 2010, 345, 586. [DOI] [PubMed] [Google Scholar]
- TLC analysis of radiolabeled products suggests octyl Rha-α(1,3)-GlcNAc is a glycosyl acceptor in microsomal preparations of cells producing GlfT1 with added radiolabeled UDP-Galp and the mutase UGM (ref (17)). Using purified GlfT1 and S6 (see Supporting Information), no glycosylation was detected.
- DeGraw A. J.; Zhao Z.; Strickland C. L.; Taban A. H.; Hsieh J.; Jefferies M.; Xie W.; Shintani D. K.; McMahan C. M.; Cornish K.; Distefano M. D. J. Org. Chem. 2007, 72, 4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft M. B.; Martinez Farias M. A.; Kiessling L. L. J. Org. Chem. 2013, 78, 2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M. M.; Weerapana E.; Ciepichal E.; Stupak J.; Reid C. W.; Swiezewska E.; Imperiali B. Biochemistry 2007, 46, 14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen C. M.; Nordstrøm L. U.; Bols M. J. Am. Chem. Soc. 2007, 129, 9222. [DOI] [PubMed] [Google Scholar]
- Mootoo D. R.; Konradsson P.; Udodong U. E.; Fraser-Reid B. J. Am. Chem. Soc. 1988, 110, 5583. [Google Scholar]
- Sim M. M.; Kondo H.; Wong C.-H. J. Am. Chem. Soc. 1993, 115, 2260. [Google Scholar]
- Caruthers M. H.; Barone A. D.; Beaucage S. L.; Dodds D. R.; Fisher E. F.; McBride L. J.; Matteucci M.; Stabinsky Z.; Tang J. Y. In Methods in Enzymology, Vol. 154; Wu R., Grossman L., Eds.; Academic Press: 1987; pp 287–313. [DOI] [PubMed] [Google Scholar]
- Wittmann V.; Wong C.-H. J. Org. Chem. 1997, 62, 2144. [DOI] [PubMed] [Google Scholar]
- Marlow A. L.; Kiessling L. L. Org. Lett. 2001, 3, 2517. [DOI] [PubMed] [Google Scholar]
- Completo G. C.; Lowary T. L. J. Org. Chem. 2008, 73, 4513. [DOI] [PubMed] [Google Scholar]
- Levengood M. R.; Splain R. A.; Kiessling L. L. J. Am. Chem. Soc. 2011, 133, 12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May J. F.; Levengood M. R.; Splain R. A.; Brown C. D.; Kiessling L. L. Biochemistry 2012, 51, 1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel L.; Vidugiris G.; Goueli S. Glycobiology 2013, 23, 1340. [Google Scholar]
- Rose N. L.; Completo G. C.; Lin S.-J.; McNeil M.; Palcic M. M.; Lowary T. L. J. Am. Chem. Soc. 2006, 128, 6721. [DOI] [PubMed] [Google Scholar]
- Lebar M. D.; Lupoli T. J.; Tsukamoto H.; May J. M.; Walker S.; Kahne D. J. Am. Chem. Soc. 2013, 135, 4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faridmoayer A.; Fentabil M. A.; Mills D. C.; Klassen J. S.; Feldman M. F. J. Bacteriol. 2007, 189, 8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley M. D.; Morrison M. J.; Aas F. E.; Børud B.; Koomey M.; Imperiali B. Biochemistry 2011, 50, 4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.