

Abstract
Hydrogenases are oxygen-sensitive enzymes that catalyze the conversion between protons and hydrogen. Water-soluble subcomplexes of membrane-bound [NiFe]-hydrogenases (MBH) have been extensively studied for applications in hydrogen–oxygen fuel cells as they are relatively tolerant to oxygen, although even these catalysts are still inactivated in oxidative conditions. Here, the full heterotrimeric MBH of Ralstonia eutropha, including the membrane-integral cytochrome b subunit, was investigated electrochemically using electrodes modified with planar tethered bilayer lipid membranes (tBLM). Cyclic voltammetry and chronoamperometry experiments show that MBH, in equilibrium with the quinone pool in the tBLM, does not anaerobically inactivate under oxidative redox conditions. In aerobic environments, the MBH is reversibly inactivated by O2, but reactivation was found to be fast even under oxidative redox conditions. This enhanced resistance to inactivation is ascribed to the oligomeric state of MBH in the lipid membrane.
Hydrogenases are complex microbial metalloenzymes which catalyze the reversible oxidation of H2 to protons at rates comparable to those normally achieved by Pt.1 They are widespread in the microbial world where they are used to dispose of excess reducing power (H2 production) or to produce energy (H2 oxidation).2 Their ability to selectively interconvert H2 to H+s makes them ideal catalysts for H2/O2 biofuel cells. When they are used in conjunction with a selective catalyst for O2 reduction, like laccase or bilirubin oxidase, it is no longer necessary to separate the anode from the cathode with a gas impermeable membrane as required in Pt-based fuel cells.3 Hydrogenases have thus been intensively studied both as an inspiration to design inorganic catalysts and as biocatalysts themselves.4 Based on the metal content of the active site, H+-H2 interconverting hydrogenases have been classified into [NiFe]- and [FeFe]-hydrogenases. [FeFe]-hydrogenases have high turnover frequencies for H2 production, but they are inactivated by trace amounts of O2.5 [NiFe]-hydrogenases, by contrast, are generally less sensitive to O2 inactivation and biased toward H2 oxidation.6
[NiFe]-hydrogenases have been further subdivided into “standard” (O2-sensitive) and O2-tolerant hydrogenases, and many studies have focused on the elucidation of the origins of the O2 tolerance.7 The membrane-bound hydrogenase (MBH) from the β-proteobacterium Ralstonia eutropha is one of the best studied O2-tolerant [NiFe]-hydrogenases.8 It is an uptake hydrogenase that links H2 oxidation to quinone reduction and has an outstanding O2 tolerance, capable of maintaining a high level of activity in the presence of air supplemented with low H2 concentrations.6b,9 As many other uptake [NiFe]-hydrogenases, it consists of three subunits, one of which is an integral membrane protein (Figure 1). The [NiFe] active center is located in the large subunit (α), and three [FeS] clusters are aligned in the small subunit (β) forming an electron relay.10 The third subunit is a diheme cytochrome b562, which anchors the protein complex to the cytoplasmic membrane and transfers electrons from H2 oxidation to the respiratory chain via the quinone pool.11 Both ubiquinone and menaquinone have been proposed to be the native substrate.8b
Figure 1.
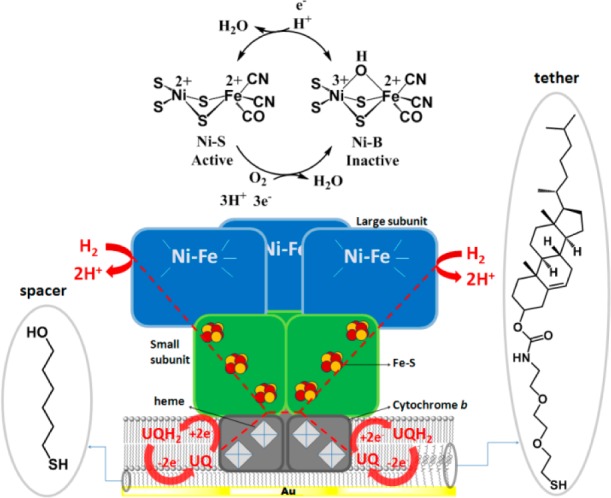
Heterotrimeric MBH is incorporated in the tBLM adsorbed on a mixed self-assembled monolayer made of EO3-cholesteryl (tether) and 6-mercaptohexan-1-ol (spacer) on the surface of a gold electrode. The main reactions catalyzed by the enzyme are shown in red. The ubiquinone (UQ) added to the tBLM is reduced by the cytchrome b562 to ubiquinol (UQH2) which is reoxidized at the electrode. The oxidative conversion of the [NiFe] site from the Ni–S state to the inactive Ni–B state is depicted at the top.
The O2 tolerance of the MBH and of closely related enzymes has been attributed to the “special” design of the unusual proximal [4Fe-3S] cluster, which has the ability to provide two electrons in a narrow potential range, helping to keep a reducing environment when the MBH reacts with oxygen.2b,7c,7f,10,12 When O2 reacts at the active site, this cluster ensures the formation of the so-called Ni–B state (Figure 1), also designated as a “ready inactive” state, which is rapidly reactivated (in a matter of seconds) under a H2 atmosphere. In contrast, O2-sensitive [NiFe]-hydrogenases, which have a conventional [4Fe-4S] cluster at the proximal site, form in addition to Ni–B the inactive Ni–A state upon reacting with O2, which usually requires nonphysiological negative potentials to reactivate.9b,13 In addition to crystallography and spectroscopy, protein film electrochemistry (PFE) has been invaluable in elucidating the inactivation–reactivation mechanism.4e,7g,14 PFE provides a direct way for monitoring catalytic turnover by adsorbing the protein on the surface of an electrode (typically a graphite electrode) and controlling the enzymatic activity via the electrode potential. PFE studies have shown that [NiFe]-hydrogenases are inactivated at high potentials in anaerobic conditions and that inactivation is faster at low H2 concentrations (in the low μM range).5,6a,6c,7a,13b The H2 oxidation activity drops as the potential is raised, and it recovers when the active site is reduced at low potentials. The inactive state formed at high redox potentials was determined to be the Ni–B state.6c,13b This anaerobic inactivation mechanism has also been observed with nonphysiological oxidants.16 Almost all PFE studies on MBH have employed the hydrophilic αβ subcomplex.5,6a,6b,7g,9a,9b,14e Here, we describe a different approach, in which the full heterotrimeric MBH is immobilized at an electrode interface using a so-called tethered bilayer lipid membrane (tBLM) (Figure 1).20 Cytoplasmic membrane extracts from R. eutropha, containing MBH, are tethered to an electrode surface using cholesterol-based anchor molecules. The full heterotrimeric structure is retained as the MBH remains in a native-like membrane environment. By incorporating ubiquinone in the tBLM, the native catalytic function of the MBH, namely H2-ubiquinone oxidoreduction, can be studied, where the redox state of the quinone pool is controlled by the potential applied to the electrode (Figure 1). We show that the full heterotrimeric MBH in a lipid environment does not display anaerobic (oxidative) inactivation, as the hydrophilic subcomplexes do, and that O2-inactivated MBH rapidly reactivates under oxidative conditions even when the quinone pool is fully oxidized.
Experiments examining the influence of pH and temperature on enzyme activity were carried out to determine the optimum conditions for monitoring the catalytic activity (Figure 2). The optimum pH value for H2 oxidation activity seems to lie in the range of 7 to 8, unlike the heterodimeric αβ subcomplex for which an optimum between 4.5 and 6.5 was determined (Figure 2a).5,6a,9a A similar difference in pH optimum was reported in a study employing spectrophotometric assays.21 However, we note that at lower pH, the oxidation wave shifts to higher potentials, preventing us to reach potentials at which MBH is fully active as the tBLM system is damaged by potentials higher than 0.6 V. The shift in potential is a consequence of the pH dependence of the ubiquinol oxidation potential. We also propose that the absence of clear current plateaus at high potential (Figure 2) is due to the particular kinetic properties of the electrochemical oxidation of ubiquinol, which is coupled or “gated” by slow proton release in the lipid bilayer.22 Above pH 8 the enzymatic activity sharply drops, and no recovery is observed when the pH is subsequently lowered, suggesting denaturation of MBH at high pH. The MBH in the tBLM is stable at temperatures up to 50 °C, although some loss of activity is observed on time-scales in the order of hours at temperatures above 30 °C (Figure 2b). Consequently all the following experiments were performed at 30 °C and pH 7.4. Control experiments were carried out by recording cyclic voltammograms (CVs) of tBLMs prepared from cytoplasmic membranes lacking the αβ subcomplex of the MBH, which showed no H2 oxidation activity.
Figure 2.

CVs showing the influence of (a) pH, (b) temperature, or (c, d) quinone pool on enzyme activity in anaerobic conditions. H2 concentrations as indicated. All experiments were performed at 10 mV/s, pH 7.4, 30 °C unless stated otherwise. Inset of (c) was measured at 1 mV/s. The arrows in (c) indicate the direction of scan. The CVs in (b) were measured with less MBH in the tBLM compared to (a, c, and d) (see the Experimental Section in SI). (a–c) were measured using 1% (w/w) ubiquinone-10 to lipid ratio in the membrane and (d) was measured with 2% (w/w) menaquinone-7. Abbreviations: SHE, standard hydrogen electrode; Re, Ralstonia eutropha.
CVs recorded under 100% N2 (Figure 2, gray lines) show no catalytic oxidation waves, instead uncovering the oxidation and reduction signals of the ubiquinone pool in the tBLM. The large peak separation of the ubiquinone electrochemistry has previously been studied in detail and is caused by the coupling of the electron transfer with protonation/deprotonation steps, which are slow due to the lipid bilayer environment.22 The onset of H2 oxidation (black line) coincides with ubiquinol oxidation (Figure 2c), confirming the fact that the electron transfer between the MBH and the electrode takes place via the quinone pool. A clear feature in all the catalytic oxidation waves in Figure 2 is the absence of any anaerobic inactivation at high potential. Previous studies have shown that the anaerobic inactivation of the heterodimeric αβ subcomplex of MBH is more pronounced at low substrate concentrations and slow scan rates.6a,14e Therefore, CVs were recorded at 1 mV/s under 0.5% (4.0 μM) and 0.1% (0.8 μM) H2 (insert in Figure 2c and Figure S1). In either condition, no decrease in current is observed as the potential is swept toward positive values, which confirms that the heterotrimeric MBH in equilibrium with the quinone pool displays little or no anaerobic inactivation even in substrate limiting conditions.
To confirm that also menaquinone can act as electron acceptor of MBH, as previously proposed,8b experiments were performed with added menaquinone (Figure 2d, see SI for details). Menaquinone is oxidized at a lower potential than ubiquinone, and consequently the onset of hydrogen oxidation is visible from about −0.1 V onward, about 0.3 V lower. The reduction potential of menaquinone is only ∼0.2 V lower than ubiquinone. However, as already mentioned, the quinone oxidation in tBLMs is coupled to the deprotonation of the quinol.22 Apparently, differences between deprotonation rates and/or pKas between menaquinone and ubiquinone cause an additional 0.1 V shift.
In Figure 2d, a slight shoulder around 0.3 V is visible, which is due to trace amounts of ubiquinone-8 present in the tBLM. This ubiquinone originates from the Escherichia coli lipid extract as well as the cytoplasmic membrane extracts from R. eutropha that were used to prepare the tBLM (see SI for details). Similar to the experiments with ubiquinone, no inactivation is observed at high potentials.
Using a method developed by Léger et al.,23 the apparent Michaelis–Menten constant for hydrogen (KM(app)) was calculated with ubiquinone as electron acceptor. The method involves the addition of a H2-saturated aliquot to a stirred working solution which is continuously flushed with N2, while the working electrode potential is maintained at a fixed positive value (Figure 3a). The value of KM(app) can be calculated by analyzing the sigmoidal current decay as the gases (in this case H2) are removed from solution following an exponential decay. The exponential time-dependency of gas removal from solution was confirmed through independent experiments (see Figure S2). Table 1 shows that KM(app) increases with the applied electrode potential, suggesting that at high H2 concentrations ubiquinol/ubiquinone cycling might be limiting the rate of H2 oxidation, especially at potentials below 0.5 V. CVs were measured at increasing H2 concentration to further support the KM(app) values in Table 1 (see Figure S3). As previously reported with PFE, a delay is observed in Figure 3a between the time of H2 injection and the time at which the maximum activity is attained (∼50s).9a The origin of this delay remains unclear, and studies are ongoing to test if this delay is due to reactivation of MBH that has been inactivated in the absence of hydrogen.
Figure 3.
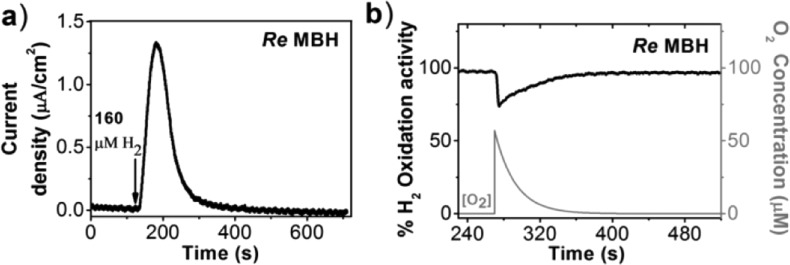
(a) Chronoamperogram showing the evolution of the MBH activity after the injection of a H2-saturated aliquot of buffer into the cell solution flushed with N2 (+0.497 V vs SHE; 30 °C, pH 7.4). (b) Chronoamperometry of the MBH (+0.397 V vs SHE, 100% H2, pH 7.4, 30 °C). The current is used to determine the hydrogen oxidation activity, which is normalized to 100% at the start of the experiment. An aliquot (one-fourth volume of the final cell volume) of air-saturated buffer was inserted into the electrochemical cell at 270 s. The exponential decay of the O2 concentration was plotted according to the equation: C(t) = C(0) exp(−t/τ), C is concentration, t is time), and τ is the time constant for exponential gas removal and was determined to be 22 s under these conditions (see Figure S2).
Table 1. Value of KM(app) (± SEM) at Different Potentialsa.
| potential (V vs SHE) | +0.397 | +0.497 | +0.597 |
|---|---|---|---|
| KM(app) (μM) | 1.5 ± 0.3 (n = 3) | 2.1 ± 0.9 (n = 8) | 9.2 ± 2.7 (n = 5) |
Number of independent experiments is given by n.
Aerobic inactivation was investigated using chronoamperometry by adding an aliquot of air-equilibrated buffer to the stirred working solution while the current resulting from H2 oxidation is monitored in time (Figure 3b). The electrode potential is held at a constant positive value at which the ubiquinone remains almost fully oxidized, ensuring a sufficiently high H2 oxidation activity of the MBH. We note that ubiquinol oxidases are also present in the cytoplasmic membrane extracts and will oxidize ubiquinol in the presence of O2. This will reduce the observed catalytic current due to H2 oxidation. The catalytic activity of ubiquinol oxidases can be directly observed at low potentials in CVs recorded in the presence of 10% O2 (Figure S4). After addition of 57 μM O2 (equivalent to one-fourth of the ambient concentration of dissolved O2), the H2 oxidation current drops very fast to ∼75 ± 1.4% (n = 12) of the initial current due to the oxidative conversion of the [NiFe] active site to the Ni–B state and possibly due to competing oxygen reduction (Figure 3b). The same behavior was observed at the higher potentials of 0.5 V. In the presence of 5% O2 (50 μM), MBH thus maintains at least three-quarters of its initial activity and begins to recover activity immediately after the injection, long before all O2 is flushed out of the cell by H2. The observed recovery in current seems to mirror that of the predicted O2 concentration in the cell, suggesting that activation and inactivation kinetics of MBH are in equilibrium during the experiment or that the decrease in current is solely due to ubiquinol oxidase activity. Importantly, the recovery of activity does not require less oxidative potentials, indicating that the “ready” inactive state (Ni–B) can be reduced back to the active state even when the ubiquinone pool is almost fully oxidized.
In conclusion, the present study shows that, unlike the hydrophilic heterodimeric subcomplex, the heterotrimeric MBH, in equilibrium with the quinone pool, does not undergo anaerobic inactivation under oxidative redox conditions. This implies that when the cytochrome b562 subunit is in equilibrium with its substrate (the ubiquinone pool) and acts as a primary electron acceptor of the hydrophilic αβ heterodimer, the [NiFe] active site is protected from permanently resting in the Ni–B state, even when H2 is scarce and the ubiquinone pool almost fully oxidized. In addition, although MBH is inactivated by O2 it immediately recovers activity even under highly oxidative redox conditions. We cannot exclude the possibility that reactivation (reduction of the Ni–B state) is driven by ubiquinol oxidation by a reversed electron flow from cytochrome b562 to the [NiFe] active site. However, at the extreme potentials applied (up to 0.6 V), we expect that only diminishing amounts of reduced ubiquinol are present in the tBLM. Instead, we propose that reactivation is related to the occurrence of higher oligomeric states of the MBH in the membrane. We have previously provided evidence to indicate that MBH forms tripartite supercomplexes of heterotrimeric complexes in the native lipid membrane and proposed that one heterotrimer donates electrons from H2 oxidation to reactivate a neighboring heterotrimer which is in the Ni–B state.8b Recently, a crystal structure of a dimer of a MBH subcomplex from E. coli (hydrogenase 1) showed that the distance between two distal [FeS] clusters is short, supporting the hypothesis that intermolecular electron transfer is possible.24
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant no. 280518 (V.R. and L.J.C.J.) and from the DFG cluster of Excellence “Unifying Concepts in Catalysis” (S.F. and O.L.). We gratefully thank Dr. Alison Parkin for the help provided with setting up the gas flow equipment.
Supporting Information Available
Experimental details and supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Jones A. K.; Sillery E.; Albracht S. P. J.; Armstrong F. A. Chem. Commun. 2002, 866. [DOI] [PubMed] [Google Scholar]
- a Vignais P. M.; Billoud B. Chem. Rev. 2007, 107, 4206. [DOI] [PubMed] [Google Scholar]; b Fritsch J.; Lenz O.; Friedrich B. Nature Rev. Microbiol. 2013, 11, 106. [DOI] [PubMed] [Google Scholar]
- Vincent K. A.; Cracknell J. A.; Clark J. R.; Ludwig M.; Lenz O.; Friedrich B.; Armstrong F. A. Chem. Commun. 2006, 5033. [DOI] [PubMed] [Google Scholar]
- a Ciaccafava A.; De Poulpiquet A.; Techer V.; Giudici-Orticoni M. T.; Tingry S.; Innocent C.; Lojou E. Electrochem. Commun. 2012, 23, 25. [Google Scholar]; b De Poulpiquet A.; Ciaccafava A.; Gadiou R.; Gounel S.; Giudici-Orticoni M. T.; Mano N.; Lojou E. Electrochem. Commun. 2014, 42, 72. [Google Scholar]; c Wait A. F.; Parkin A.; Morley G. M.; dos Santos M.; Armstrong F. A. J. Phys. Chem. C 2010, 114, 12003. [Google Scholar]; d Xu L.; Armstrong F. A. Energy Environ. Sci. 2013, 6, 2166. [Google Scholar]; e Hambourger M.; Gervaldo M.; Svedruzic D.; King P. W.; Gust D.; Ghirardi M.; Moore A. L.; Moore T. M. J. Am. Chem. Soc. 2008, 130, 2015. [DOI] [PubMed] [Google Scholar]; f Le Goff A.; Artero V.; Jousselme B.; Tran P. D.; Guillet N.; Métayé R.; Fihri A.; Palacin S.; Fontecave M. Science 2009, 326, 1384. [DOI] [PubMed] [Google Scholar]; g Song L.-C.; Li J.-P.; Xie Z.-J.; Song H.-B. Inorg. Chem. 2013, 52, 11618–11626. [DOI] [PubMed] [Google Scholar]; h Chambers G. M.; Angamuthu R.; Gray D. L.; Rauchfuss T. B. Organometallics 2013, 32, 6324. [Google Scholar]; i Roy S.; Groy T. L.; Jones A. K. Dalton Trans. 2013, 42, 3843. [DOI] [PubMed] [Google Scholar]
- a Vincent K. A.; Parkin A.; Lenz O.; Albracht S. P. J.; Fontecilla-Camps J. C.; Cammack R.; Friedrich B.; Armstrong F. A. J. Am. Chem. Soc. 2005, 127, 18179. [DOI] [PubMed] [Google Scholar]; b Liebgott P. P.; Leroux F.; Burlat B.; Dementin S.; Baffert C.; Lautier T.; Fourmond V.; Ceccaldi P.; Cavazza C.; Meynial-Salles I.; Soucaille P.; Fontecilla-Camps J. C.; Guigliarelli B.; Bertrand P.; Rousset M.; Leger C. Nat. Chem. Biol. 2010, 6, 63. [DOI] [PubMed] [Google Scholar]
- a Goldet G.; Wait A. F.; Cracknell J. A.; Vincent K. A.; Ludwig M.; Lenz O.; Friedrich B.; Armstrong F. A. J. Am. Chem. Soc. 2008, 130, 11106. [DOI] [PubMed] [Google Scholar]; b Vincent K. A.; Cracknell J. A.; Lenz O.; Zebger I.; Friedrich B.; Armstrong F. A. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 16951. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pandelia M.-E.; Fourmond V.; Tron-Infossi P.; Lojou E.; Bertrand P.; Leger C.; Giudici-Orticoni M.-T.; Lubitz W. J. Am. Chem. Soc. 2010, 132, 6991. [DOI] [PubMed] [Google Scholar]; d Friedrich B.; Fritsch J.; Lenz O. Curr. Opin. Biotechnol. 2011, 22, 358. [DOI] [PubMed] [Google Scholar]
- a Evans R. M.; Parkin A.; Roessler M. M.; Murphy B. J.; Adamson H.; Lukey M. J.; Sargent F.; Volbeda A.; Fontecilla-Camps J. C.; Armstrong F. A. J. Am. Chem. Soc. 2013, 135, 2694. [DOI] [PubMed] [Google Scholar]; b Abou Hamdan A.; Dementin S.; Liebgott P. P.; Gutierrez-Sanz O.; Richaud P.; De Lacey A. L.; Roussett M.; Bertrand P.; Cournac L.; Leger C. J. Am. Chem. Soc. 2012, 134, 8368. [DOI] [PubMed] [Google Scholar]; c Lukey M. J.; Roessler M. M.; Parkin A.; Evans R. M.; Davies R. A.; Lenz O.; Friedrich B.; Sargent F.; Armstrong F. A. J. Am. Chem. Soc. 2011, 133, 16881. [DOI] [PubMed] [Google Scholar]; d Dementin S.; Belle V.; Bertrand P.; Guigliarelli B.; Adryanczyk-Perrier G.; De Lacey A. L.; Fernandez V. M.; Rousset M.; Leger C. J. Am. Chem. Soc. 2006, 128, 5209. [DOI] [PubMed] [Google Scholar]; e Dementin S.; Leroux F.; Cournac L.; de Lacey A. L.; Volbeda A.; Leger C.; Burlat B.; Martinez N.; Champ S.; Martin L.; Sanganas O.; Haumann M.; Fernandez V. M.; Guigliarelli B.; Fontecilla-Camps J. C.; Rousset M. J. Am. Chem. Soc. 2009, 131, 10156. [DOI] [PubMed] [Google Scholar]; f Roessler M. M.; Evans R. M.; Davies R. A.; Harmer J.; Armstrong F. A. J. Am. Chem. Soc. 2012, 134, 15581. [DOI] [PubMed] [Google Scholar]; g Vincent K. A.; Parkin A.; Armstrong F. A. Chem. Rev. 2007, 107, 4366. [DOI] [PubMed] [Google Scholar]; h Cracknell J. A.; Vincent K. A.; Armstrong F. A. Chem. Rev. 2008, 108, 2439. [DOI] [PubMed] [Google Scholar]; i Liebgott P. P.; Dementin S.; Leger C.; Rousset M. Energy Environ. Sci. 2011, 4, 33. [Google Scholar]; j Abou Hamdan A.; Burlat B.; Gutierrez-Sanz O.; Liebgott P. P.; Baffert C.; De Lacey A. L.; Rousset M.; Guigliarelli B.; Leger C.; Dementin S. Nat. Chem. Biol. 2013, 9, 15. [DOI] [PubMed] [Google Scholar]; k Lubitz W.; Ogata H.; Rüdiger O.; Reijerse E. Chem. Rev. 2014, 114, 4081. [DOI] [PubMed] [Google Scholar]
- a Lenz O.; Ludwig M.; Schubert T.; Bürstel I.; Ganskow S.; Goris T.; Schwarze A.; Friedrich B. ChemPhysChem 2010, 11, 1107. [DOI] [PubMed] [Google Scholar]; b Frielingsdorf S.; Schubert T.; Pohlmann A.; Lenz O.; Friedrich B. Biochemistry 2011, 50, 10836. [DOI] [PubMed] [Google Scholar]
- a Ludwig M.; Cracknell J. A.; Vincent K. A.; Armstrong F. A.; Lenz O. J. Biol. Chem. 2009, 284, 465. [DOI] [PubMed] [Google Scholar]; b Cracknell J. A.; Wait A. F.; Lenz O.; Friedrich B.; Armstrong F. A. Proc. Natl. Acad. Sci. U.S.A. 2009, 1064920681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch J.; Scheerer P.; Frielingsdorf S.; Kroschinsky S.; Friedrich B.; Lenz O.; Spahn C. M. T. Nature 2011, 479, 249. [DOI] [PubMed] [Google Scholar]
- Bernhard M.; Benell B.; Hochkoeppler A.; Zannoni D.; Friedrich B. Eur. J. Biochem. 1997, 248, 179. [DOI] [PubMed] [Google Scholar]
- a Goris T.; Wait A. F.; Saggu M.; Fritsch J.; Heidary N.; Stein M.; Zebger I.; Lendzian F.; Armstrong F. A.; Friedrich B.; Lenz O. Nat. Chem. Biol. 2011, 7, 310. [DOI] [PubMed] [Google Scholar]; b Shomura Y.; Yoon K.-S.; Nishihara H.; Higuchi Y. Nature 2011, 479, 253. [DOI] [PubMed] [Google Scholar]; c Pandelia M. E.; Nitschke W.; Infossi P.; Giudici-Orticoni M. T.; Bill E.; Lubitz W. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ogata H.; Hirota S.; Nakahara A.; Komori H.; Shibata N.; Kato T.; Kano K.; Higuchi Y. Structure 2005, 13, 1635. [DOI] [PubMed] [Google Scholar]; b Jones A. K.; Lamle S. E.; Pershad H. R.; Vincent K. A.; Albracht S. P. J.; Armstrong F. A. J. Am. Chem. Soc. 2003, 125, 8505. [DOI] [PubMed] [Google Scholar]; c Saggu M.; Zebger I.; Ludwig M.; Lenz O.; Friedrich B.; Hildebrandt P.; Lendzian F. J. Biol. Chem. 2009, 284, 16264. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lamle S. E.; Albracht S. P. J.; Armstrong F. A. J. Am. Chem. Soc. 2004, 126, 14899. [DOI] [PubMed] [Google Scholar]; e Lamle S. E.; Albracht S. P. J.; Armstrong F. A. J. Am. Chem. Soc. 2004, 127, 6595. [DOI] [PubMed] [Google Scholar]
- a McIntosh C. L.; Germer F.; Schulz R.; Appel J.; Jones A. K. J. Am. Chem. Soc. 2011, 133, 11308. [DOI] [PubMed] [Google Scholar]; b Leger C.; Bertrand P. Chem. Rev. 2008, 108, 2379. [DOI] [PubMed] [Google Scholar]; c Lojou E.; Giudici-Orticoni M.-T.; Bianco P. J. Electroanal. Chem. 2005, 577, 79. [Google Scholar]; d Armstrong F. A.; Belsey N. A.; Cracknell J. A.; Goldet G.; Parkin A.; Reisner E.; Vincent K. A.; Wait A. F. Chem. Soc. Rev. 2009, 38, 36. [DOI] [PubMed] [Google Scholar]
- a Ciaccafava A.; Infossi P.; Ilbert M.; Guiral M.; Lecomte S.; Giudici-Orticoni M.-T.; Lojou E. Angew. Chem., Int. Ed. 2012, 51, 953. [DOI] [PubMed] [Google Scholar]; b Rudiger O.; Abad J. M.; Hatchikian E. C.; Fernandez V. M.; De Lacey A. L. J. Am. Chem. Soc. 2005, 127, 16008. [DOI] [PubMed] [Google Scholar]; c Gutierrez-Sanz O.; Marques M.; Pereira I. A. C.; De Lacey A. L.; Lubitz W.; Rudiger O. J. Phys. Chem. Lett. 2013, 4, 2794. [Google Scholar]
- a Jeuken L. J. C.; Connell S. D.; Henderson P. J. F.; Gennis R. B.; Evans S. D.; Bushby R. J. J. Am. Chem. Soc. 2006, 128, 1711. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weiss S. A.; Bushby R. J.; Evans S. D.; Henderson P. J. F.; Jeuken L. J. C. Biochem. J. 2009, 417, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schink B.; Schlegel H. G. Biochim. Biophys. Acta 1979, 567, 315. [DOI] [PubMed] [Google Scholar]
- a Gordillo G. J.; Schiffrin D. J. Faraday Discuss. 2000, 116, 89. [DOI] [PubMed] [Google Scholar]; b Jeuken L. J. C.; Bushby R. J.; Evans S. D. Electrochem. Commun. 2007, 9, 610. [Google Scholar]
- Leger C.; Dementin S.; Bertrand P.; Rousset M.; Guigliarelli B. J. Am. Chem. Soc. 2004, 126, 12162. [DOI] [PubMed] [Google Scholar]
- Volbeda A.; Darnault C.; Parkin A.; Sargent F.; Armstrong F. A.; Fontecilla-Camps J. C. Structure 2013, 21, 184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.