

Abstract
We have previously shown that hydrogen sulfide (H2S) reduces myogenic tone and causes relaxation of phenylephrine (PE)-constricted mesenteric arteries. This effect of H2S to cause vasodilation and vascular smooth muscle cell (VSMC) hyperpolarization was mediated by large-conductance Ca2+-activated potassium channels (BKCa). Ca2+ sparks are ryanodine receptor (RyR)-mediated Ca2+-release events that activate BKCa channels in VSMCs to cause membrane hyperpolarization and vasodilation. We hypothesized that H2S activates Ca2+ sparks in small mesenteric arteries. Ca2+ sparks were measured using confocal microscopy in rat mesenteric arteries loaded with the Ca2+ indicator fluo-4. VSMC membrane potential (Em) was measured in isolated arteries using sharp microelectrodes. In PE-constricted arteries, the H2S donor NaHS caused vasodilation that was inhibited by ryanodine (RyR blocker), abluminal or luminal iberiotoxin (IbTx, BKCa blocker), endothelial cell (EC) disruption, and sulfaphenazole [cytochrome P-450 2C (Cyp2C) inhibitor]. The H2S donor NaHS (10 μmol/l) increased Ca2+ sparks but only in the presence of intact EC and this was blocked by sulfaphenazole or luminal IbTx. Inhibiting cystathionine γ-lyase (CSE)-derived H2S with β-cyano-l-alanine (BCA) also reduced VSMC Ca2+ spark frequency in mesenteric arteries, as did EC disruption. However, excess CSE substrate homocysteine did not affect spark activity. NaHS hyperpolarized VSMC Em in PE-depolarized mesenteric arteries with intact EC and also hyperpolarized EC Em in arteries cut open to expose the lumen. This hyperpolarization was prevented by ryanodine, sulfaphenazole, and abluminal or luminal IbTx. BCA reduced IbTx-sensitive K+ currents in freshly dispersed mesenteric ECs. These results suggest that H2S increases Ca2+ spark activity in mesenteric artery VSMC through activation of endothelial BKCa channels and Cyp2C, a novel vasodilatory pathway for this emerging signaling molecule.
Keywords: cytochrome P-450 epoxygenase, sodium hydrosulfide, membrane potential
hydrogen sulfide (H2S) is a recently described vasodilator produced in the vasculature by the enzymes cystathionine γ-lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3MST). H2S has been proposed to cause vasodilation through a variety of mechanisms (1, 3, 26, 33) and genetic knockout of the CSE gene causes hypertension (32). We previously reported that inhibiting CSE or disrupting the endothelium enhances myogenic tone in small mesenteric arteries from rats and that exogenous H2S hyperpolarizes vascular smooth muscle cell (VSMC) membrane potential (Em) and dilates arteries through activation of large-conductance Ca2+-activated K+ (BKCa) channels (7).
Ca2+ sparks are spatially and temporally limited Ca2+-release events from ryanodine receptor Ca2+-release channels (RyR) in the sarcoplasmic reticulum (SR) of VSMCs and have been shown to activate VSMC BKCa channels to cause hyperpolarization, leading to a reduced open probability of L-type voltage-gated Ca2+ channels (VGCCs) and a decrease in cytosolic [Ca2+] (21). Ca2+ spark frequency can be increased by activation of transient receptor protein (TRP) channels (4) and has been hypothesized to act as an intrinsic negative-feedback mechanism to regulate stretch-induced VSMC depolarization and myogenic tone (10). Although RyR activity can be directly modulated in VSMCs (12), endothelial cell (EC)-derived factors can also increase VSMC Ca2+ spark activity (4, 8, 18), supporting a role for the endothelium in regulating VSMC RyR. Both nitric oxide (NO) and carbon monoxide (CO), two endothelium-derived dilators that function similarly to H2S in many tissues, have been shown to modulate spark activity (20).
Based on the previous description of VSMC BKCa channel activation by Ca2+ sparks downstream of CO and NO and our observations of EC-dependent dilation by H2S, we hypothesized that H2S activates Ca2+ sparks in an EC-dependent manner to contribute to VSMC hyperpolarization and vasodilation.
METHODS
Animals.
Male Sprague-Dawley rats (275–325 g) were used for all studies. On the day of the experiments, animals were euthanized with pentobarbital sodium (200 mg/kg ip) and mesenteric arteries were isolated for dilation, Ca2+ imaging, and Em studies. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine and conform to National Institutes of Health guidelines for animal use.
Isolated vessel preparation.
Intestinal arcades were removed and placed in a Silastic-coated Petri dish containing chilled physiological saline solution (PSS; in mmol/l: 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 0.43 NaH2PO4, 19 NaHCO3, 1.8 CaCl2, and 5.5 glucose). Fourth- or fifth-order artery segments (inner diameter < 100 μm) were dissected from the mesenteric vascular arcade and transferred to a vessel chamber (Living Systems Instrumentation). Segments (1–2 mm in length) were cannulated onto glass micropipettes and secured with silk ligatures. The arteries were pressurized to 100 mmHg with PSS using a servocontrolled peristaltic pump (Living Systems Instrumentation) and superfused with warmed, oxygenated PSS at a rate of 5 ml/min.
Vasodilation studies.
Arterial inner diameter was recorded in cannulated arteries using edge-detection software (IonOptix). Arteries were equilibrated at 37°C in warmed, oxygenated PSS for 30 min prior to the start of the experiment. Following equilibration, arteries were preconstricted to 50% resting diameter using phenylephrine (PE) and vasodilation was measured during cumulative addition of the H2S donor NaHS. Arteries were incubated with Ca2+-free PSS (in mmol/l: 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 0.43 NaH2PO4, 19 NaHCO3, 3.7 tetrasodium EGTA, and 5.5 glucose) to determine passive diameter at the end of the experiment.
Fluo-4 imaging.
Arteries used for VSMC Ca2+ spark imaging studies were incubated in a fluo-4 AM (10 μmol/l, Invitrogen) solution containing 0.25% pluronic acid in HEPES buffer (in mmol/l: 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 0.026 EDTA, and 10 glucose) for 60 min at 28°C prior to cannulation. After loading with fluo-4, arteries were transferred to a vessel chamber and cannulated as described above. To measure endothelial Ca2+ events, arteries were first cannulated and then the endothelium was selectively loaded with fluo-4 AM through the lumen of the artery. After 10 min equilibration in oxygenated PSS at 32°C, arteries were excited at 488 nm by a solid-state laser, and emitted light > 500 nm was collected using an Olympus IX71 microscope with a 60X water-immersion lens and a spinning-disk confocal scanning unit (Andor). A 75 × 50-μm area was imaged at 50–60 Hz using laser power of 30%. All Ca2+ spark imaging studies were conducted in pressurized arteries in the absence of exogenous PE.
Spark analysis.
Collected images were analyzed using SparkAn software, developed by A. D. Bonev and M. T. Nelson (University of Vermont). Ten images without spark activity were averaged to determine background fluorescence levels (F0). A minimum F/F0 of 1.2 was required for an event to be considered a spark. Each spark site was analyzed as a region of interest (ROI; 25 pixels2 or 3 μm2). Each image contained 15–25 cells, and spark frequency, amplitude, and duration were averaged for all ROIs in all visible cells to provide a single value for each of these parameters for each artery. These values were then averaged for all arteries in the study.
Em recordings.
Small mesenteric arteries were dissected and cannulated as described above. VSMCs were impaled with glass microelectrodes (tip resistance 40–120 MΩ) filled with 1 mol/l KCl. Endothelial Em was measured by impaling ECs in arteries cut open to expose the luminal surface. A Neuroprobe amplifier (A-M Systems) was used for recording Em. Analog output from the amplifier was low pass filtered at 1 kHz and recorded and analyzed using Axoscope software (Axon Instruments). Criteria for acceptance of Em recordings were 1) an abrupt negative deflection of potential as the microelectrode was advanced into a cell; 2) stable membrane potential for at least 1 min; and 3) an abrupt change in potential to ∼0 mV after the electrode was retracted from the cell.
Whole cell patch-clamp studies in isolated ECs.
Mesenteric arteries were cut into 2-mm segments and exposed to mild digestion solution containing 0.2 mg/ml dithiothreitol and 0.2 mg/ml papain in HEPES buffer for 45 min at 37°C. Arteries were removed from the digestion solution and placed in 1 ml of HEPES buffer containing 2 mg/ml BSA. Single ECs were released by gentle trituration with a small-bore Pasteur pipette and were stored at 4°C until use (for up to 5 h). One to two drops of the resulting cell suspension were seeded on a glass cover slip mounted on an inverted fluorescence microscope (Olympus IX71) for 30 min prior to superfusion. Single ECs were identified by the selective uptake of the fluorescently labeled acylated low-density lipoprotein Ac-LDL-Dil using a rhodamine filter (29) prior to each electrophysiological experiment. ECs were superfused under constant flow (2 ml/min) at room temperature (22–23°C) in an extracellular solution (in mmol/l: 141 NaCl, 4.0 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 10 glucose, buffered to pH 7.4 with NaOH). Whole cell current data were generated using an Axopatch 200B amplifier (Axon Instruments). Biophysical criteria (seal resistance > 1 GΩ, series resistance < 25 MΩ) were checked following membrane rupture and monitored throughout the course of the experiment. Cells were held at −60 mV and were dialyzed for 5 min with an intracellular solution (in mmol/l: 140 KCl, 0.5 MgCl2, 5 Mg2ATP, 10 HEPES, 1 EGTA adjusted to pH 7.2 with KOH). CaCl2 was added to yield a free-Ca2+ concentration of 1 μmol/l, as calculated using WinMAXC chelator software.
Immunohistochemistry.
Intestinal arcades were removed and placed in ice-cold HEPES buffer. Vessels were dilated with 100 μM papaverine for 10 min at room temperature and then frozen in Tissue-Tek O.C.T. Compound (VWR International). Frozen tissue was cut in 10-μm cross-sections and thaw mounted on slide. Slides were air dried for 20 min and then fixed with 2% formalin for 15 min at room temperature. Following a 5 min wash in phosphate-buffered saline (PBS; Mediatech) slides were blocked and permeabilized with PBS + 0.1% Triton X-100 (J. T. Baker) + 2% donkey serum (Sigma) for 25 min at room temperature. Slides were stained overnight at 4°C with primary antibodies against cystathionase (1:100; AbCam) and BKCa (1:50; Alamone Labs) in PBS + 2% donkey serum + 0.1% Tween-20 (Bio-Rad). DyLight 549 donkey anti-mouse and DyLight 649 donkey anti-rabbit secondary antibodies (1:200; Jackson Immunoresearch) used to detect cystathionase and BKCa, respectively, were applied to slides for 1.5 h at room temperature. Nuclei were stained with Sytox Green (1:5,000; Invitrogen) for 15 min at room temperature.
Statistical analysis.
Data are presented as means ± SE and were analyzed using one-way or two-way ANOVA as appropriate with Student-Newman-Keuls post hoc analysis for differences between groups, concentrations, and interactions for studies with multiple groups (SigmaStat). Paired data were analyzed using a paired Student's t-test. P < 0.05 was considered statistically significant for all analyses.
RESULTS
H2S vasodilation.
The H2S donor NaHS produced a robust vasodilation in PE-constricted arteries, and this dilation was significantly reduced by EC disruption (Fig. 1A). Due to recent evidence that H2S can activate EC BKCa channels (34), we measured vasodilation in arteries following BKCa channel blockade with IbTx administered in the superfusion (abluminal) or in the lumen (Fig. 1B). Both routes of IbTx administration abolished dilation to NaHS. To assess the EC-specific effect of luminal IbTx application, VSMC Em was measured in isolated arteries with luminal or abluminal IbTx in the absence of PE. VSMC Em was depolarized by abluminal IbTx whereas luminal IbTx had no significant effect (Fig. 1C), suggesting that luminal IbTx does not cross the EC layer.
Fig. 1.
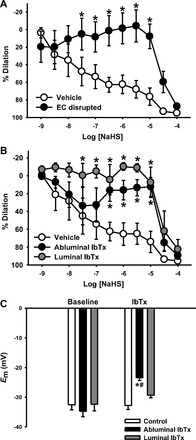
Small mesenteric artery dilation to increasing concentrations of the H2S donor NaHS in endothelial cell (EC)-intact and EC-disrupted arteries (A) and the presence of vehicle or iberiotoxin (IbTx) (100 nmol/l) administered abluminally or luminally (B). Arteries were pressurized to 100 mmHg and preconstricted to ∼50% with PE. C: vascular smooth muscle cell (VSMC) membrane potential (Em) measured by sharp microelectrode impalement in the absence of PE, with and without luminal or abluminal IbTx application. n = 5–6 per group. *P < 0.05 vs. baseline, #P < 0.05 vs. luminal IbTx.
Endogenous and exogenous H2S regulation of Ca2+ sparks.
Importantly, endogenous H2S produced by CSE opposes myogenic tone in small mesenteric arteries (7). To determine whether endogenous H2S activates Ca2+ sparks, the CSE inhibitor BCA was applied to arteries and Ca2+ spark frequency was assessed. Example traces of Ca2+ spark activity under basal and NaHS-stimulated conditions are shown in Fig. 2, A and B, respectively. BCA reduced Ca2+ spark frequency, as did EC disruption, and these effects were not additive (Fig. 2C). Conversely, adding exogenous NaHS (10 μmol/l) to mesenteric arteries significantly increased spark frequency (Fig. 2C). To determine whether the decreased spark frequency observed following BCA treatment could be an effect of homocysteine accumulation due to CSE inhibition, Ca2+ sparks were measured in the presence of exogenous homocysteine. Addition of homocysteine did not decrease spark frequency in a manner parallel to CSE inhibition with BCA (Fig. 2D). Although 3-MST is expressed in these arteries (data not shown), BCA completely mimics H2S scavenging, suggesting that vasoactive H2S is synthesized primarily by CSE in these small mesenteric arteries.
Fig. 2.
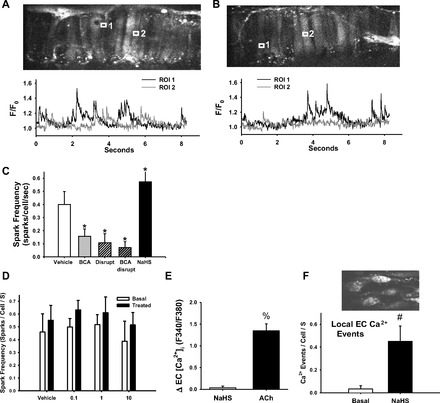
Example traces of Ca2+ spark activity in two regions of interest (ROI) in VSMC of mesenteric arteries loaded with fluo-4 and pressurized to 100 mmHg in the presence of vehicle (A) or β-cyano-l-alanine (BCA) (B). C: summary data showing Ca2+ spark frequency in arteries under EC-intact vehicle conditions or with BCA (100 μmol/l; EC intact), disrupted EC, both BCA and EC disruption, or NaHS (10 μmol/l; EC intact). ROIs = 1.7 μm per side. D: Ca2+ spark frequency before and after addition of exogenous homocysteine. E: the change in endothelial cell Ca2+ as measured by fura-2 AM in response to NaHS (10 μmol/l) or acetylcholine (10 μmol/l). F: the frequency of endothelial Ca2+ events before and after addition of NaHS (10 μmol/l) measured with the Ca2+ indicator fluo-4. n = 5–6 per group. *P < 0.05 vs. vehicle, #P < 0.05 vs. basal, %P < 0.05 vs. NaHS.
EC Ca2+, H2S vasodilation, and Ca2+ sparks.
It has been proposed that EC K+ channel activation increases EC [Ca2+], which can activate several EC enzymes to produce vasodilators (27). The effect of NaHS on EC [Ca2+] was assessed in small mesenteric arteries. NaHS did not elicit a detectable change in global EC [Ca2+] as measured with the Ca2+ indicator fura-2 AM (Fig. 2E). Conversely, acetylcholine caused a robust change in EC [Ca2+]. Localized Ca2+ events were also measured in EC of mesenteric arteries in response to NaHS. There were few EC Ca2+ events under basal conditions, but frequency of these events was increased upon addition of NaHS (Fig. 2F).
EC-derived vasodilators such as 11,12-epoxyeicosatrienoic acid (11,12-EET) produced by cytochrome P-450 epoxygenase can activate VSMC Ca2+ sparks (4). Therefore, NaHS vasodilation was measured in the presence of the Cyp2C inhibitor sulfaphenazole (10 μmol/l); inhibition of Cyp2C reduced vasodilation (Fig. 3A) using a concentration that we have previously demonstrated decreases synthesis of 11,12-EET without affecting synthesis of other EET products (5). Additionally, adding ryanodine at a concentration that eliminates Ca2+ sparks (10 μmol/l; Fig. 3B) diminished vasodilation to the H2S donor NaHS (Fig. 3C).
Fig. 3.
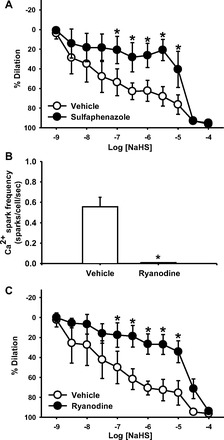
A: NaHS dilation curves in the presence of vehicle and the cytochrome P-450 2C (Cyp2C) inhibitor sulfaphenazole (10 μmol/l) in arteries preconstricted to ∼50% with PE. B: effect of 10 μmol/l ryanodine on Ca2+ spark frequency in cannulated small EC-intact mesenteric arteries loaded with fluo-4. C: NaHS dilation curves in the presence of vehicle and the RyR inhibitor ryanodine (10 μmol/l) in arteries preconstricted to ∼50% with PE. n = 5–6 per group. *P < 0.05 vs. vehicle.
H2S-induced Ca2+ spark activity.
The mechanism of NaHS-induced VSMC Ca2+ spark activity was assessed. The increase in spark frequency elicited by NaHS was blocked by EC disruption, sulfaphenazole, or luminal IbTx (Fig. 4A). NaHS actually reduced Ca2+ spark frequency in EC-disrupted arteries. NaHS did not affect Ca2+ spark amplitude (Fig. 4B) or duration (Fig. 4C), except in the presence of luminal IbTx, in this case reducing amplitude and extending duration. EC disruption alone reduced spark amplitude, and amplitude was not affected by the presence of NaHS in the EC-disrupted group (Fig. 4B).
Fig. 4.
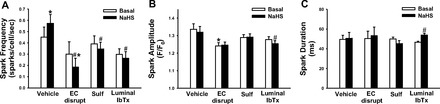
A: summary data of Ca2+ spark frequency at 100 mmHg in the presence of vehicle or NaHS (10 μmol/l) under control conditions (EC intact), EC disruption, sulfaphenazole (Sulf; 10 μmol/l; EC intact), or luminal IbTx (100 nmol/l; EC intact). Sparks were recorded before and after addition of NaHS in the same artery. Spark amplitude (B) and duration (C) at half-maximal amplitude under the same conditions as in A. n = 5–7 per group. *P < 0.05 vs. vehicle, #P < 0.05 vs. control within treatment.
H2S regulation of Em and EC K+ current.
Ca2+ sparks mediate vasodilation through activation of VSMC BKCa channels and subsequent Em hyperpolarization of VSMC (21). We therefore assessed the effect of NaHS on VSMC Em in small mesenteric arteries depolarized with 1 μmol/l PE. Representative recordings are shown in Fig. 5A. NaHS caused a hyperpolarization of mesenteric artery VSMC Em measured by sharp electrodes (Fig. 5B). This Em hyperpolarizing effect of NaHS was prevented with ryanodine, sulfaphenazole, abluminal IbTx, or luminal IbTx (Fig. 5B). The effect of luminal IbTx suggested an effect of H2S on EC BKCa channels; therefore, whole cell outward K+ currents were measured in freshly dispersed mesenteric artery EC to directly assess this possibility. These cells demonstrate an IbTx-sensitive current (Fig. 5C), demonstrating a contribution of BKCa channels to the K+ current. Inhibition of either BKCa or CSE reduced K+ currents. The combination of IbTx and BCA also decreased currents but currents in the presence of both inhibitors were not different from either BCA or IbTx alone. The effect of NaHS on EC Em was also assessed in the absence and presence of IbTx and is shown in Fig. 6. IbTx depolarized EC Em under basal conditions. NaHS hyperpolarized EC Em, but this was prevented in the presence of IbTx.
Fig. 5.
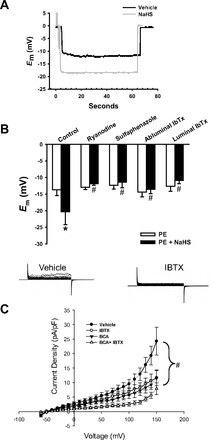
A: example recordings of sharp microelectrode impalements of VSMC in arteries pressurized to 100 mmHg and depolarized with 1 μmol/l PE in the presence of vehicle or NaHS under control conditions. Negative deflection of trace indicates cell impalement; positive deflection to 0 mV indicates retraction of electrode. B: summary data of Em recordings in EC-intact arteries in the presence of vehicle or NaHS under control conditions or in the presence of ryanodine (10 μmol/l), sulfaphenazole (10 μmol/l), or IbTx (100 nmol/l) in superfusate (abluminal) or in the artery lumen. n = 4–5 per group. C: effect of BCA (100 μmol/l) and IbTx (100 nmol/l) or IbTx + BCA on whole cell K+ currents in freshly dispersed mesenteric EC. n = 5–7 per group. *P < 0.05 vs. PE (B); #P < 0.05 vs. control or vehicle (B and C).
Fig. 6.
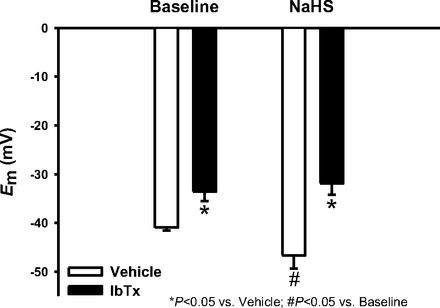
EC Em measured in the absence or presence of NaHS (10 μmol/l) with and without IbTx (100 nmol/l) pretreatment in mesenteric arteries cut open to expose the endothelium. n = 6–7 per group. *P < 0.05 vs. vehicle, #P < 0.05 vs. baseline.
Vascular expression of CSE and BKCa.
CSE and BKCa expression were assessed in the mesenteric vasculature by immunohistochemistry. CSE expression appeared to be more localized to the endothelial and adventitial layers (Fig. 7A), whereas BKCa staining was apparent throughout the vascular wall (Fig. 7B). Overlay of images from the same vessel revealed that both CSE and BKCa are expressed in the endothelium (Fig. 7, C and D).
Fig. 7.
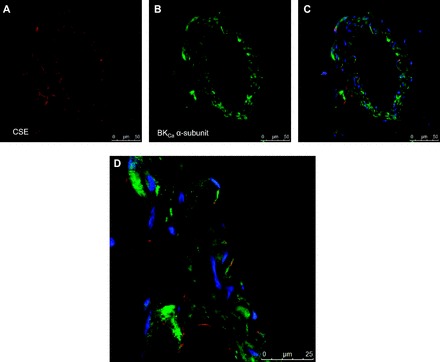
Representative images of cystathionine γ-lyase (CSE) (A), large-conductance Ca2+-activated potassium channels (BKCa) (B), overlay of the images in A and B (C), and mgnification of C (D).
DISCUSSION
Our findings show that H2S dilates small mesenteric arteries through activation of EC BKCa channels and Cyp2C with downstream activation of VSMC Ca2+ sparks. In addition, a Cyp2C product is necessary for H2S-induced dilation. H2S is a recently described vasodilator and novel modes of action are being described at a rapid pace, but this is the first demonstration of an H2S-initiated endothelium-dependent mechanism mediated in part by activation of VSMC Ca2+ sparks. These results are consistent with our previous observation in the same vascular bed that dilation and VSMC Em hyperpolarization evoked by the H2S donor NaHS require BKCa channels (7). It is also consistent with a recent study in vas deferens smooth muscle showing that H2S can directly activate BKCa channels through a redox mechanism (14), and a study in cerebral arteries demonstrating activation of VSMC Ca2+ sparks by H2S (15).
The effect of H2S on the activity of BKCa channels is a contentious issue. As noted above, some studies suggest H2S activates the channel (7, 15, 17, 28, 34), whereas others find H2S inhibits BKCa activity (13, 30, 31). Because the cell types in these two groups of studies do not overlap, it is possible that tissue-specific expression of subunits or other associated proteins modify the effect of H2S on BKCa channels. The demonstration that H2S activates EC BKCa channels (34) led us to test whether EC BKCa channels were the IbTx-sensitive target of H2S in mesenteric arteries. We used luminal application of IbTx to specifically target EC BKCa channels as previously described (6). Importantly, luminal application of IbTx did not significantly affect VSMC Em although abluminal application of the same concentration depolarized VSMC (Fig. 1C). In contrast, luminal application of IbTx blocked NaHS-induced dilations similarly to abluminal application of the BKCa blocker (Fig. 1B). Furthermore, EC disruption also blocked NaHS dilation (Fig. 1A). We observed expression of both BKCa and the H2S-producing enzyme CSE in the endothelium of the mesenteric vasculature (Fig. 7). Thus multiple functional tests suggest EC BKCa channels are critically important in the vasodilatory response to H2S in small mesenteric arteries and that EC contain the primary target for this compound.
K+ channel activation has been demonstrated to increase EC [Ca2+] (11, 27). This finding, however, is a controversial issue and the opposite relationship, that of EC [Ca2+] activating K+ channels, has also been demonstrated (29). Recent studies demonstrated that NaHS increases microvascular and aortic EC [Ca2+] (19, 22); our findings are consistent with these studies (Fig. 2F). In conjunction with the current finding that NaHS-induced vasodilation requires intact endothelium and, specifically, EC BKCa channels, one of these two mechanisms, or perhaps both, appear to contribute to H2S mediated dilation. In either case, increased EC [Ca2+] should elevate the production of endothelial dilators, several of which have been shown to affect Ca2+ spark signaling. For example, NO increases spark frequency and EC denudation decreases basal spark activity in cerebral artery VSMC (18). The EC product CO also increases spark frequency and enhances coupling between sparks and “spontaneous transient outward currents” (STOCs) in cerebral artery VSMCs (8). Finally, the EC-derived Cyp2C product 11,12-EET increases spark frequency downstream of VSMC TRP vanilloid type 4 (TRPV4) channel activation in cerebral arteries (4), and cytochrome P-450 enzymes contribute to NaHS-mediated relaxation in the perfused mesenteric bed (3). Consistent with these last two observations, our data demonstrate that inhibiting Cyp2C with sulfaphenazole blocks NaHS-induced dilation (Fig. 3A) and hyperpolarization of VSMC (Fig. 5B). However, mesenteric arteries display extensive myoendothelial electrical coupling through gap junctions (25). Thus it is possible that the endothelium-dependent vasodilation seen in these experiments occurs without a diffusible factor released from the EC. Furthermore, the Cyp2C product 11,12-EET could act on EC TRPV4 channels to mediate its effect (29), suggesting the need for future experiments to address these questions.
A major downstream target for Cyp2C products is VSMC BKCa channel activation by Ca2+ sparks, and we further investigated this pathway. Rather than increasing global intracellular [Ca2+], Ca2+ sparks increase [Ca2+] locally in the subsarcolemmal space to increase the open probability of BKCa channels, measured electrophysiologically as STOCs (21). Therefore, the net effect of Ca2+ sparks is Em hyperpolarization, reduced open probability of VGCCs, and a reduction in global intracellular [Ca2+] (9, 21).
We observed that concentrations of NaHS that cause dilation also increase VSMC Ca2+ spark activity (Fig. 2C) in pressurized arteries but this activation is absent in arteries with abraded EC (Fig. 4A). This is consistent with H2S releasing an endothelial factor that subsequently activates Ca2+ spark activity rather than acting directly on the VSMC as recently described in cerebral arteries (15). Sulfaphenazole administration and luminal IbTx administration abolished NaHS stimulation of increased spark frequency (Fig. 4A). Thus NaHS dilation in small mesenteric arteries appears to be at least partly mediated through enhanced VSMC Ca2+ spark activity dependent on EC BKCa channels and Cyp2C activation. In EC-disrupted arteries, NaHS in fact reduced Ca2+ spark frequency, suggesting H2S may inhibit sparks in mesenteric VSMC through a mechanism separate from effects on EC BKCa channels and Cyp2C. This is in contrast to observations in cerebral arteries where a portion of the H2S-induced dilation appears to be caused by direct effects on VSMC SR to increase spark activity (15). However, similar to the studies in cerebral arteries, H2S minimally affected spark kinetics (Fig. 4, B and C). Importantly, both ryanodine and IbTx largely blocked NaHS-induced vasodilation (Figs. 1 and 3), as expected if Ca2+ spark activation of VSMC BKCa channels contributes to the H2S effect. However, dilation induced by concentrations of NaHS above 10 μmol/l is not inhibited by these treatments, similar to our previous observations (7) suggesting these concentrations of NaHS exert nonspecific effects.
We also observed a pressure effect on NaHS-induced dilation. In the current study arteries were held at 100 mmHg luminal pressure and preconstricted to ∼50% of resting diameter with PE. Under these conditions, NaHS induced a robust dilation (∼70%) at relatively low concentrations (1 μmol/l), whereas in arteries pressurized to 60 mmHg luminal pressure, NaHS elicited a much smaller dilation (7). This enhanced H2S-induced vasodilation with increasing luminal pressure is consistent with H2S acting as a hyperpolarizing agent and an endogenous inhibitor of myogenic tone (7, 15). That is, since increasing luminal pressure stimulates VSMC depolarization and myogenic tone, H2S-induced vasodilation is amplified at higher pressures, perhaps due to enhanced spark-hyperpolarization coupling at these pressures.
NaHS hyperpolarized VSMC Em by ∼7 mV (Fig. 5B). Consistent with this effect being mediated by Ca2+ spark activation, superfusing with ryanodine to block Ca2+ spark generation or with IbTx to block VSMC BKCa channels abolished the hyperpolarization (Fig. 5B). Luminal IbTx, sulfaphenazole and EC inactivation also prevented hyperpolarization, suggesting both EC and VSMC BKCa channels participate in H2S-induced VSMC hyperpolarization in intact arteries. The observation that outward K+ currents in EC were greatly diminished by BCA (Fig. 5C) provides additional support that endogenous H2S activates EC BKCa channels in an autocrine manner. Indeed, endogenous H2S activation of EC BKCa channels appears to account for about half of the resting BKCa current in mesenteric artery EC. This is consistent with recent reports that H2S can activate BKCa channels by a redox method (14) and that H2S increases the open probability of BKCa in cultured mesenteric artery endothelial cells (34).
Based on these studies, we propose that in small mesenteric arteries, H2S activates EC BKCa channels, increasing EC [Ca2+] to activate Cyp2C and TRPV4 to increase Ca2+ spark activity in VSMC (4). VSMC sparks then activate VSMC BKCa channels to promote vasodilation. This hypothesized pathway explains several apparently disparate observations of H2S dilation in small arteries. Specifically, it has been observed that H2S 1) activates BKCa channels in microvascular EC (34); 2) increases microvascular EC [Ca2+] (22); 3) dilates perfused mesenteric beds in a cytochrome P-450, KCa channel, and EC-dependent manner (2, 3, 3); and 4) hyperpolarizes small mesenteric artery VSMC Em in a BKCa-dependent manner (7). In this model, EC BKCa channels are the primary target of H2S in small mesenteric arteries.
It is not clear why abluminal NaHS activates EC BKCa channels but does not directly activate VSMC BKCa channels. We have described differences in Ca2+ sensitivity of BKCa channels in EC and VSMC (23, 24), directly demonstrating potential differences in regulation of BKCa channels in these two cell types. Specifically, EC BKCa channels display a greater Ca2+ sensitivity despite the apparent lack of functional BK-β1 subunits, and H2S may affect the moiety responsible for this difference. Future studies investigating the mechanism of H2S activation of EC BKCa channels and the molecular differences between VSMC and EC BKCa channels are needed to resolve these questions.
In conclusion, the present work demonstrates for the first time that exogenous and endogenous H2S act on the endothelium to activate VSMC Ca2+ sparks to elicit vasodilation, adding to and complementing the growing list of vascular effects of this molecule.
GRANTS
O. Jackson-Weaver was supported by National Heart, Lung, and Blood Institute (NHLBI) Grant HL-07736 and American Heart Association (AHA) Grant 10-PRE-4050028; J. M. Osmond by NHLBI Grant HL-07736 and AHA Grant 12-POST-8690008; M. A. Riddle by NHLBI Grant HL-07736; L. V. Gonzalez Bosc by NHLBI Grant HL-088151; B. R. Walker by NHLBI Grant HL-95640; and N. L. Kanagy by NHLBI Grant HL-82799.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: O.J.-W., J.O., L.V.G.B., B.R.W., and N.L.K. conception and design of research; O.J.-W., J.O., M.A.R., and J.S.N. performed experiments; O.J.-W., J.O., M.A.R., J.S.N., and N.L.K. analyzed data; O.J.-W., J.O., M.A.R., J.S.N., L.V.G.B., B.R.W., and N.L.K. interpreted results of experiments; O.J.-W., J.O., M.A.R., J.S.N., and N.L.K. prepared figures; O.J.-W. drafted manuscript; O.J.-W., J.O., J.S.N., L.V.G.B., B.R.W., and N.L.K. edited and revised manuscript; O.J.-W., J.O., M.A.R., J.S.N., L.V.G.B., B.R.W., and N.L.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to Carolyn Pace and Tamara Howard for conducting immunohistochemistry experiments.
REFERENCES
- 1. Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, Roviezzo F, Brancaleone V, Cirino G. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol 30: 1998–2004, 2010 [DOI] [PubMed] [Google Scholar]
- 2. Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol 287: H2316–H2323, 2004 [DOI] [PubMed] [Google Scholar]
- 3. d'Emmanuele d V, Sorrentino R, Coletta C, Mitidieri E, Rossi A, Vellecco V, Pinto A, Cirino G, Sorrentino R. Hydrogen sulfide-induced dual vascular effect involves arachidonic acid cascade in rat mesenteric arterial bed. J Pharmacol Exp Ther 337: 59–64, 2011 [DOI] [PubMed] [Google Scholar]
- 4. Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 97: 1270–1279, 2005 [DOI] [PubMed] [Google Scholar]
- 5. Earley S, Pastuszyn A, Walker BR. Cytochrome p-450 epoxygenase products contribute to attenuated vasoconstriction after chronic hypoxia. Am J Physiol Heart Circ Physiol 285: H127–H136, 2003 [DOI] [PubMed] [Google Scholar]
- 6. Hughes JM, Riddle MA, Paffett ML, Gonzalez Bosc LV, Walker BR. Novel role of endothelial BKCa channels in altered vasoreactivity following hypoxia. Am J Physiol Heart Circ Physiol 299: H1439–H1450, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jackson-Weaver O, Paredes DA, Gonzalez Bosc LV, Walker BR, Kanagy NL. Intermittent hypoxia in rats increases myogenic tone through loss of hydrogen sulfide activation of large-conductance Ca2+-activated potassium cChannels. Circ Res 108: 1439–1447, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jaggar JH, Leffler CW, Cheranov SY, Tcheranova D, ES , Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+-activated K+ channels. Circ Res 91: 610–617, 2002 [DOI] [PubMed] [Google Scholar]
- 9. Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 278: C235–C256, 2000 [DOI] [PubMed] [Google Scholar]
- 10. Ji G, Barsotti RJ, Feldman ME, Kotlikoff MI. Stretch-induced calcium release in smooth muscle. J Gen Physiol 119: 533–544, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kamouchi M, Droogmans G, Nilius B. Membrane potential as a modulator of the free intracellular Ca2+ concentration in agonist-activated endothelial cells. Gen Physiol Biophys 18: 199–208, 1999 [PubMed] [Google Scholar]
- 12. Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol 2: a003996, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang LH, Rong W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid Redox Signal 12: 1179–1189, 2010 [DOI] [PubMed] [Google Scholar]
- 14. Li Y, Zang Y, Fu S, Zhang H, Gao L, Li J. H2S relaxes vas deferens smooth muscle by modulating the large-conductance Ca2+-activated K+ (BKCa) channels via a redox mechanism. J Sex Med 9: 2806–2813, 2012 [DOI] [PubMed] [Google Scholar]
- 15. Liang GH, Xi Q, Leffler CW, Jaggar JH. Hydrogen sulfide activates Ca2+ sparks to induce cerebral arteriole dilation. J Physiol 590: 2709–2720, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y, Kalogeris T, Wang M, Zuidema MY, Wang Q, Dai H, Davis MJ, Hill MA, Korthuis RJ. Hydrogen sulfide preconditioning or neutrophil depletion attenuates ischemia-reperfusion-induced mitochondrial dysfunction in rat small intestine. Am J Physiol Gastrointest Liver Physiol 302: G44–G54, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mandala M, Heppner TJ, Bonev AD, Nelson MT. Effect of endogenous and exogenous nitric oxide on calcium sparks as targets for vasodilation in rat cerebral artery. Nitric Oxide 16: 104–109, 2007 [DOI] [PubMed] [Google Scholar]
- 19. Moccia F, Bertoni G, Pla AF, Dragoni S, Pupo E, Merlino A, Mancardi D, Munaron L, Tanzi F. Hydrogen sulfide regulates intracellular Ca2+ concentration in endothelial cells from excised rat aorta. Curr Pharm Biotechnol 12: 1416–1426, 2011 [DOI] [PubMed] [Google Scholar]
- 20. Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal 2: ra72, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science 270: 633–637, 1995 [DOI] [PubMed] [Google Scholar]
- 22. Pupo E, Pla AF, Avanzato D, Moccia F, Cruz JE, Tanzi F, Merlino A, Mancardi D, Munaron L. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Radic Biol Med 51: 1765–1773, 2011 [DOI] [PubMed] [Google Scholar]
- 23. Riddle MA, Hughes JM, Walker BR. Role of caveolin-1 in endothelial BKCa channel regulation of vasoreactivity. Am J Physiol Cell Physiol 301: C1404–C1414, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Riddle MA, Walker BR. Regulation of endothelial BK channels by heme oxygenase-derived carbon monoxide and caveolin-1. Am J Physiol Cell Physiol 303: C92–C101, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ Res 90: 1108–1113, 2002 [DOI] [PubMed] [Google Scholar]
- 26. Schleifenbaum J, Kohn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T, Crean CS, Luft FC, Huang Y, Schubert R, Gollasch M. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens 28: 1875–1882, 2010 [DOI] [PubMed] [Google Scholar]
- 27. Sheng JZ, Ella S, Davis MJ, Hill MA, Braun AP. Openers of SKCa and IKCa channels enhance agonist-evoked endothelial nitric oxide synthesis and arteriolar vasodilation. FASEB J 23: 1138–1145, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sitdikova GF, Weiger TM, Hermann A. Hydrogen sulfide increases calcium-activated potassium (BK) channel activity of rat pituitary tumor cells. Pflügers Arch 459: 389–397, 2010 [DOI] [PubMed] [Google Scholar]
- 29. Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336: 597–601, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Telezhkin V, Brazier SP, Cayzac S, Muller CT, Riccardi D, Kemp PJ. Hydrogen sulfide inhibits human BKCa channels. Adv Exp Med Biol 648: 65–72, 2009 [DOI] [PubMed] [Google Scholar]
- 31. Telezhkin V, Brazier SP, Cayzac SH, Wilkinson WJ, Riccardi D, Kemp PJ. Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir Physiol Neurobiol 172: 169–178, 2010 [DOI] [PubMed] [Google Scholar]
- 32. Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322: 587–590, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J 20: 6008–6016, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zuidema MY, Yang Y, Wang M, Kalogeris T, Liu Y, Meininger CJ, Hill MA, Davis MJ, Korthuis RJ. Antecedent hydrogen sulfide elicits an anti-inflammatory phenotype in postischemic murine small intestine: role of BK channels. Am J Physiol Heart Circ Physiol 299: H1554–H1567, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]